Pichia pastoris is a Suitable Host for the Heterologous Expression of Predicted Class I and Class II Hydrophobins for Discovery, Study, and Application in Biotechnology

Abstract
1. Introduction
2. Materials and Methods
2.1. Protein Sequence Analyses
2.2. Strain Construction and Expression Level Screening
2.3. Flask Protein Production
2.4. Bioreactor Protein Production
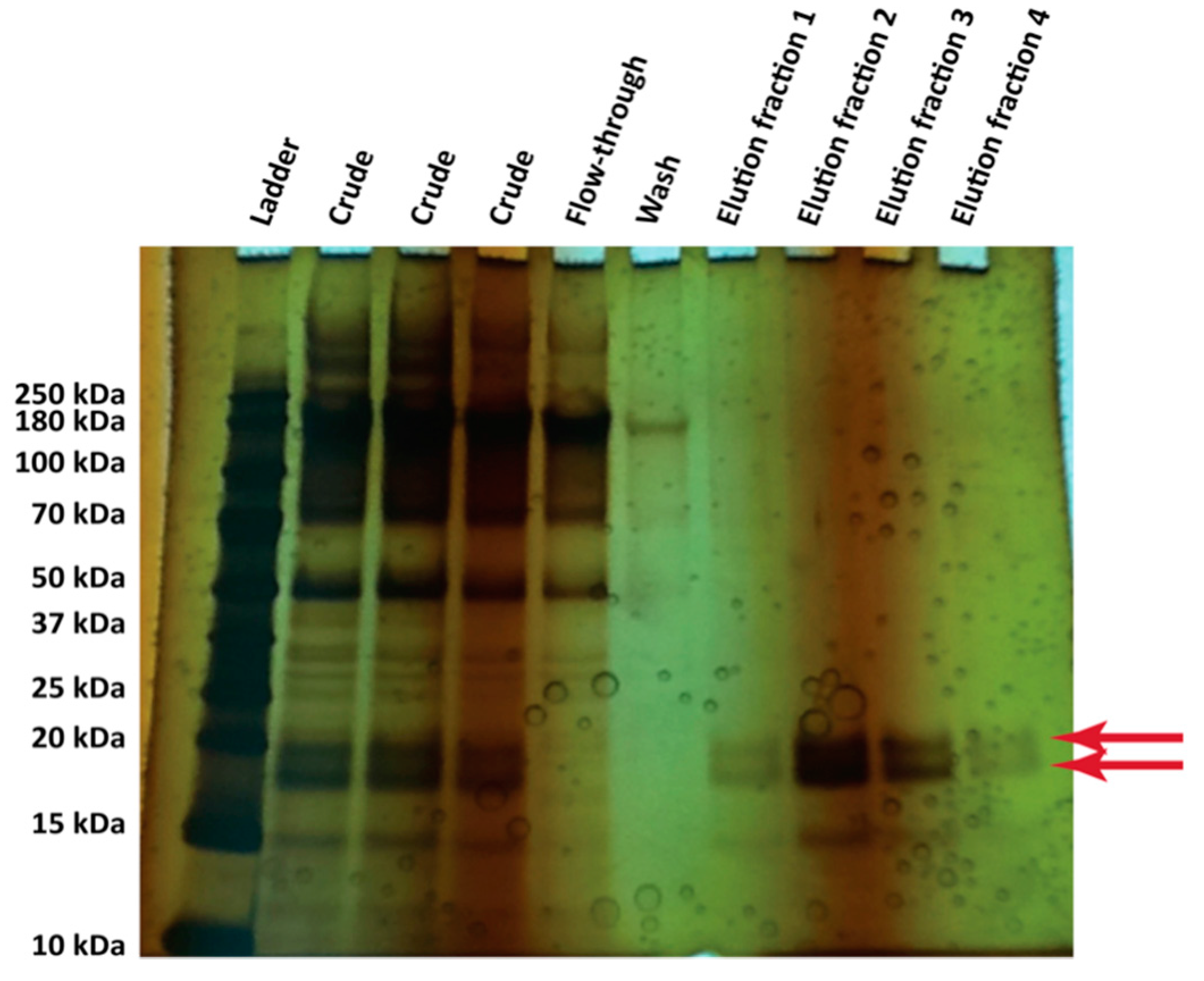
2.5. Nickel Affinity Purification of Hydrophobins
2.6. SDS-PAGE
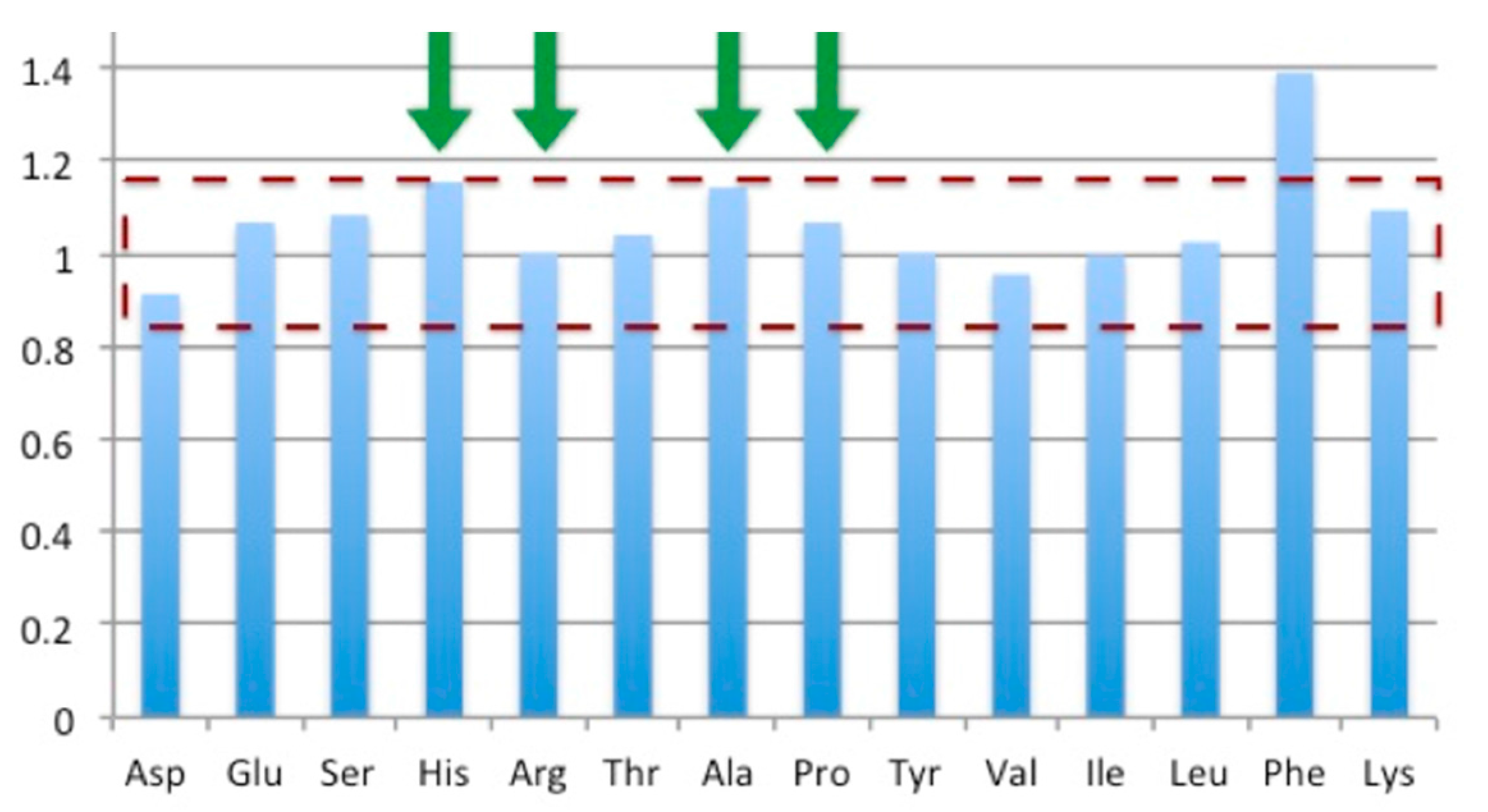
2.7. Protein Concentration Determination
3. Results and Discussion
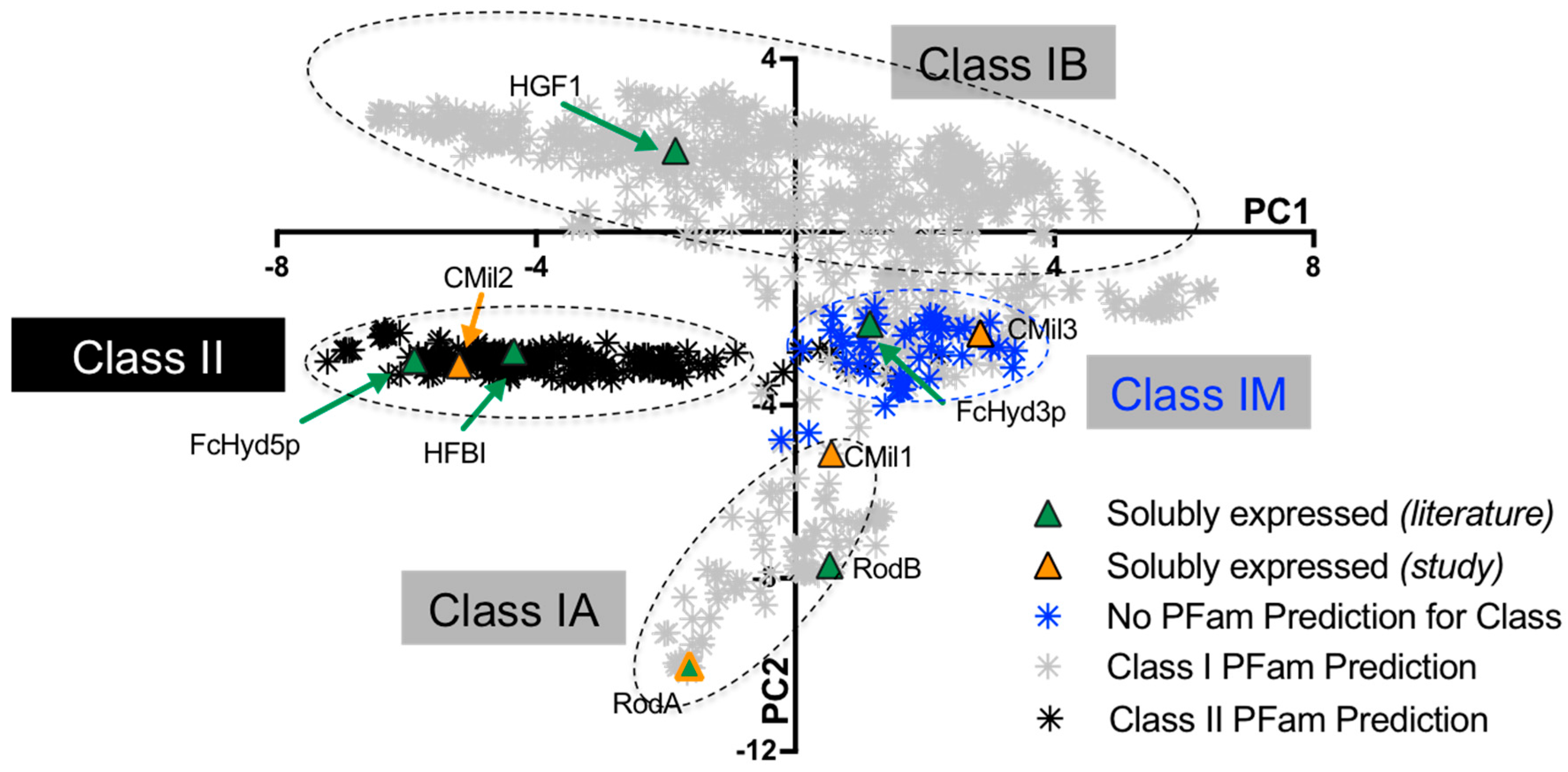
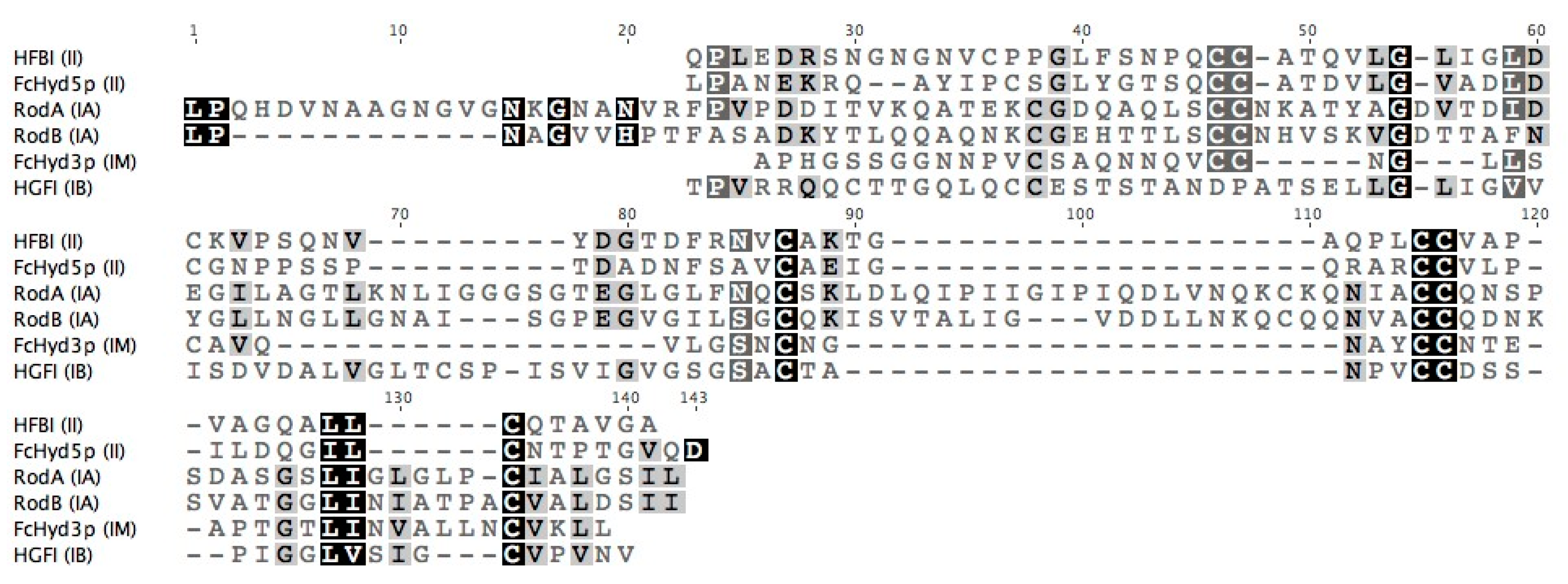
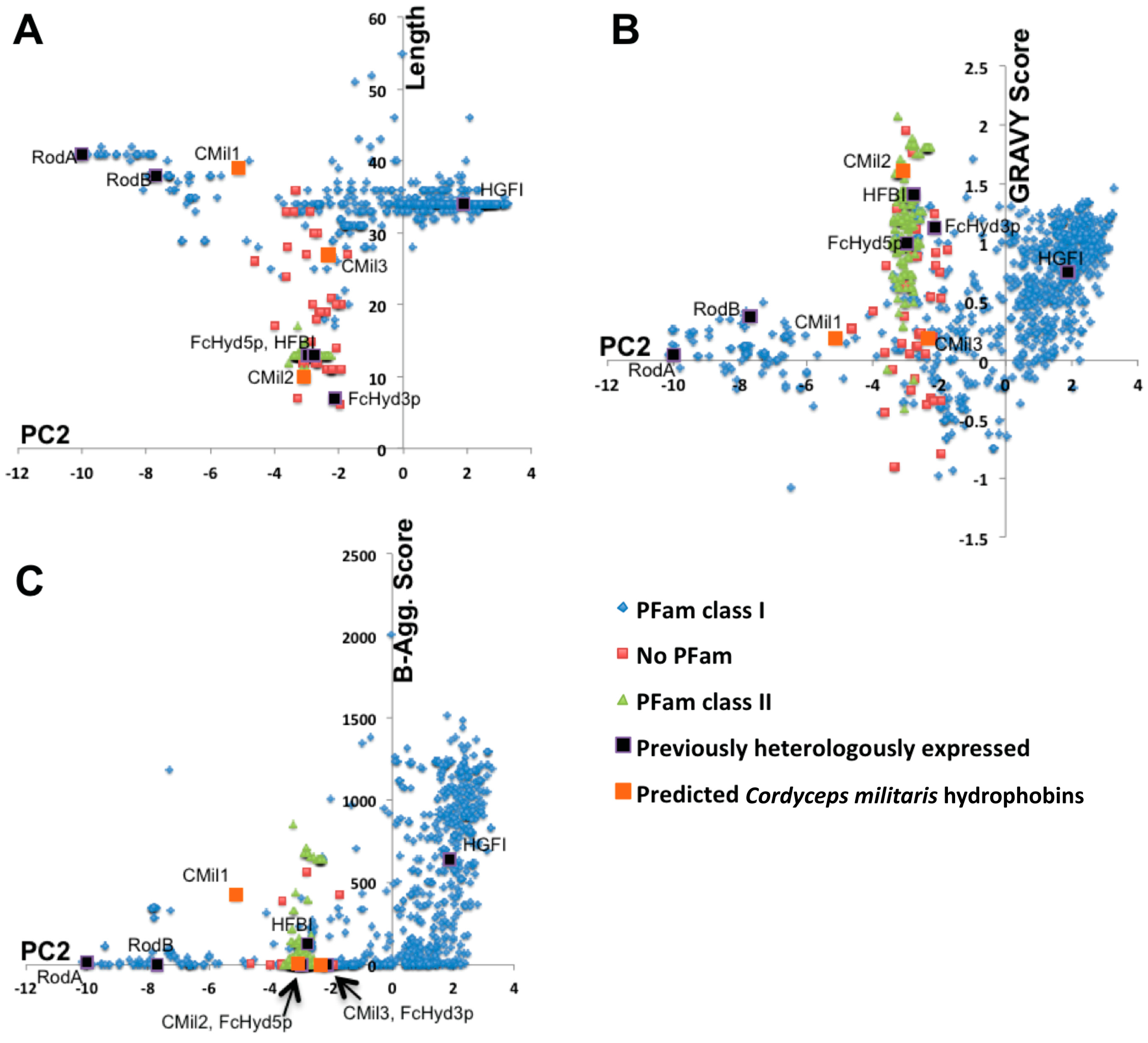
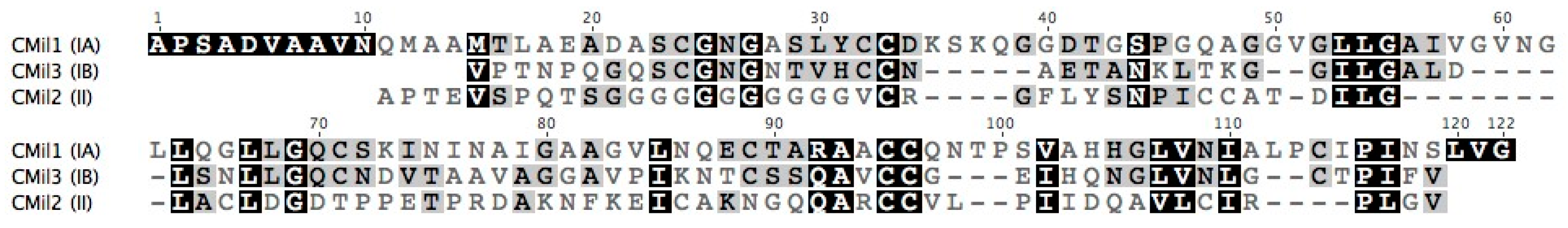
3.1. Selecting Hydrophobin Targets for Expression in P. pastoris
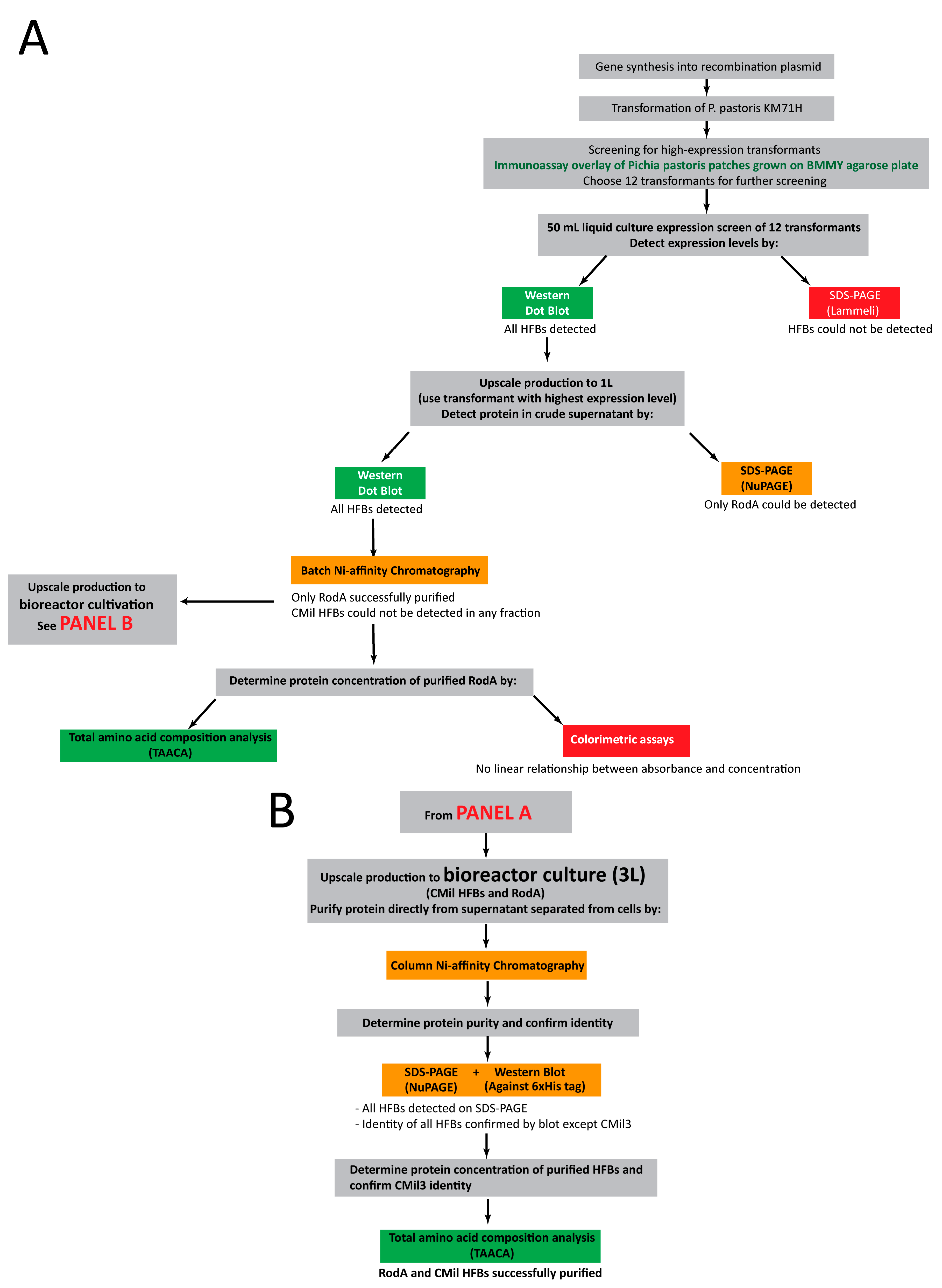
3.2. Approach to Recombinant Production of Selected Hydrophobins
3.3. Initial Screens of Hydrophobin Production are Enabled through Dot Blot Analyses
3.4. Protein Production in 500 mL Cultures in Flasks
3.5. Bioreactor Production
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Chambergo, F.S.; Valencia, E.Y. Fungal biodiversity to biotechnology. Appl. Microbiol. Biotechnol. 2016, 100, 2567–2577. [Google Scholar] [CrossRef] [PubMed]
- Saloheimo, M.; Paloheimo, M.; Hakola, S.; Pere, J.; Swanson, B.; Nyyssonen, E.; Bhatia, A.; Ward, M.; Penttilä, M. Swollenin, a Trichoderma reesei protein with sequence similarity to the plant expansins, exhibits disruption activity on cellulosic materials. Eur. J. Biochem. 2002, 269, 4202–4211. [Google Scholar] [CrossRef] [PubMed]
- Andberg, M.; Penttilä, M.; Saloheimo, M. Swollenin from Trichoderma reesei exhibits hydrolytic activity against cellulosic substrates with features of both Endoglucanases and Cellobiohydrolases. Bioresour. Technol. 2015, 181, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Gourlay, K.; Hu, J.; Arantes, V.; Penttilä, M.; Saddler, J.N. The use of carbohydrate binding modules (CBMs) to monitor changes in fragmentation and cellulose fiber surface morphology during cellulase- and swollenin-induced deconstruction of lignocellulosic substrates. J. Biol. Chem. 2015, 290, 2938–2945. [Google Scholar] [CrossRef] [PubMed]
- Baccelli, I.; Luti, S.; Bernardi, R.; Scala, A.; Pazzagli, L. Cerato-platanin shows expansin-like activity on cellulosic materials. Appl. Microbiol. Biotechnol. 2014, 98, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Quiroz-Castañeda, R.E.; Martínez-Anaya, C.; Cuervo-Soto, L.I.; Segovia, L.; Folch-Mallol, J.L. Loosenin, a novel protein with cellulose-disrupting activity from Bjerkandera adusta. Microb. Cell Fact. 2011, 10. [Google Scholar] [CrossRef] [PubMed]
- Stringer, M.A.; Dean, R.A.; Sewall, T.C.; Timberlake, W.E. Rodletless, a new Aspergillus developmental mutant induced by directed gene inactivation. Genes Dev. 1991, 5, 1161–1171. [Google Scholar] [CrossRef] [PubMed]
- Thau, N.; Monod, M.; Crestani, B.; Rolland, C.; Tronchin, G.; Latge, J.P.; Paris, S. Rodletless mutants of Aspergillus fumigatus. Infect. Immun. 1994, 62, 4380–4388. [Google Scholar] [PubMed]
- Wessels, J.G.H.; de Vries, O.M.H.; Asgeirsdottir, S.A.; Schuren, F.H.J. Hydrophobin genes involved in formation of aerial hyphae and fruit bodies in Schizophyllum. Plant Cell 1991, 3, 793–799. [Google Scholar] [CrossRef] [PubMed]
- Wösten, H.A.; Schuren, F.H.; Wessels, J.G. Interfacial self-assembly of a hydrophobin into an amphipathic protein membrane mediates fungal attachment to hydrophobic surfaces. EMBO J. 1994, 13, 5848–5854. [Google Scholar] [PubMed]
- Wösten, H.A.B.; Van Wetter, M.A.; Lugones, L.G.; Van der Mei, H.C.; Busscher, H.J.; Wessels, J.G.H. How a fungus escapes the water to grow into the air. Curr. Biol. 1999, 9, 85–88. [Google Scholar] [CrossRef]
- Pham, C.L.L.; Rey, A.; Lo, V.; Soulès, M.; Ren, Q.; Meisl, G.; Knowles, T.P.J.; Kwan, A.H.; Sunde, M. Self-assembly of MPG1, a hydrophobin protein from the rice blast fungus that forms functional amyloid coatings, occurs by a surface-driven mechanism. Sci. Rep. 2016, 6, 25288. [Google Scholar] [CrossRef] [PubMed]
- Khalesi, M.; Gebruers, K.; Derdelinckx, G. Recent advances in fungal hydrophobin towards using in industry. Protein J. 2015. [Google Scholar] [CrossRef] [PubMed]
- Piscitelli, A.; Cicatiello, P.; Gravagnuolo, A.M.; Sorrentino, I.; Pezzella, C.; Giardina, P. Applications of functional amyloids from fungi: Surface modification by class I hydrophobins. Biomolecules 2017, 7, 45. [Google Scholar] [CrossRef] [PubMed]
- Wösten, H.A.B.; Scholtmeijer, K. Applications of hydrophobins: Current state and perspectives. Appl. Microbiol. Biotechnol. 2015, 99, 1587–1597. [Google Scholar] [CrossRef] [PubMed]
- Fang, G.; Tang, B.; Liu, Z.; Gou, J.; Zhang, Y.; Xu, H.; Tang, X. Novel hydrophobin-coated docetaxel nanoparticles for intravenous delivery: In vitro characteristics and in vivo performance. Eur. J. Pharm. Sci. 2014, 60, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Bimbo, L.M.; Mäkilä, E.; Raula, J.; Laaksonen, T.; Laaksonen, P.; Strommer, K.; Kauppinen, E.I.; Salonen, J.; Linder, M.B.; Hirvonen, J.; et al. Functional hydrophobin-coating of thermally hydrocarbonized porous silicon microparticles. Biomaterials 2011, 32, 9089–9099. [Google Scholar] [CrossRef] [PubMed]
- Cox, A.R.; Cagnol, F.; Russell, A.B.; Izzard, M.J. Surface properties of class II hydrophobins from Trichoderma Reesei and influence on bubble stability. Langmuir 2007, 23, 7995–8002. [Google Scholar] [CrossRef] [PubMed]
- Cox, A.R.; Aldred, D.L.; Russell, A.B. Exceptional stability of food foams using class II hydrophobin HFBII. Food Hydrocoll. 2009, 23, 366–376. [Google Scholar] [CrossRef]
- Li, X.; Hou, S.; Feng, X.; Yu, Y.; Ma, J.; Li, L. Patterning of neural stem cells on Poly(lactic-Co-Glycolic Acid) film modified by hydrophobin. Colloids Surf. B Biointerfaces 2009, 74, 370–374. [Google Scholar] [CrossRef] [PubMed]
- Qin, M.; Wang, L.; Feng, X.; Yang, Y.; Wang, R.; Wang, C.; Yu, L.; Shao, B.; Qiao, M. Bioactive surface modification of mica and poly (Dimethylsiloxane) with hydrophobins for protein immobilization. Langmuir 2007, 23, 4465–4471. [Google Scholar] [CrossRef] [PubMed]
- Weickert, U.; Wiesend, F.; Subkowski, T.; Eickhoff, A.; Reiss, G. Optimizing biliary stent patency by coating with hydrophobin alone or hydrophobin and antibiotics or heparin: An in vitro proof of principle study. Adv. Med. Sci. 2011, 56, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Bayry, J.; Aimanianda, V.; Guijarro, J.I.; Sunde, M.; Latgé, J.P. Hydrophobins-unique fungal proteins. PLoS Pathog. 2012, 8, e6–e9. [Google Scholar] [CrossRef] [PubMed]
- Sunde, M.; Kwan, A.H.Y.; Templeton, M.D.; Beever, R.E.; Mackay, J.P. Structural analysis of Hydrophobins. Micron 2008, 39, 773–784. [Google Scholar] [CrossRef] [PubMed]
- Gandier, J.-A.; Langelaan, D.N.; Won, A.; O’Donnell, K.; Grondin, J.L.; Spencer, H.L.; Wong, P.; Tillier, E.; Yip, C.; Smith, S.P.; et al. Characterization of a basidiomycota hydrophobin reveals the structural basis for a high-similarity Class I subdivision. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Zampieri, F.; Wösten, H.A.B.; Scholtmeijer, K. Creating surface properties using a palette of hydrophobins. Materials 2010, 3, 4607–4625. [Google Scholar] [CrossRef] [PubMed]
- Wohlleben, W.; Subkowski, T.; Bollschweiler, C.; von Vacano, B.; Liu, Y.; Schrepp, W.; Baus, U. Recombinantly produced hydrophobins from fungal analogues as highly surface-active performance proteins. Eur. Biophys. J. 2010, 39, 457–468. [Google Scholar] [CrossRef] [PubMed]
- Daly, R.; Hearn, M.T.W. Expression of heterologous proteins in Pichia pastoris: A useful experimental tool in protein engineering and production. J. Mol. Recognit. 2005, 18, 119–138. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, H.; Engelbrecht, J.; Brunak, S.; Von Heijne, G. Identification of prokaryotic and eukaryotic signal peptides and prediction of their cleavage sites artificial neural networks have been used for many biological. Protein Eng. 1997, 10, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Mistry, J.; Mitchell, A.L.; Potter, S.C.; Punta, M.; Qureshi, M.; Sangrador-Vegas, A.; et al. The Pfam protein Families database: Towards a more sustainable future. Nucleic Acids Res. 2016, 44, D279–D285. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.A.; Clamp, M.; Barton, G.J. Jalview Version 2-A multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [PubMed]
- Drummond, A.J.; Ashton, B.; Buxton, S.; Cheung, M.; Cooper, A.; Duran, C.; Field, M.; Heled, J.; Kearse, M.; Markowitz, S.; et al. Geneious v5.6. Available online: http://www.geneious.com (accessed on 3 April 2017).
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Duvaud, S.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. The Proteomics Protocols Handbook—Chapter 52: Protein Identification and Analysis Tools on the ExPASy Server; Humana Press: New York, NY, USA, 2005. [Google Scholar]
- Pande, V.S. A Universal TANGO? Nat. Biotechnol. 2004, 22, 1240–1241. [Google Scholar] [CrossRef] [PubMed]
- Stübner, M.; Lutterschmid, G.; Vogel, R.F.; Niessen, L. Heterologous expression of the hydrophobin FcHyd5p from Fusarium culmorum in Pichia pastoris and evaluation of its surface activity and contribution to gushing of carbonated beverages. Int. J. Food Microbiol. 2010, 141, 110–115. [Google Scholar] [CrossRef] [PubMed]
- Lutterschmid, G.; Muranyi, M.; Stübner, M.; Vogel, R.F.; Niessen, L. Heterologous expression of surface-active proteins from barley and filamentous fungi in Pichia pastoris and characterization of their contribution to beer gushing. Int. J. Food Microbiol. 2011, 147, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Kottmeier, K.; Günther, T.J.; Weber, J.; Kurtz, S.; Ostermann, K.; Rödel, G.; Bley, T. Constitutive expression of hydrophobin HFB1 from Trichoderma reesei in Pichia pastoris and its pre-purification by foam separation during cultivation. Eng. Life Sci. 2012, 12, 162–170. [Google Scholar] [CrossRef]
- Pedersen, M.H.; Borodina, I.; Moresco, J.L.; Svendsen, W.E.; Frisvad, J.C.; Søndergaard, I. High-yield production of hydrophobins RodA and RodB from Aspergillus fumigatus in Pichia pastoris. Appl. Microbiol. Biotechnol. 2011, 90, 1923–1932. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Feng, S.; Huang, Y.; Li, S.; Xu, H.; Zhang, X.; Bai, Y.; Qiao, M. Expression and characterization of a Grifola frondosa hydrophobin in Pichia pastoris. Protein Expr. Purif. 2010, 72, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Invitrogen. Easy Select Pichia Expression Kit User Manual; Invitrogen: Carlsbad, CA, USA, 2010. [Google Scholar]
- Mollerup, F.; Master, E. Influence of a Family 29 carbohydrate binding module on the recombinant production of Galactose oxidase in Pichia pastoris. Data Br. 2016, 6, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Scopes, R.K.; John, A.S. Current Protocols in Molecular Biology—Analysis of Proteins; Ausubel, F., Brend, R., Kingtson, R.E., Moore, D.D., Seidman, J.G., Smith, J.A., Struhl, K., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2006. [Google Scholar]
- Zheng, P.; Xia, Y.; Xiao, G.; Xiong, C.; Hu, X.; Zhang, S.; Zheng, H.; Huang, Y.; Zhou, Y.; Wang, S.; et al. Genome sequence of the insect pathogenic fungus Cordyceps militaris, a valued traditional Chinese medicine. Genome Biol. 2011, 12, R116. [Google Scholar] [CrossRef] [PubMed]
- Wichadakul, D.; Kobmoo, N.; Ingsriswang, S.; Tangphatsornruang, S.; Chantasingh, D.; Luangsa-Ard, J.J.; Eurwilaichitr, L. Insights from the Genome of Ophiocordyceps polyrhachis-furcata to pathogenicity and host specificity in insect fungi. BMC Genom. 2015, 16, 881. [Google Scholar] [CrossRef] [PubMed]
- Paifer, E.; Margolles, E.; Cremata, J.; Montesino, R.; Herrera, L.; Delgado, J.M. Efficient expression and secretion of recombinant Alpha amylase in Pichia pastoris using two different signal sequences. Yeast 1994, 10, 1415–1419. [Google Scholar] [CrossRef] [PubMed]
- Kwan, A.H.Y.; Winefield, R.D.; Sunde, M.; Matthews, J.M.; Haverkamp, R.G.; Templeton, M.D.; Mackay, J.P. Structural basis for rodlet assembly in fungal hydrophobins. Proc. Natl. Acad. Sci. USA 2006, 103, 3621–3626. [Google Scholar] [CrossRef] [PubMed]
- Hakanpää, J.; Paananen, A.; Askolin, S.; Nakari-Setälä, T.; Parkkinen, T.; Penttilä, M.; Linder, M.B.; Rouvinen, J. Atomic resolution structure of the HFBII hydrophobin, a self-assembling amphiphile. J. Biol. Chem. 2004, 279, 534–539. [Google Scholar] [CrossRef] [PubMed]
- Hakanpää, J.; Szilvay, G.R.; Kaljunen, H.; Maksimainen, M.; Linder, M.; Rouvinen, J. Two crystal structures of Trichoderma reesei hydrophobin HFBI—The structure of a protein amphiphile with and without detergent interaction. Protein Sci. 2006, 15, 2129–2140. [Google Scholar] [CrossRef] [PubMed]
- Ren, Q.; Kwan, A.H.; Sunde, M. Solution structure and interface-driven self-assembly of NC2, a New member of the class II hydrophobin proteins. Proteins Struct. Funct. Bioinform. 2014, 82, 990–1003. [Google Scholar] [CrossRef] [PubMed]
- Gersten, D.M.; George, D.G.; Johnston, D.A.; Zapolski, E.J. On the relationship of amino acid composition to silver staining of proteins in electrophoresis gels. Pept. Seq. Anal. 1991, 11, 409–414. [Google Scholar]
- Chevallet, M.; Luche, S.; Rabilloud, T. Silver staining of proteins in polyacrylamide gels. Nat. Protoc. 2006, 1, 1852–1858. [Google Scholar] [CrossRef] [PubMed]
- Sapan, C.V.; Lundblad, R.L.; Price, N.C. Colorimetric protein assay techniques. Biotechnol. Appl. Biochem. 1999, 29, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Gandier, J.A.; Master, E.R. Silver Staining of Hydrophobin Stacking Gels; University of Toronto Department of Chemical Engineering and Applied Chemistry: Toronto, ON, Canada, 2014. [Google Scholar]
- De Vries, O.M.H.; Fekkes, M.P.; Wösten, H.A.B.; Wessels, J.G.H. Insoluble hydrophobin complexes in the walls of Schizophyllum commune and other filamentous fungi. Arch. Microbiol. 1993, 159, 330–335. [Google Scholar] [CrossRef]
- Kurien, B.T.; Scofield, R.H. Western blotting. Methods 2015, 38, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Graham, D.R.M.; Garnham, C.P.; Fu, Q.; Robbins, J.; Van Eyk, J.E. Improvements in two-dimensional gel electrophoresis by utilizing a low cost “in-house” neutral pH sodium dodecyl sulfate-polyacrylamide gel electrophoresis system. Proteomics 2005, 5, 2309–2314. [Google Scholar] [CrossRef] [PubMed]
- Hamby, S.E.; Hirst, J.D. Prediction of glycosylation sites using random forests. BMC Bioinform. 2008, 9, 500. [Google Scholar] [CrossRef] [PubMed]
- Gandier, J.A.; Master, E.R. SDS-PAGE Gel of the Crude Supernatant of a Pichia pastoris Strain Expressing Secreted Hydrophobin Proteins; University of Toronto Department of Chemical Engineering and Applied Chemistry: Toronto, ON, Canada, 2014. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Class | Organism/Phylum | Yield * | Strain | Promoter | Tags | Culture Type **** | Notes on Protein Detection | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| FcHyd5p | II | Fusarium culmorum/ Ascomycota | Not reported | X33 | AOX1 ** | No tag C-terminal 6His | Flask | Protein could not be readily detected without immunoassay against 6His tag | [35] |
| FcHyd3p | IM | Fusarium culmorum/ Ascomycota | Not reported | X33 | AOX1 | No tag | Flask | Transformants were screened by RT-PCR for high expression as protein could not be readily detected on gel | [36] |
| HFBI | II | Trichoderma reesei/ Ascomycota | Not reported 120 mg/L | SMD1168H GS115 | GAP *** AOX1 | No tag No tag | Bioreactor Flask | Proteins detected by silver-staining of SDS-PAGE gels | [37] |
| RodA | IA | Aspergillus fumigatus/ Ascomycota | 329 mg/L | X33 | AOX1 | C-terminal 6His | Bioreactor | [38] | |
| RodB | IA | Aspergillus fumigatus/ Ascomycota | 262 mg/L | X33 | AOX1 | C-terminal 6His | Bioreactor | [38] | |
| HGFI | IB | Grifola frondosa/ Basidiomycota | 86 mg/L | GS115 | AOX1 | No tag | Flask | [39] |
| HFBI (II) | FcHyd5p (II) | RodA (IA) | RodB (IA) | FcHyd3p (IM) | HGFI (IB) | |
|---|---|---|---|---|---|---|
| HFBI (II) | - | 37 | 14 | 12 | 17 | 17 |
| FcHyd5p (II) | 37 | - | 13 | 12 | 13 | 11 |
| RodA (IA) | 14 | 13 | - | 39 | 14 | 13 |
| RodB (IA) | 12 | 12 | 39 | - | 18 | 17 |
| FcHyd3p (IM) | 17 | 13 | 14 | 18 | - | 16 |
| HGFI (IB) | 17 | 11 | 13 | 17 | 16 | - |
| CMil1 (IA) | CMil3 (IB) | CMil2 (II) | |
|---|---|---|---|
| CMil1 (IA) | - | 30 | 14 |
| CMil3 (IM) | 30 | - | 24 |
| CMil2 (II) | 14 | 24 | - |
| Protein | Yield * |
|---|---|
| RodA | 4.6 mg/L |
| CMil1 | 3.6 mg/L |
| CMil2 | 4.1 mg/L |
| CMil3 | 8.3 mg/L |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gandier, J.-A.; Master, E.R. Pichia pastoris is a Suitable Host for the Heterologous Expression of Predicted Class I and Class II Hydrophobins for Discovery, Study, and Application in Biotechnology. Microorganisms 2018, 6, 3. https://doi.org/10.3390/microorganisms6010003
Gandier J-A, Master ER. Pichia pastoris is a Suitable Host for the Heterologous Expression of Predicted Class I and Class II Hydrophobins for Discovery, Study, and Application in Biotechnology. Microorganisms. 2018; 6(1):3. https://doi.org/10.3390/microorganisms6010003
Chicago/Turabian StyleGandier, Julie-Anne, and Emma R. Master. 2018. "Pichia pastoris is a Suitable Host for the Heterologous Expression of Predicted Class I and Class II Hydrophobins for Discovery, Study, and Application in Biotechnology" Microorganisms 6, no. 1: 3. https://doi.org/10.3390/microorganisms6010003
APA StyleGandier, J.-A., & Master, E. R. (2018). Pichia pastoris is a Suitable Host for the Heterologous Expression of Predicted Class I and Class II Hydrophobins for Discovery, Study, and Application in Biotechnology. Microorganisms, 6(1), 3. https://doi.org/10.3390/microorganisms6010003