Transcriptomic Complexity of Aspergillus terreus Velvet Gene Family under the Influence of Butyrolactone I



Abstract
:1. Introduction
2. Materials and Methods
2.1. Strain and Chemicals
2.2. Culture Conditions
2.3. Addition of Butyrolactone I
2.4. RNA Extraction
2.5. Gene Expression Analysis Using Microarrays
2.6. Strand-Specific Transcriptome Sequencing
2.7. The Assembly of Strand-Specific RNA Sequence Data
2.8. Further Analysis of the Transcriptome Sequence Data
2.9. Accession Numbers
3. Results
3.1. Strand-Specific Whole Transcriptome Sequencing and Refinement of Genome-Wide Gene Expression Analysis of A. terreus Strain MUCL 38669
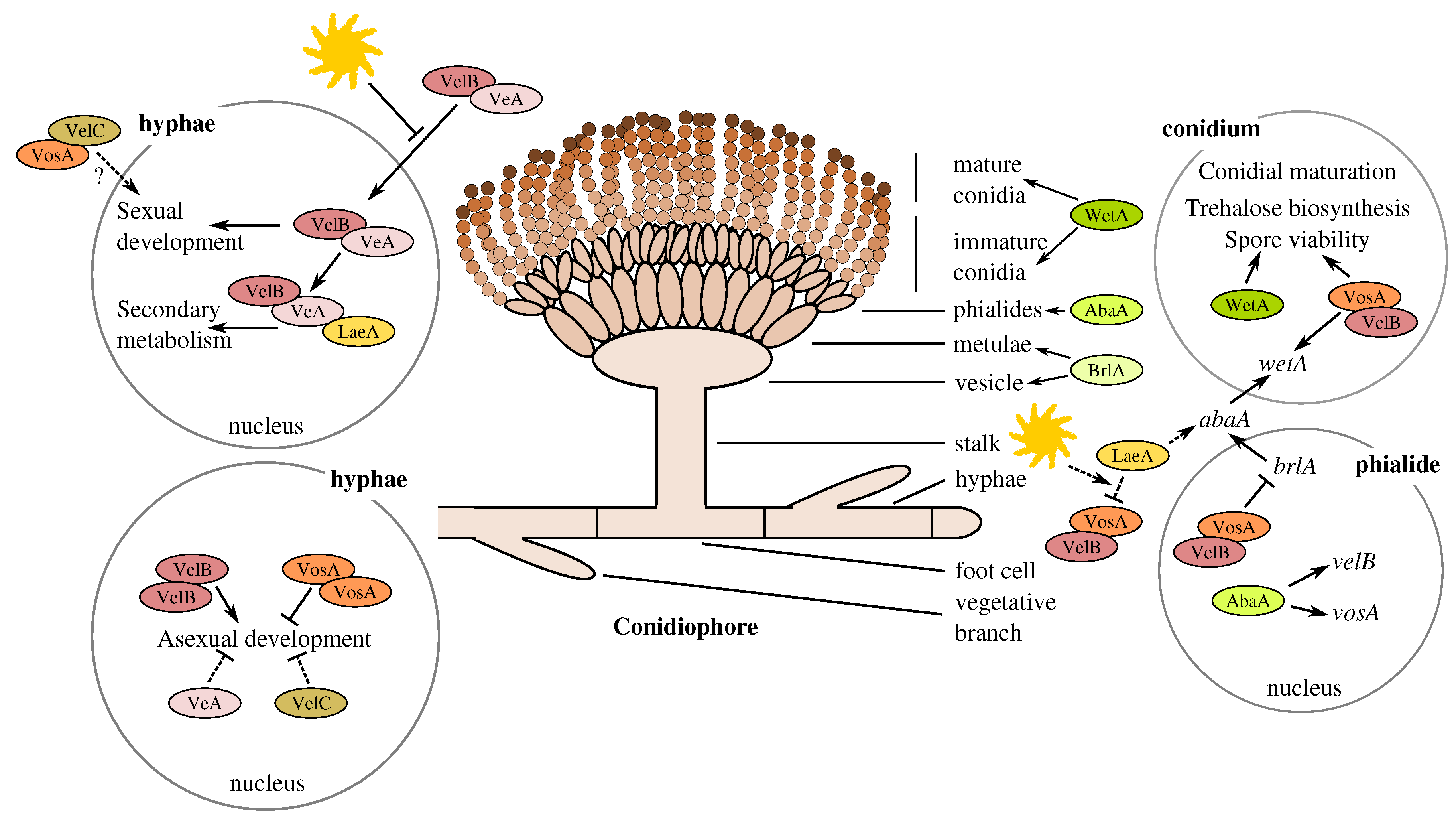
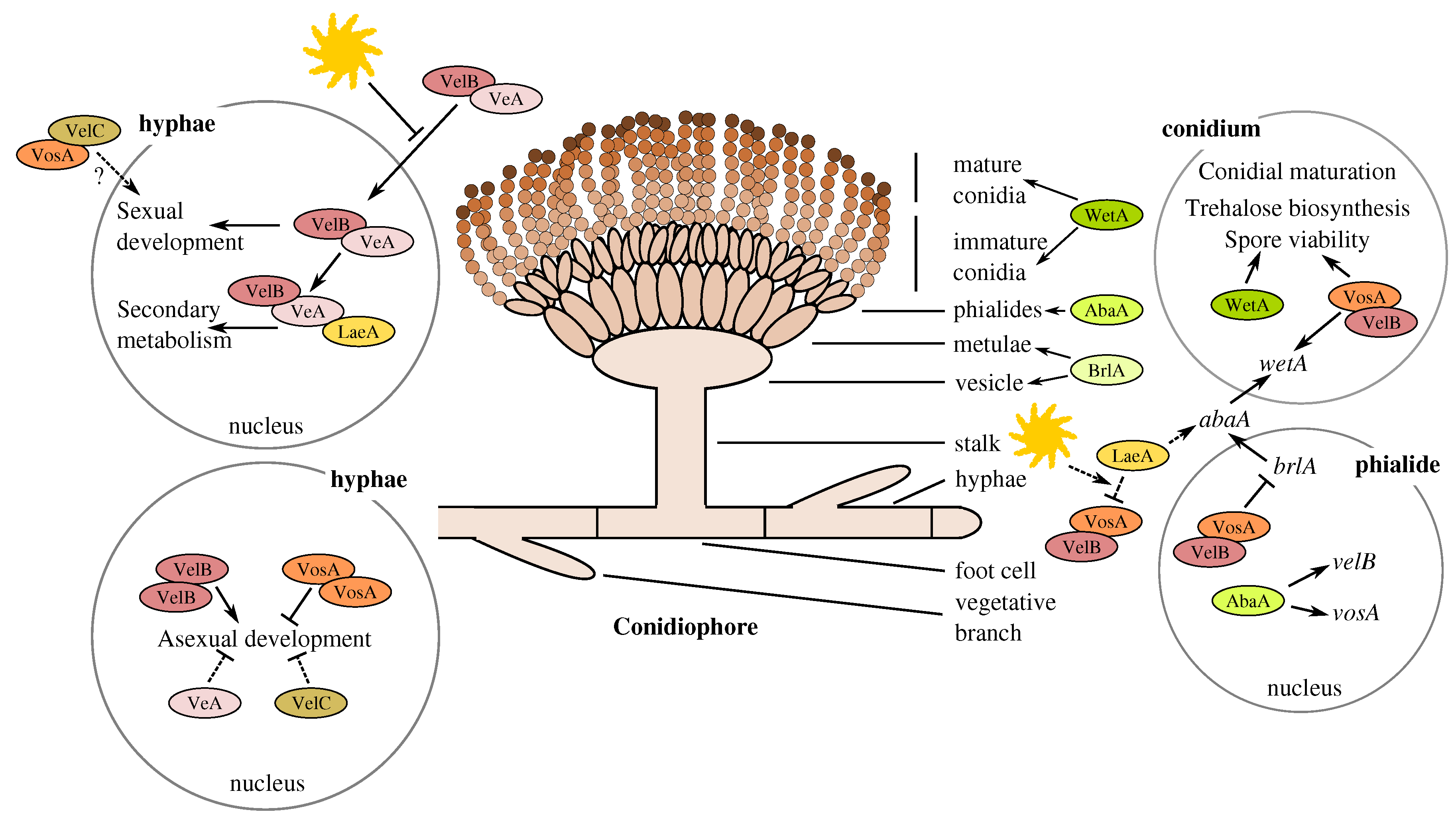
3.2. The Members of the A. terreus Velvet Family
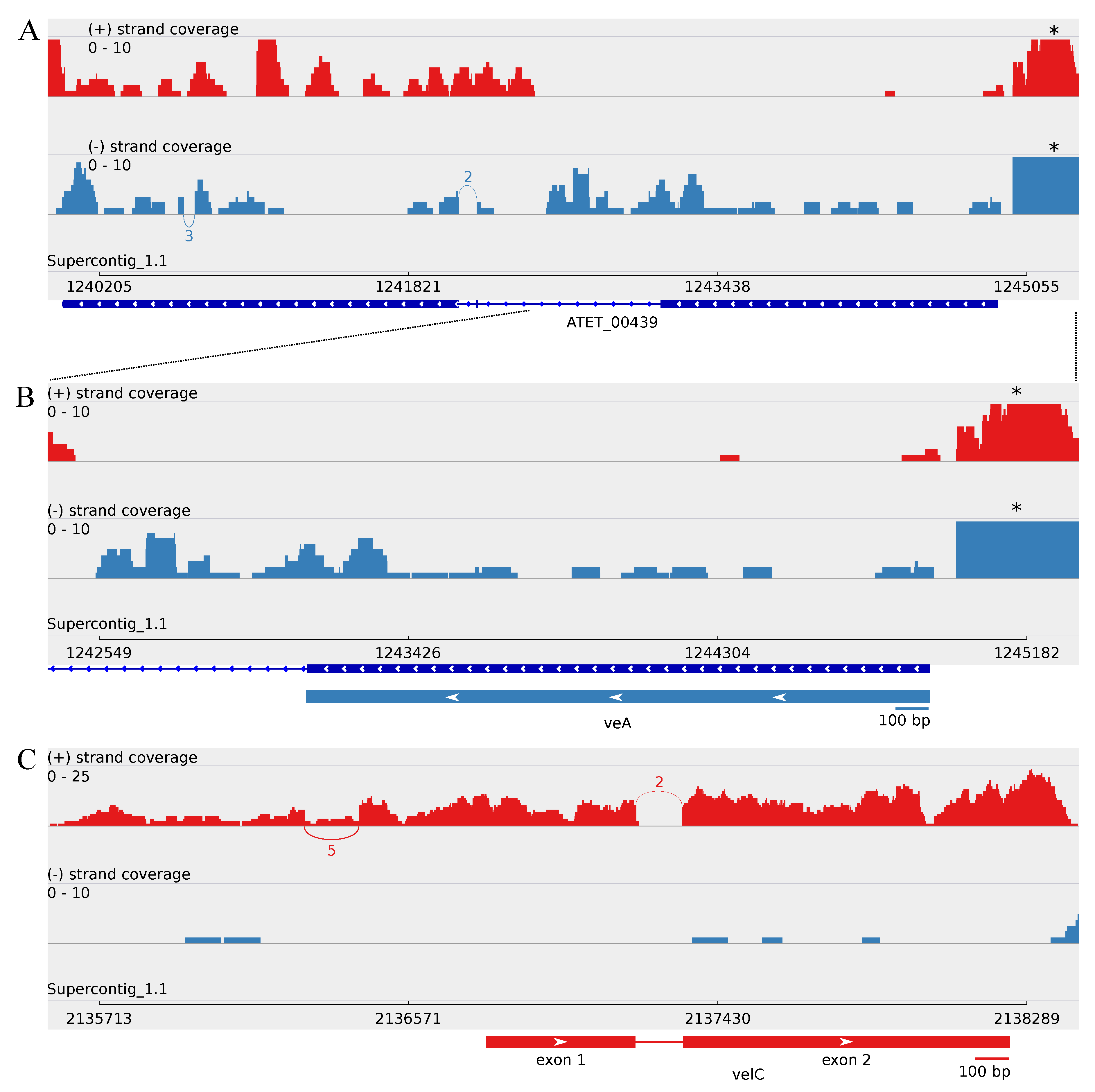
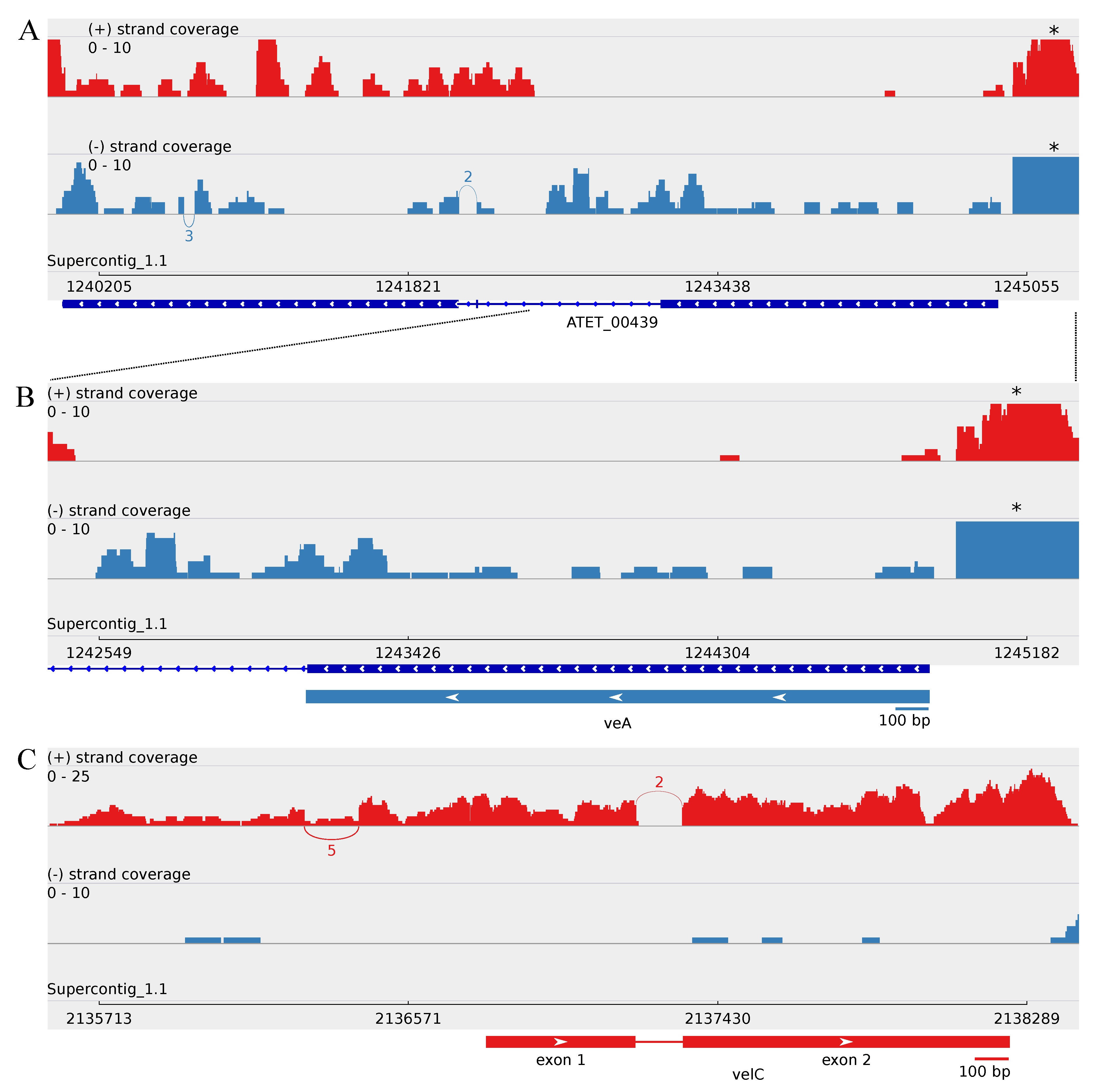
3.2.1. Revealing the A. terreus velC Gene
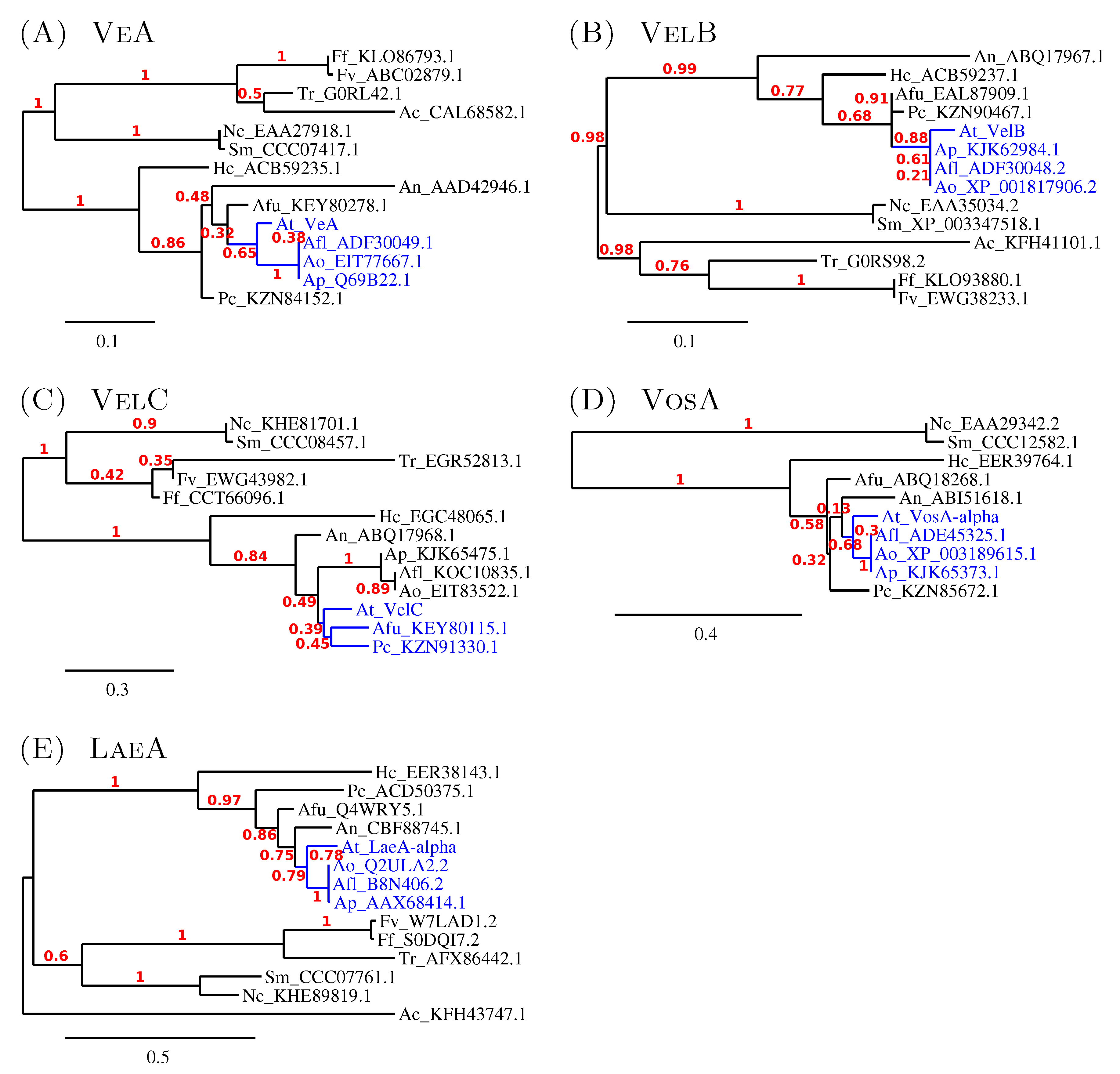
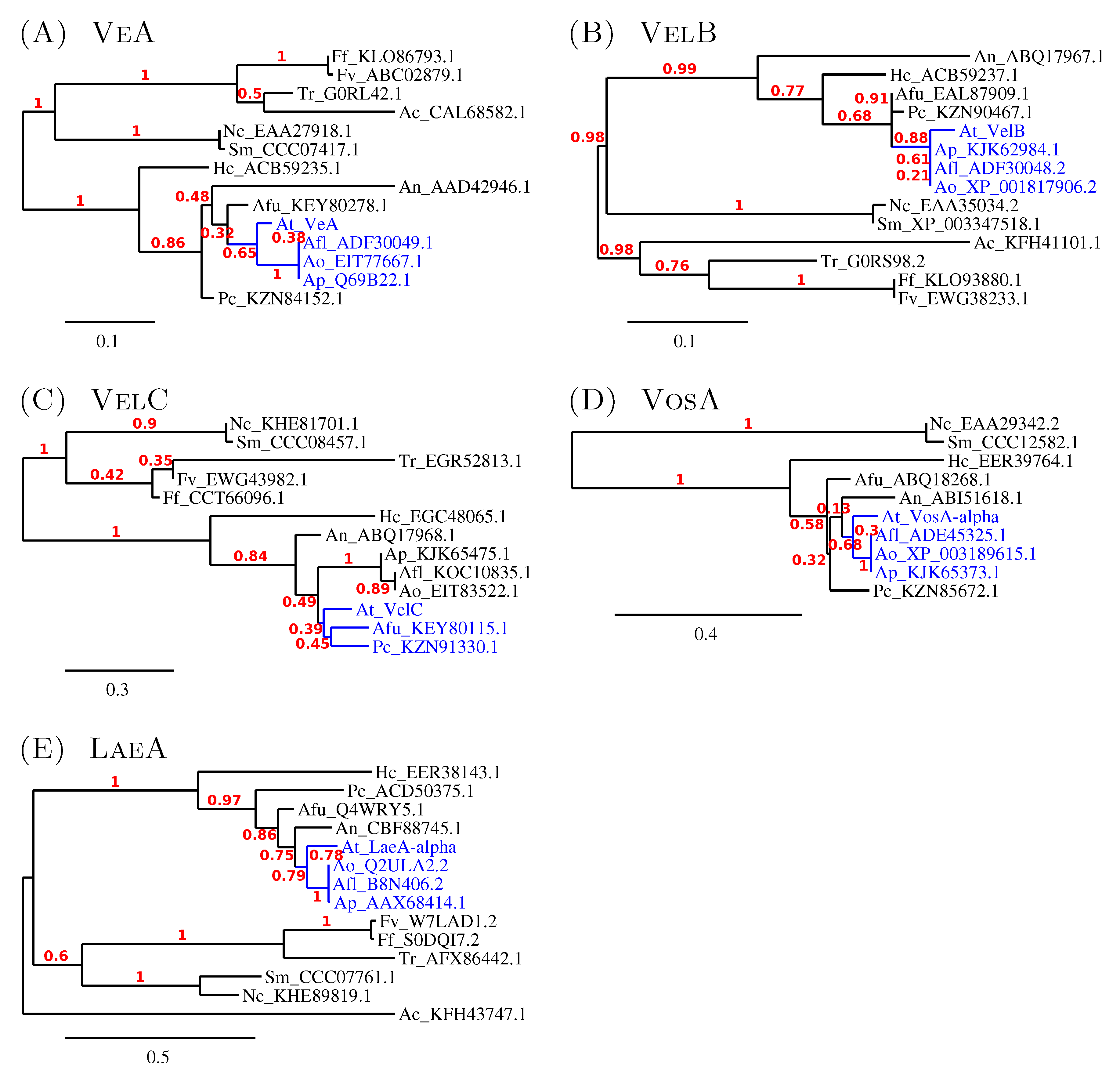
3.2.2. The Similarity amongst Velvet Complex Members of Some Ascomycota Fungi
3.3. Minor Updates of the A. terreus veA and velB Genes
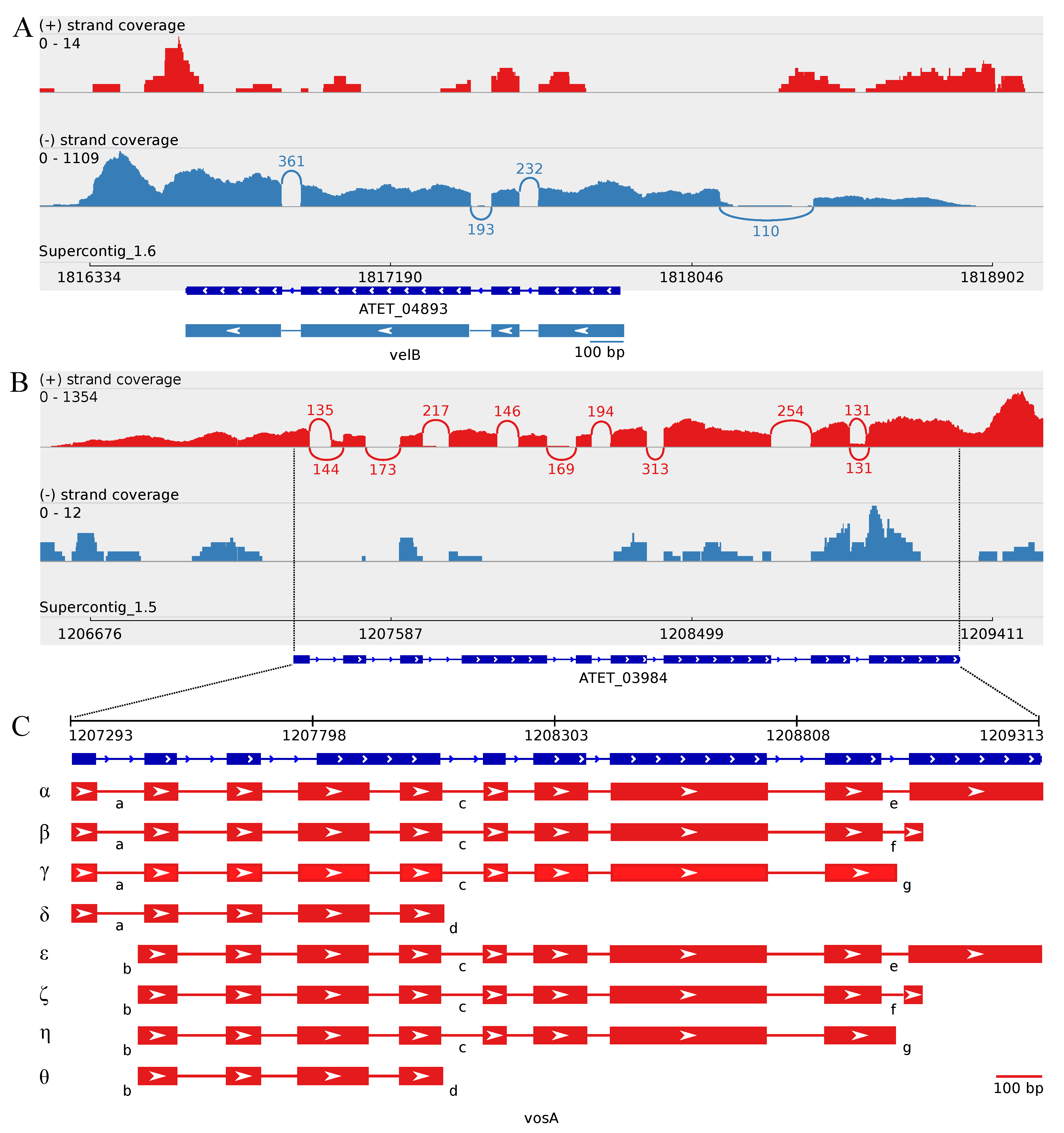
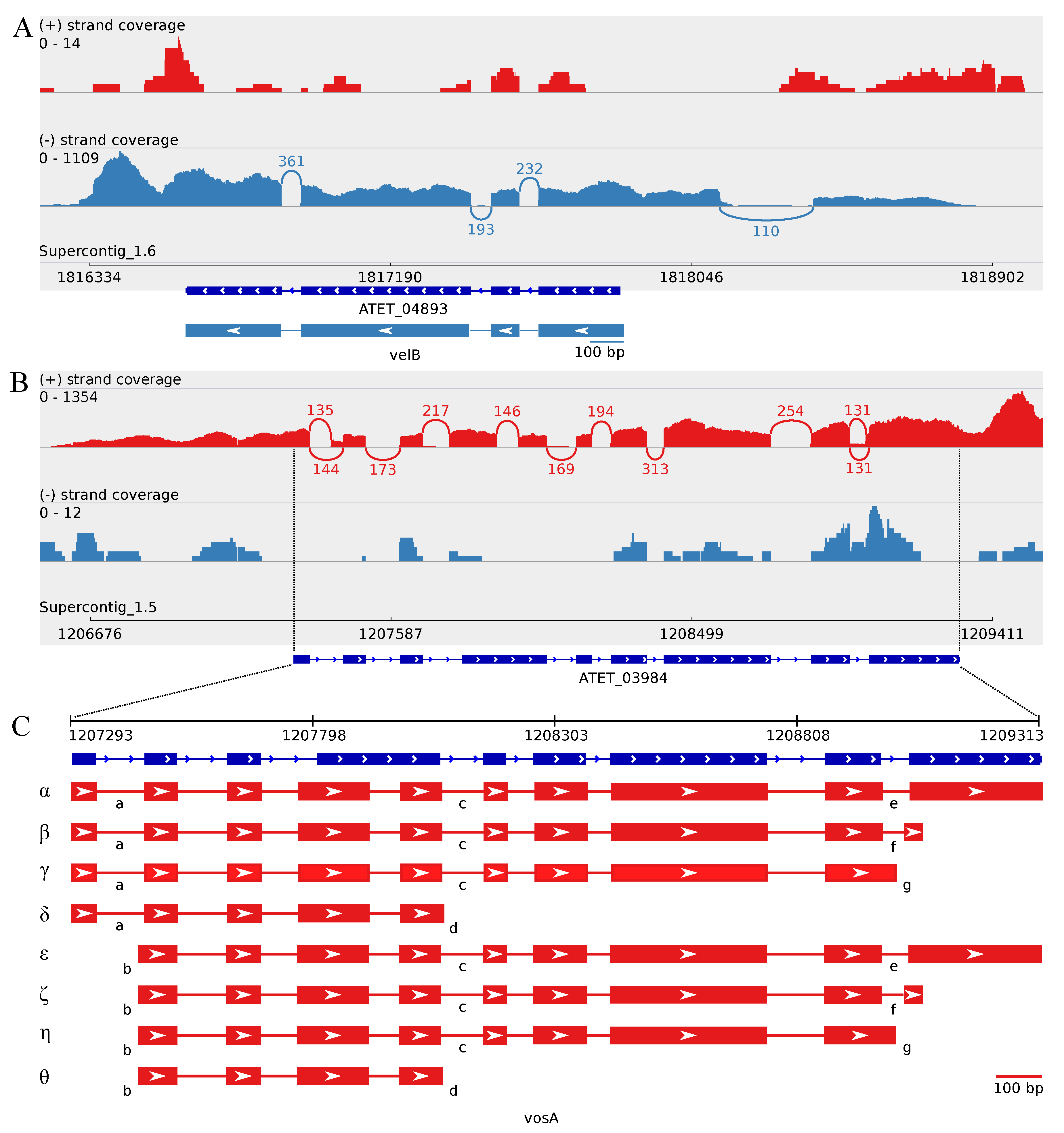
3.4. The Transcriptome Sequencing Revealed Numerous Splice Variants for vosA and laeA Orthologs during Enhanced Butyrolactone I Biogenesis
3.4.1. Splice Variants of the A. terreus vosA Ortholog
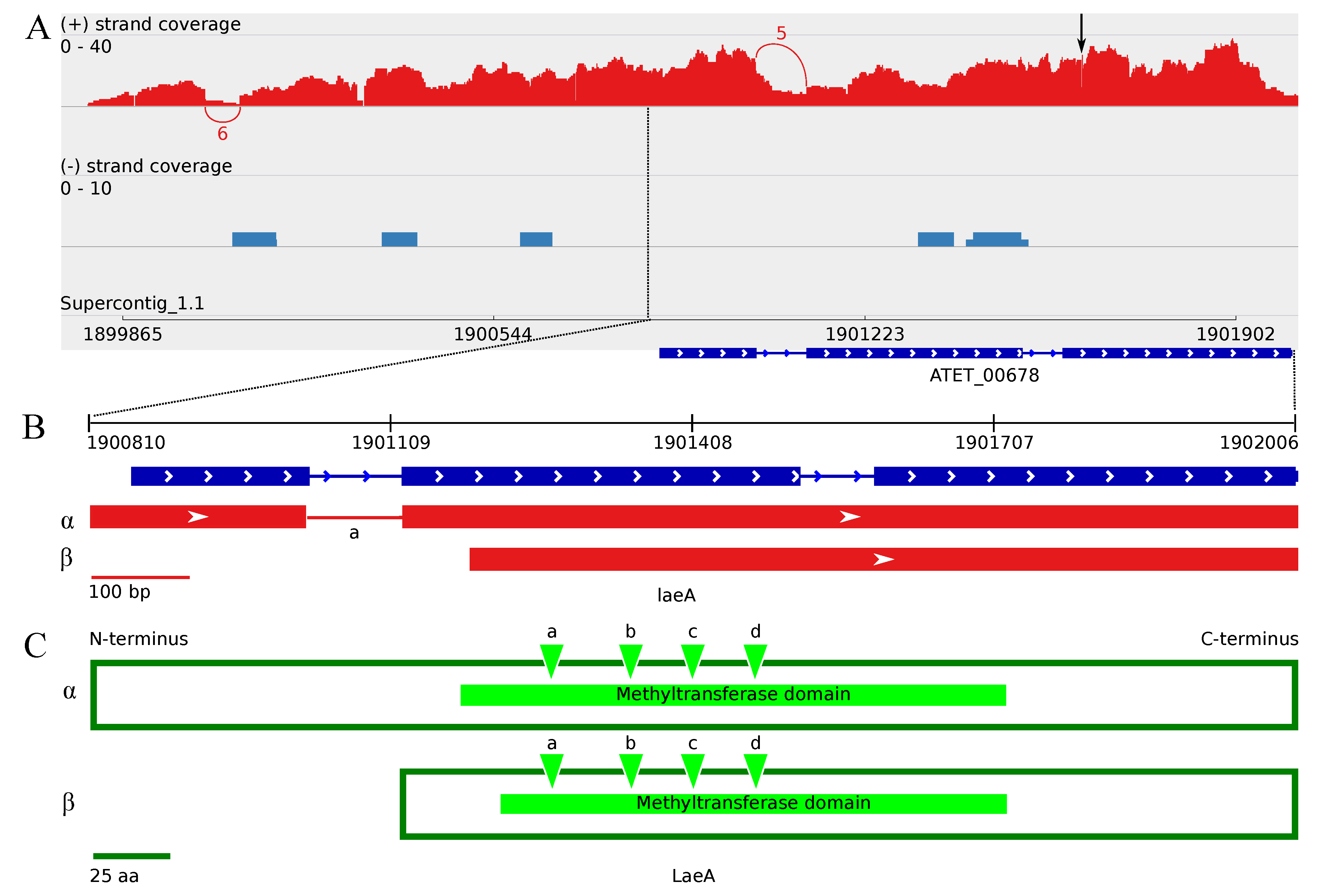
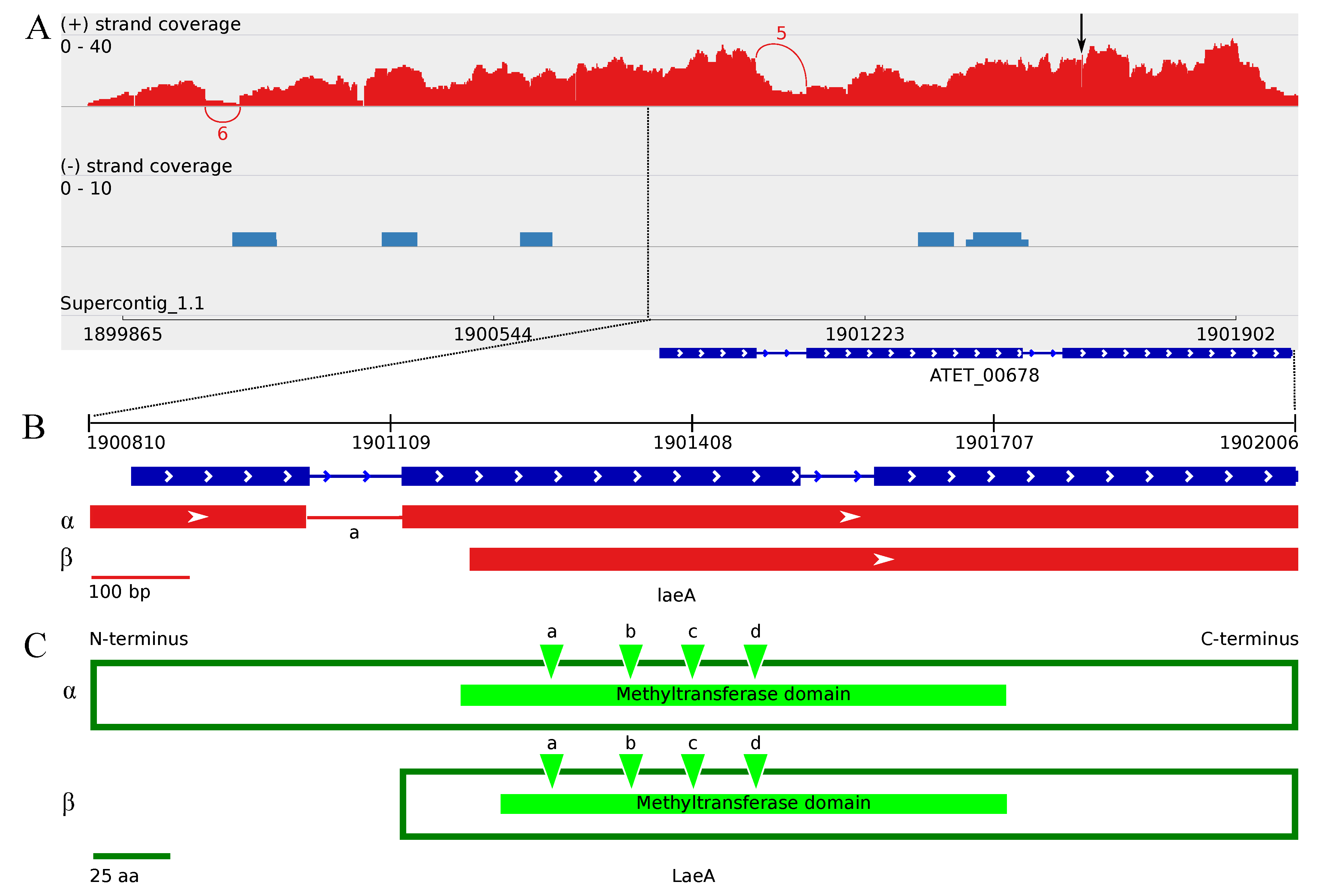
3.4.2. The Two Encoded Splice Variants of the A. terreus laeA Ortholog
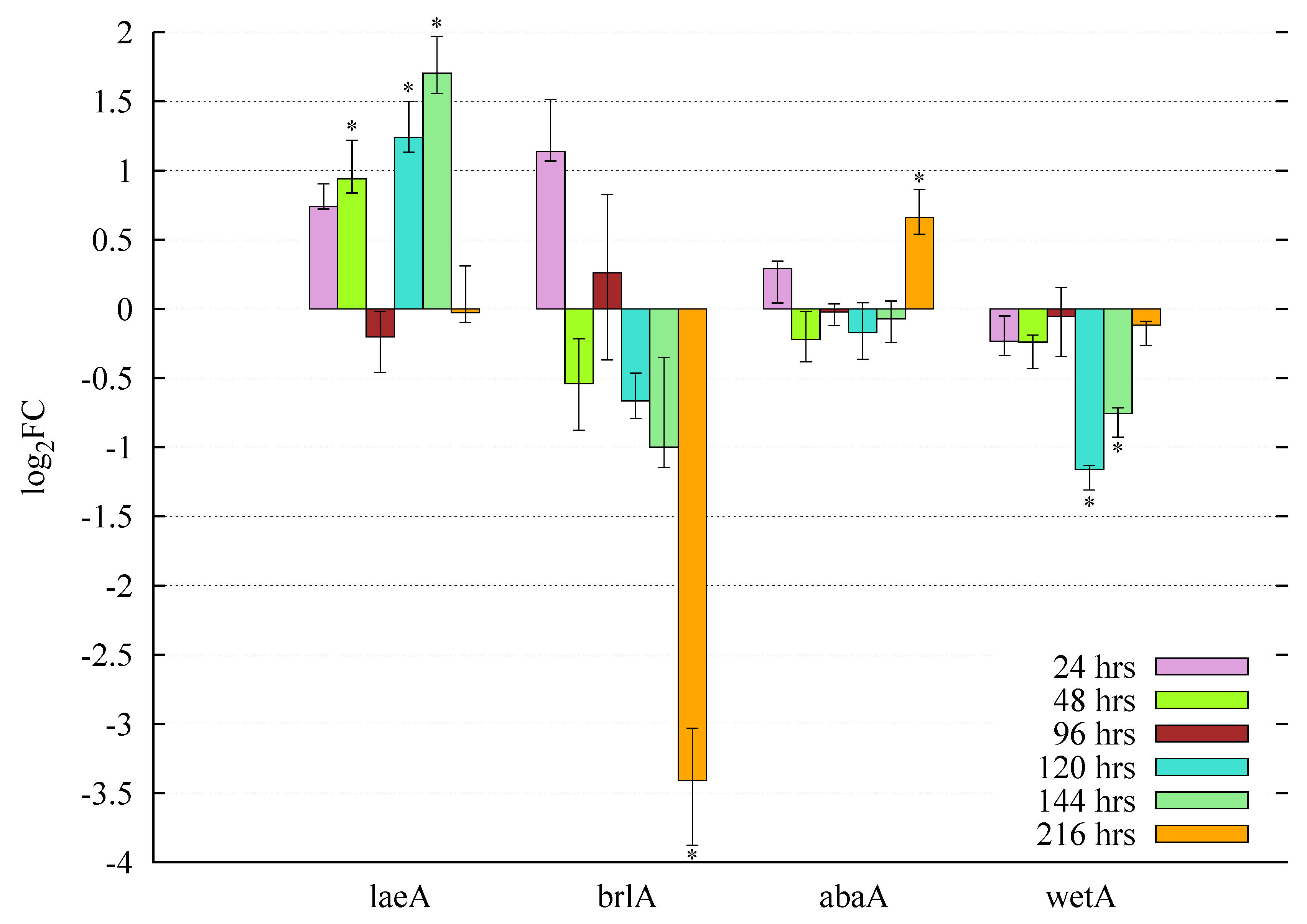
3.5. Gene Expression Profiles and Pooled Transcript Accumulation of the Velvet Family, laeA and Conidiation Core Regulators under Butyrolactone I Influence
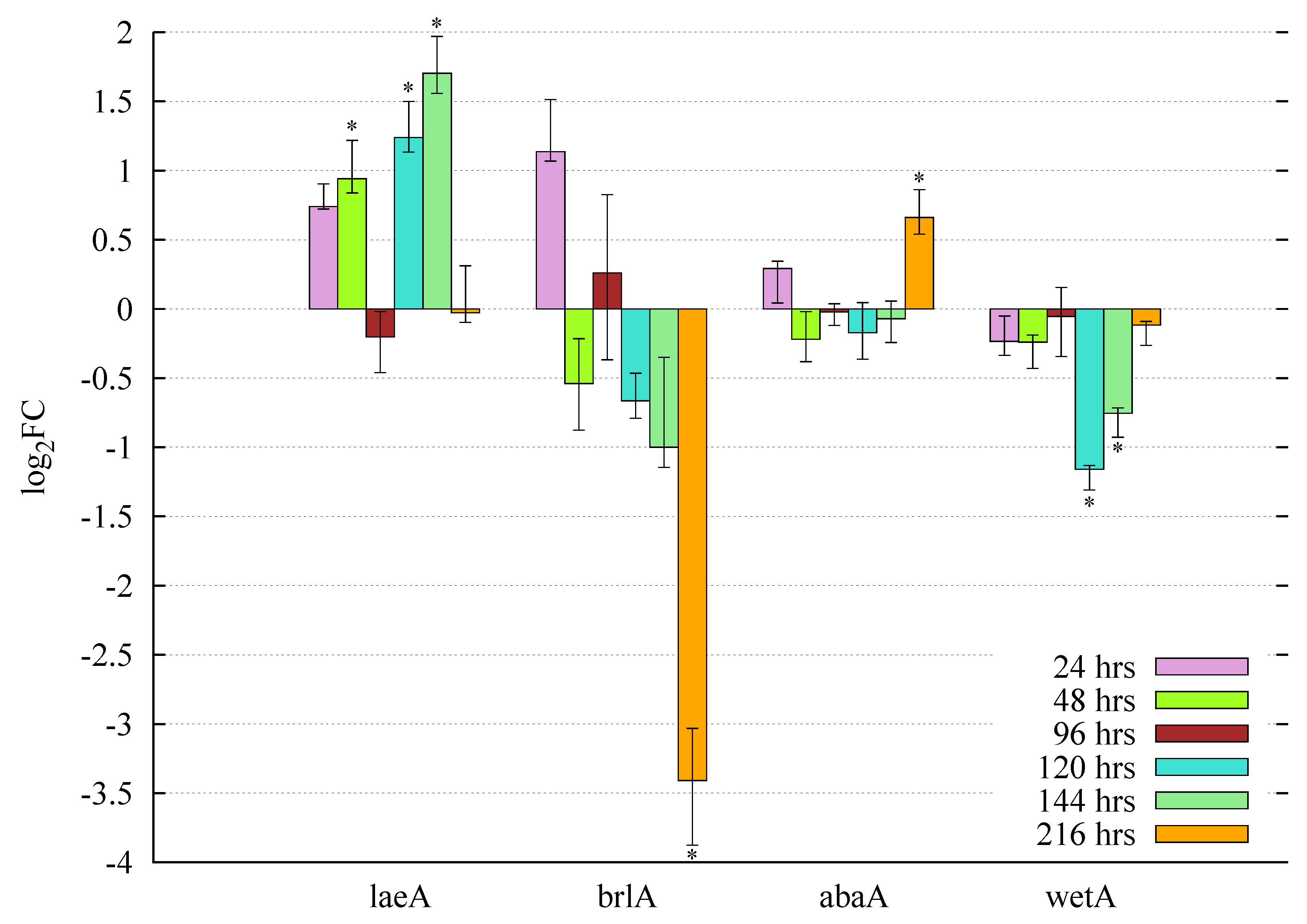
3.5.1. Butyrolactone I Upregulates the Global Regulator laeA Ortholog
3.5.2. Butyrolactone I Plays a Role in the Control of the Core Regulators of Conidiation
3.5.3. Accumulation Levels of the Pooled, Sequenced Transcripts
4. Discussion
4.1. Involvement of Butyrolactone I in the Control of Asexual Sporulation through LaeA
4.2. The Divergent Variants of the Global Regulator laeA under Submerged Culture
4.3. Accumulation of vosA, velB and velC Pooled Transcripts under Influence of Enhanced Butyrolactone I Biogenesis
4.4. Upstream Splice Variants of vosA, velC and laeA under the Influence of Butyrolactone I
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| FPKM | Fragments per kilobase of exon per million reads mapped |
| ORF | open reading frame |
| p.i. | post inoculation |
| FC | fold change |
References
- He, J.; Wijeratne, E.M.K.; Bashyal, B.P.; Zhan, J.; Seliga, C.J.; Liu, M.X.; Pierson, E.E.; Pierson, L.S.; VanEtten, H.D.; Gunatilaka, A.A.L. Cytotoxic and other metabolites of Aspergillus inhabiting the rhizosphere of Sonoran Desert plants. J. Nat. Prod. 2004, 67, 1985–1991. [Google Scholar] [CrossRef] [PubMed]
- Wijeratne, E.M.K.; Turbyville, T.J.; Zhang, Z.; Bigelow, D.; Pierson, L.S.; VanEtten, H.D.; Whitesell, L.; Canfield, L.M.; Gunatilaka, A.A.L. Cytotoxic constituents of Aspergillus terreus from the rhizosphere of Opuntia versicolor of the Sonoran Desert. J. Nat. Prod. 2003, 66, 1567–1573. [Google Scholar] [CrossRef] [PubMed]
- Reddy, C.; Singh, R. Enhanced production of itaconic acid from corn starch and market refuse fruits by genetically manipulated Aspergillus terreus SKR10. Bioresour. Technol. 2002, 85, 69–71. [Google Scholar] [CrossRef]
- Kuenz, A.; Gallenmüller, Y.; Willke, T.; Vorlop, K.D. Microbial production of itaconic acid: Developing a stable platform for high product concentrations. Appl. Microbiol. Biotechnol. 2012, 96, 1209–1216. [Google Scholar] [CrossRef] [PubMed]
- Yadav, R.P.; Saxena, R.K.; Gupta, R.; Davidson, W.S. Purification and characterization of a regiospecific lipase from Aspergillus terreus. Biotechnol. Appl. Biochem. 1998, 28, 243–249. [Google Scholar] [PubMed]
- Narra, M.; Dixit, G.; Divecha, J.; Madamwar, D.; Shah, A.R. Production of cellulases by solid state fermentation with Aspergillus terreus and enzymatic hydrolysis of mild alkali-treated rice straw. Bioresour. Technol. 2012, 121, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Alberts, A.W.; Chen, J.; Kuron, G.; Hunt, V.; Huff, J.; Hoffman, C.; Rothrock, J.; Lopez, M.; Joshua, H.; Harris, E.; et al. Mevinolin: A highly potent competitive inhibitor of hydroxymethylglutaryl-coenzyme A reductase and a cholesterol-lowering agent. Proc. Natl. Acad. Sci. USA 1980, 77, 3957–3961. [Google Scholar] [CrossRef] [PubMed]
- Sankawa, U.; Ebizuka, Y.; Noguchi, H.; Isikawa, Y.; Kitaghawa, S.; Yamamoto, Y.; Kobayashi, T.; Iitak, Y.; Seto, H. Biosynthesis of citrinin in Aspergillus terreus. Tetrahedron 1983, 39, 3583–3591. [Google Scholar] [CrossRef]
- Couch, R.D.; Gaucher, G.M. Rational elimination of Aspergillus terreus sulochrin production. J. Biotechnol. 2004, 108, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Lewis, R.E.; Wiederhold, N.P.; Chi, J.; Han, X.Y.; Komanduri, K.V.; Kontoyiannis, D.P.; Prince, R.A. Detection of gliotoxin in experimental and human aspergillosis. Infect. Immun. 2005, 73, 635–637. [Google Scholar] [CrossRef] [PubMed]
- Lewis, R.E.; Wiederhold, N.P.; Lionakis, M.S.; Prince, R.A.; Kontoyiannis, D.P. Frequency and species distribution of gliotoxin-producing Aspergillus isolates recovered from patients at a tertiary-care cancer center. J. Clin. Microbiol. 2005, 43, 6120–6122. [Google Scholar] [CrossRef] [PubMed]
- Kiriyama, N.; Nitta, K.; Sakaguchi, Y.; Taguchi, Y.; Yamamoto, Y. Studies on the metabolic products of Aspergillus terreus. III. Metabolites of the strain IFO 8835. (1). Chem. Pharm. Bull. (Tokyo) 1977, 25, 2593–2601. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Nishimura, K.I.; Kiriyama, N. Studies on the metabolic products of Aspergillus terreus. I. Metabolites of the strain IFO 6123. Chem. Pharm. Bull. (Tokyo) 1976, 24, 1853–1859. [Google Scholar] [CrossRef]
- Arai, K.; Masuda, K.; Kiriyama, N.; Nitta, K.; Yamamoto, Y.; Shimizu, S. Metabolic products of Aspergillus terreus. IV. Metabolites of the strain IFO 8835. (2.) The isolation and chemical structure of indolyl benzoquinone pigments. Chem. Pharm. Bull. (Tokyo) 1981, 29, 961–969. [Google Scholar] [CrossRef]
- Nitta, K.; Fujita, N.; Yoshimura, T.; Arai, K.; Yamamoto, Y. Metabolic products of Aspergillus terreus. IX. Biosynthesis of butyrolactone derivatives isolated from strains IFO 8835 and 4100. Chem. Pharm. Bull. (Tokyo) 1983, 31, 1528–1533. [Google Scholar] [CrossRef]
- Rao, K.V.; Sadhukhan, A.K.; Veerender, M.; Ravikumar, V.; Mohan, E.V.S.; Dhanvantri, S.D.; Sitaramkumar, M.; Babu, J.M.; Vyas, K.; Reddy, G.O. Butyrolactones from Aspergillus terreus. Chem. Pharm. Bull. (Tokyo) 2000, 48, 559–562. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.G.; Fujii, I.; Ebizuka, Y.; Sankawa, U. Purification and characterization of emodinanthrone oxygenase from Aspergillus terreus. Phytochemistry 1995, 38, 299–305. [Google Scholar] [CrossRef]
- Schimmel, T.G.; Coffman, A.D.; Parsons, S.J. Effect of butyrolactone I on the producing fungus, Aspergillus terreus. Appl. Environ. Microbiol. 1998, 64, 3707–3712. [Google Scholar] [PubMed]
- Raina, S.; de Vizio, D.; Palonen, E.K.; Odell, M.; Brandt, A.M.; Soini, J.T.; Keshavarz, T. Is quorum sensing involved in lovastatin production in the filamentous fungus Aspergillus terreus? Process. Biochem. 2012, 47, 843–852. [Google Scholar] [CrossRef]
- Palonen, E.K.; Neffling, M.R.; Raina, S.; Brandt, A.; Keshavarz, T.; Meriluoto, J.; Soini, J. Butyrolactone I quantification from lovastatin producing Aspergillus terreus using tandem mass spectrometry—Evidence of signalling functions. Microorganisms 2014, 2, 111–127. [Google Scholar] [CrossRef] [PubMed]
- Lass-Flörl, C.; Griff, K.; Mayr, A.; Petzer, A.; Gastl, G.; Bonatti, H.; Freund, M.; Kropshofer, G.; Dierich, M.P.; Nachbaur, D. Epidemiology and outcome of infections due to Aspergillus terreus: 10-year single centre experience. Br. J. Haematol. 2005, 131, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Slesiona, S.; Gressler, M.; Mihlan, M.; Zaehle, C.; Schaller, M.; Barz, D.; Hube, B.; Jacobsen, I.D.; Brock, M. Persistence versus escape: Aspergillus terreus and Aspergillus fumigatus employ different strategies during interactions with macrophages. PLoS ONE 2012, 7, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Fischer, R.; Kües, U. Asexual sporulation in mycelial fungi. In Growth, Differentiation and Sexuality, 2nd ed.; Kües, U., Fischer, R., Eds.; Springer: Berlin/Heidelberg, Germany, 2006; Volume 1, pp. 263–292. [Google Scholar]
- Boylan, M.T.; Mirabito, P.M.; Willett, C.E.; Zimmerman, C.R.; Timberlake, W.E. Isolation and physical characterization of three essential conidiation genes from Aspergillus nidulans. Mol. Cell Biol. 1987, 7, 3113–3118. [Google Scholar] [CrossRef] [PubMed]
- Mirabito, P.M.; Adams, T.H.; Timberlake, W.E. Interactions of three sequentially expressed genes control temporal and spatial specificity in Aspergillus development. Cell 1989, 57, 859–868. [Google Scholar] [CrossRef]
- Adams, T.H.; Boylan, M.T.; Timberlake, W.E. brlA is necessary and sufficient to direct conidiophore development in Aspergillus nidulans. Cell 1988, 54, 353–362. [Google Scholar] [CrossRef]
- Sewall, T.C.; Mims, C.W.; Timberlake, W.E. abaA controls phialide differentiation in Aspergillus nidulans. Plant Cell 1990, 2, 731–739. [Google Scholar] [CrossRef] [PubMed]
- Sewall, T.; Mims, C.; Timberlake, W. Conidium differentiation in Aspergillus nidulans wild-type and wet-white (wetA) mutant strains. Dev. Biol. 1990, 138, 499–508. [Google Scholar] [CrossRef]
- Marshall, M.A.; Timberlake, W.E. Aspergillus nidulans wetA activates spore-specific gene expression. Mol. Cell Biol. 1991, 11, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Fillinger, S.; Chaveroche, M.K.; van Dijck, P.; de Vries, R.; Ruijter, G.; Thevelein, J.; d’Enfert, C. Trehalose is required for the acquisition of tolerance to a variety of stresses in the filamentous fungus Aspergillus nidulans. Microbiology 2001, 147, 1851–1862. [Google Scholar] [CrossRef] [PubMed]
- Ni, M.; Yu, J.H. A novel regulator couples sporogenesis and trehalose biogenesis in Aspergillus nidulans. PLoS ONE 2007, 2, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Sarikaya Bayram, Ö.; Bayram, Ö.; Valerius, O.; Park, H.S.; Irniger, S.; Gerke, J.; Ni, M.; Han, K.H.; Yu, J.H.; Braus, G.H. LaeA control of velvet family regulatory proteins for light-dependent development and fungal cell-type specificity. PLoS Genet. 2010, 6, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Park, H.S.; Ni, M.; Jeong, K.C.; Kim, Y.H.; Yu, J.H. The role, interaction and regulation of the velvet regulator VelB in Aspergillus nidulans. PLoS ONE 2012, 7, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Park, H.S.; Yu, J.H. Genetic control of asexual sporulation in filamentous fungi. Curr. Opin. Microbiol. 2012, 15, 669–677. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Han, K.Y.; Kim, K.J.; Han, D.M.; Jahng, K.Y.; Chae, K.S. The veA gene activates sexual development in Aspergillus nidulans. Fungal Genet. Biol. 2002, 37, 72–80. [Google Scholar] [CrossRef]
- Kato, N.; Brooks, W.; Calvo, A.M. The expression of sterigmatocystin and penicillin genes in Aspergillus nidulans is controlled by veA, a gene required for sexual development. Eukaryot. Cell 2003, 2, 1178–1186. [Google Scholar] [CrossRef] [PubMed]
- Bayram, Ö.; Krappmann, S.; Ni, M.; Bok, J.W.; Helmstaedt, K.; Valerius, O.; Braus-Stromeyer, S.; Kwon, N.J.; Keller, N.P.; Yu, J.H.; et al. VelB/VeA/LaeA complex coordinates light signal with fungal development and secondary metabolism. Science 2008, 320, 1504–1506. [Google Scholar] [CrossRef] [PubMed]
- Park, H.S.; Nam, T.Y.; Han, K.H.; Kim, S.C.; Yu, J.H. VelC positively controls sexual development in Aspergillus nidulans. PLoS ONE 2014, 9, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Bok, J.W.; Keller, N.P. LaeA, a regulator of secondary metabolism in Aspergillus spp. Eukaryot. Cell 2004, 3, 527–535. [Google Scholar] [CrossRef] [PubMed]
- Birren, B.; Lander, E.; Galagan, J.; Nusbaum, C.; Devon, K.; Henn, M.; Ma, L.J.; Jaffe, D.; Butler, J.; Alvarez, P.; et al. Annotation of the Aspergillus terreus NIH2624 Genome. Available online: http://www.broadinstitute.org/ftp/pub/annotation/fungi/aspergillus/genomes/ (accessed on 9 March 2007).
- R: A Language and Environment for Statistical Computing, 2011. Available online: http://www.r-project.org/ (accessed on 1 January 2012).
- Smyth, G.K.; Speed, T. Normalization of cDNA microarray data. Methods 2003, 31, 265–273. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. B Met. 1995, 57, 289–300. [Google Scholar]
- Smyth, G.K. Limma: Linear models for microarray. In Bioinformatics and Computational Biology Solutions Using R and Bioconductor; Gentleman, R., Carey, V.J., Huber, W., Irizarry, R.A., Dudoit, S., Eds.; Statistics for Biology and Health; Springer: New York, NY, USA, 2005; pp. 397–420. [Google Scholar]
- Smyth, G.K. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 2004, 3, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Nucleotide-Nucleotide BLAST Version 2.2.29+. Available online: ftp://ftp.ncbi.nlm.nih.gov/blast/ executables/blast+/2.2.29/ (accessed on 10 October 2014).
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: architecture and applications. BMC Bioinform. 2009, 10, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Marioni, J.C.; Mason, C.E.; Mane, S.M.; Stephens, M.; Gilad, Y. RNA-seq: An assessment of technical reproducibility and comparison with gene expression arrays. Genome Res. 2008, 18, 1509–1517. [Google Scholar] [CrossRef] [PubMed]
- Parkhomchuk, D.; Borodina, T.; Amstislavskiy, V.; Banaru, M.; Hallen, L.; Krobitsch, S.; Lehrach, H.; Soldatov, A. Transcriptome analysis by strand-specific sequencing of complementary DNA. Nucleic Acids Res. 2009, 37, e123. [Google Scholar] [CrossRef] [PubMed]
- Levin, J.Z.; Yassour, M.; Adiconis, X.; Nusbaum, C.; Thompson, D.A.; Friedman, N.; Gnirke, A.; Regev, A. Comprehensive comparative analysis of strand-specific RNA sequencing methods. Nat. Methods 2010, 7, 709–715. [Google Scholar] [CrossRef] [PubMed]
- Hannon, G.J. FASTX Toolkit, Version 0.0.13. Available online: http://hannonlab.cshl.edu/fastx_toolkit/ (accessed on 1 January 2012).
- Simon, A. FastQC: A Quality Control Tool for High Throughput Sequence Data. Version 0.10.1. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 1 January 2012).
- RNA-seq De Novo Assembly Using Trinity. Version trinityrnaseq_r2012-03-17. Available online: https://github.com/trinityrnaseq/trinityrnaseq/wiki/ (accessed on 17 March 2012).
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef] [PubMed]
- Genome-Guided Trinity De Novo Transcriptome Assembly (Beta). Version trinityrnaseq_r2013-02-25. Available online: https://github.com/trinityrnaseq/trinityrnaseq/wiki (accessed on 25 February 2013).
- Wu, T.D.; Nacu, S. Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics 2010, 26, 873–881. [Google Scholar] [CrossRef] [PubMed]
- GSNAP: Genomic Short-Read Nucleotide Alignment Program. GMAP: A Genomic Mapping and Alignment Program for mRNA and EST Sequences. GSNAP and GMAP Program Version 2012-07-20 v3. Available online: http://research-pub.gene.com/gmap (accessed on 20 July 2012).
- Wu, T.D.; Watanabe, C.K. GMAP: A genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics 2005, 21, 1859–1875. [Google Scholar] [CrossRef] [PubMed]
- Gene Structure Annotation and Analysis Using PASA. Version PASA2-r20130605. Available online: http://pasapipeline.github.io/ (accessed on 5 June 2013).
- Haas, B.J.; Delcher, A.L.; Mount, S.M.; Wortman, J.R.; Smith, R.K., Jr.; Hannick, L.I.; Maiti, R.; Ronning, C.M.; Rusch, D.B.; Town, C.D.; et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 2003, 31, 5654–5666. [Google Scholar] [CrossRef] [PubMed]
- Campbell, M.A.; Haas, B.J.; Hamilton, J.P.; Mount, S.M.; Buell, C.R. Comprehensive analysis of alternative splicing in rice and comparative analyses with Arabidopsis. BMC Genom. 2006, 7, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Standalone BLAT Fast Sequence Search Command Line Tool. Version 35x1. Available online: https://genome.ucsc.edu/FAQ/FAQblat.html#blat3 (accessed on 1 January 2012).
- Kent, W.J. BLAT—The BLAST-Like Alignment Tool. Genome Res. 2002, 12, 656–664. [Google Scholar] [CrossRef] [PubMed]
- AMOS Assembler Pipeline. Version 3.1.0. Available online: https://sourceforge.net/projects/amos/files/ (accessed on 15 December 2011).
- Treangen, T.J.; Sommer, D.D.; Angly, F.E.; Koren, S.; Pop, M. Next generation sequence assembly with AMOS. In Curr Protoc Bioinformatics; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2011. [Google Scholar]
- Integrative Genomics Viewer Software. Version IGV_2.3.26. Available online: http://software.broadinstitute.org/software/igv/ (accessed on 31 January 2014).
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [PubMed]
- Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P. Integrative genomics viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotech. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed]
- The GENSCAN Web Server at MIT. Available online: http://genes.mit.edu/GENSCAN.html (accessed on 1 January 2015).
- Burge, C.; Karlin, S. Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 1997, 268, 78–94. [Google Scholar] [CrossRef] [PubMed]
- Burge, C.B. Chapter 8 Modeling dependencies in pre-mRNA splicing signals. In Computational Methods in Molecular Biology; Salzberg, S.L., Searls, D.B., Kasif, S., Eds.; New Comprehensive Biochemistry; Elsevier: New York, NY, USA, 1998; Volume 32, pp. 129–164. [Google Scholar]
- ExPASy—Translate Tool. Available online: http://web.expasy.org/translate/ (accessed on 1 January 2015).
- Artimo, P.; Jonnalagedda, M.; Arnold, K.; Baratin, D.; Csardi, G.; de Castro, E.; Duvaud, S.; Flegel, V.; Fortier, A.; Gasteiger, E.; et al. ExPASy: SIB bioinformatics resource portal. Nucleic Acids Res. 2012, 40, W597–W603. [Google Scholar] [CrossRef] [PubMed]
- InterProScan Sequence Search. Available online: http://www.ebi.ac.uk/interpro/search/sequence-search/ (accessed on 1 January 2015).
- Mulder, N.J.; Apweiler, R.; Attwood, T.K.; Bairoch, A.; Bateman, A.; Binns, D.; Bork, P.; Buillard, V.; Cerutti, L.; Copley, R.; et al. New developments in the InterPro database. Nucleic Acids Res. 2007, 35, D224–D228. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.; Binns, D.; Chang, H.Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef] [PubMed]
- NCBI’s CDD, the Conserved Domain Database. Available online: https://www.ncbi.nlm.nih.gov/Structure/ cdd/wrpsb.cgi/ (accessed on 1 January 2015).
- Marchler-Bauer, A.; Derbyshire, M.K.; Gonzales, N.R.; Lu, S.; Chitsaz, F.; Geer, L.Y.; Geer, R.C.; He, J.; Gwadz, M.; Hurwitz, D.I.; et al. CDD: NCBI’s conserved domain database. Nucleic Acids Res. 2015, 43, D222–D226. [Google Scholar] [CrossRef] [PubMed]
- Basic Local Alignment Tool, NCBI BLAST Sequence Database. Available online: http://blast.ncbi.nlm.nih.gov/ (accessed on 1 January 2015).
- Robust Phylogenetic Analysis for the Non-Specialist. Available online: http://www.phylogeny.fr/ (accessed on 1 January 2015).
- Dereeper, A.; Guignon, V.; Blanc, G.; Audic, S.; Buffet, S.; Chevenet, F.; Dufayard, J.F.; Guindon, S.; Lefort, V.; Lescot, M.; et al. Phylogeny.fr: Robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 2008, 36, W465–W469. [Google Scholar] [CrossRef] [PubMed]
- Etxebeste, O.; Garzia, A.; Espeso, E.A.; Ugalde, U. Aspergillus nidulans asexual development: Making the most of cellular modules. Trends Microbiol. 2010, 18, 569–576. [Google Scholar] [CrossRef] [PubMed]
- Skromne, I.; Sánchez, O.; Aguirre, J. Starvation stress modulates the expression of the Aspergillus ni dulans brlA regulatory gene. Microbiology 1995, 141, 21–28. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| A. terreus | A. nidulans | A. flavus | A. fumigatus | ||||
|---|---|---|---|---|---|---|---|
| Gene | Length (aa) | ID% | Accession | ID% | Accession | ID% | Accession |
| VeA | 589 | 53 | AAD42946.1 | 70 | ADF30049.1 | 67 | KEY80278.1 |
| VelB | 360 | 67 | ABQ17967.1 | 85 | ADF30048.2 | 82 | EAL87909.1 |
| VelC | 441 | 53 | ABQ17968.1 | 57 | KOC10835.1 | 53 | KEY80115.1 |
| VosA-α | 430 | 73 | ABI51618.1 | 81 | KOC10835.1 | 78 | ABQ18268.1 |
| VosA-β | 350 | 75 | ABI51618.1 | 82 | KOC10835.1 | 80 | ABQ18268.1 |
| VosA-γ | 348 | 75 | ABI51618.1 | 83 | KOC10835.1 | 80 | ABQ18268.1 |
| VosA-δ | 140 | 79 | ABI51618.1 | 87 | KOC10835.1 | 88 | ABQ18268.1 |
| VosA-ϵ | 418 | 74 | ABI51618.1 | 82 | KOC10835.1 | 78 | ABQ18268.1 |
| VosA-ζ | 338 | 76 | ABI51618.1 | 84 | KOC10835.1 | 80 | ABQ18268.1 |
| VosA-η | 336 | 76 | ABI51618.1 | 84 | KOC10835.1 | 80 | ABQ18268.1 |
| VosA-θ | 128 | 83 | ABI51618.1 | 91 | KOC10835.1 | 88 | ABQ18268.1 |
| LaeA-α | 367 | 74 | CBF88745.1 | 80 | B8N406 e | 75 | Q4WRY5 |
| NA | NA | 87 | EED56057.1 | NA | NA | ||
| LaeA-β | 273 | 85 | CBF88745.1 | 87 | B8N406 e | 84 | Q4WRY5 |
| NA | NA | 87 | EED56057.1 | NA | NA | ||
| ORF | Gene | Transcript | Number of | Upstream | Approximate Occurrence | |
|---|---|---|---|---|---|---|
| Gene | Length (bp) | Length (bp) | Length (bp) | Exons (Introns) | Intron | of the Upstream Intron (%) |
| veA | 1770 | 1770 | NA | 1 (0) | NA | NA |
| velB | 1083 | 1250 | 3432 | 4 (4) | Yes | 88 e |
| velB | 1083 | 1250 | 3220 | 4 (3) | No | - |
| velC | 1326 | 1453 | 2545 | 2 (2) | Yes | 63 e |
| velC | 1326 | 1453 | 2696 | 2 (1) | No | - |
| vosA-α | 1293 | 2021 | 2621 | 10 (9) | No | - |
| vosA-β | 1053 | 1770 | 2632 | 10 (9) | No | - |
| vosA-γ | 1047 | 1716 | 2680 | 9 (8) | No | - |
| vosA-δ | 423 | 772 | 1429 | 5 (4) | No | - |
| vosA-ϵ | 1257 | 1884 | 2658 | 9 (9) | Yes | 48 |
| vosA-ζ | 1017 | 1633 | 2669 | 9 (9) | Yes | 48 |
| vosA-η | 1011 | 1579 | 2717 | 8 (8) | Yes | 48 |
| vosA-θ | 387 | 635 | 1466 | 4 (4) | Yes | 48 |
| laeA-α | 1104 | 1196 | 2745 | 2 (2) | Yes | 67 e |
| laeA-α | 1104 | 1196 | 2808 | 2 (1) | No | - |
| laeA-β | 822 | 822 | 2837 | 1 (1) | Yes | 67 e |
| laeA-β | 822 | 822 | 2900 | 1 (0) | No | - |
| Gene | Pooled FPKM | Pooled Counts Max | Pooled Coverage Max | ||
|---|---|---|---|---|---|
| Sense | Antisense | Sense | Antisense | ||
| veA | 0.83 | 0.45 | 0.057 | 8 | 1 |
| velB | 120 | 62 | 0.64 | 1109 | 14 |
| velC | NA | 1.3 | 0.062 | 25 | 1 |
| vosA-α | 110 | 73 | 0.66 | 1317 | 12 |
| laeA-α | 6.8 | 2.1 | 0.12 | 40 | 2 |
| brlA | 1.5 | 0.78 | 0.00 | 14 | 0 |
| abaA | 7.0 | 2.9 | 0.11 | 50 | 2 |
| wetA | 13 | 3.8 | 0.25 | 70 | 4 |
| tpsA | 270 | 77 | 0.98 | 1461 | 16 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palonen, E.K.; Raina, S.; Brandt, A.; Meriluoto, J.; Keshavarz, T.; Soini, J.T. Transcriptomic Complexity of Aspergillus terreus Velvet Gene Family under the Influence of Butyrolactone I. Microorganisms 2017, 5, 12. https://doi.org/10.3390/microorganisms5010012
Palonen EK, Raina S, Brandt A, Meriluoto J, Keshavarz T, Soini JT. Transcriptomic Complexity of Aspergillus terreus Velvet Gene Family under the Influence of Butyrolactone I. Microorganisms. 2017; 5(1):12. https://doi.org/10.3390/microorganisms5010012
Chicago/Turabian StylePalonen, Elina K., Sheetal Raina, Annika Brandt, Jussi Meriluoto, Tajalli Keshavarz, and Juhani T. Soini. 2017. "Transcriptomic Complexity of Aspergillus terreus Velvet Gene Family under the Influence of Butyrolactone I" Microorganisms 5, no. 1: 12. https://doi.org/10.3390/microorganisms5010012
APA StylePalonen, E. K., Raina, S., Brandt, A., Meriluoto, J., Keshavarz, T., & Soini, J. T. (2017). Transcriptomic Complexity of Aspergillus terreus Velvet Gene Family under the Influence of Butyrolactone I. Microorganisms, 5(1), 12. https://doi.org/10.3390/microorganisms5010012