
Tackling Drug Resistant Infection Outbreaks of Global Pandemic Escherichia coli ST131 Using Evolutionary and Epidemiological Genomics

Abstract
:
1. The Advent of Genome-Based Escherichia coli Monitoring

{kind=link}
{kind=link}
| Gene | Gene Product | Role in Virulence | Typing Use |
|---|---|---|---|
| H7 fliC | Flagellin variant | Multiple | |
| iss | Increased serum survival | Multiple | |
| malX | Pathogenicity island marker | Multiple | |
| ompT | Outer membrane protease T | Multiple | |
| traT | Serum resistance-associated | Multiple | |
| usp | Uropathogenic-specific protein | Multiple | |
| K1/K2/K5/K100 | Group 2 capsule variants | Capsule-related | |
| kpsM II | kpsM II group 2 capsule | Capsule-related | Extraintestinal E. coli definition |
| kpsMT III | Group 3 capsule | Capsule-related | Extraintestinal E. coli definition |
| afa | Afa adhesin | Adhesion | Extraintestinal E. coli definition |
| draBC | Dr-binding adhesin | Adhesion | Extraintestinal E. coli definition |
| fimH | Type 1 fimbriae | Adhesion | Extraintestinal E. coli definition |
| papC/papE/papF/papG | P fimbriae operon | Adhesion | Extraintestinal E. coli definition |
| sfa/foc | F1C fimbriae or S | Adhesion | Extraintestinal E. coli definition |
| F10 papA | P fimbriae subunit variant | Adhesion | |
| focG | F1C fimbriae adhesin | Adhesion | |
| hra | Heat-resistant agglutinin | Adhesion | |
| iha | Adhesion siderophore | Adhesion | |
| tsh | Temperature sensitive hemagglutinin | Adhesion | |
| iutA | Aerobactin receptor | Siderophore | Extraintestinal E. coli definition |
| fyuA | Yersiniabactin receptor | Siderophore | |
| ireA | Siderophore receptor | Siderophore | |
| iroN | Salmochelin receptor | Siderophore | |
| stbB | Plasmid stability | plasmid stability | |
| aac | Aminoglycoside acetyltransferases | AMR (multiple) | |
| aadA5 | Aminoglycoside 3′-adenylyltransferase | AMR (streptomycin) | |
| acrR | Repressor of acrAB genes | AMR (multiple) | |
| catB4 | Chloramphenicol acetyltransferase | AMR (chloramphenicol) | |
| dfrA7 | Dihydrofolate reductase type VII | AMR (trimethoprim) | |
| gyrA | DNA gyrase subunit A | AMR (fluoroquinolone) | |
| marR | Repressor of the marRAB operon | AMR (multiple) | |
| mphA | Macrolide 2′-phosphotransferase I | AMR (macrolide) | |
| parC | DNA topoisomerase 4 subunit A | AMR (fluoroquinolone) | |
| rpsL105 | Ribosomal gene S12 | AMR (multiple) | |
| sulI | Dihydropteroate synthase | AMR (sulfonamide) | |
| tetA | Tetracycline efflux pump | AMR (tetracycline) | |
| Tn10 | Transposon-tetracycline resistance | AMR (tetracycline) | |
| astA | Arginine succinyltransferase | Toxin | |
| cnf1 | Cytotoxic necrotizing factor | Toxin | |
| hlyD | Alpha-Hemolysin | Toxin | |
| pic | Serine protease | Toxin | |
| sat | Secreted autotransporter toxin | Toxin | |
| vat | Vacuolating toxin | Toxin | |
| dinB | DNA polymerase | Pasteur MLST system | |
| pdbB | P-aminobenzoate synthase | Pasteur MLST system | |
| polB | Polymerase PolII | Pasteur MLST system | |
| putP | Proline permease | Pasteur MLST system | |
| trpA/trpB | Tryptophan synthase subunits A, B | Pasteur MLST system | |
| uidA | Beta-glucuronidase | Pasteur MLST system | |
| icd/icdA | Isocitrate dehydrogenase | Pasteur/Achtman MLST system | |
| adk | Adenylate kinase | Achtman MLST system | |
| fumC | Fumarate hydratase | Achtman MLST system | |
| gyrB | DNA gyrase subunit B | Achtman MLST system | |
| mdh | Malate dehydrogenase | Achtman MLST system | |
| purA | Adenylosuccinate dehydrogenase | Achtman MLST system | |
| recA | ATP/GTP binding motif | Achtman MLST system |
2. Escherichia coli ST131 Is a Major Global Health Issue
3. The Genomic Landscape of Antimicrobial Resistance in E. coli
4. Antimicrobial Resistance Genetically Defines ST131
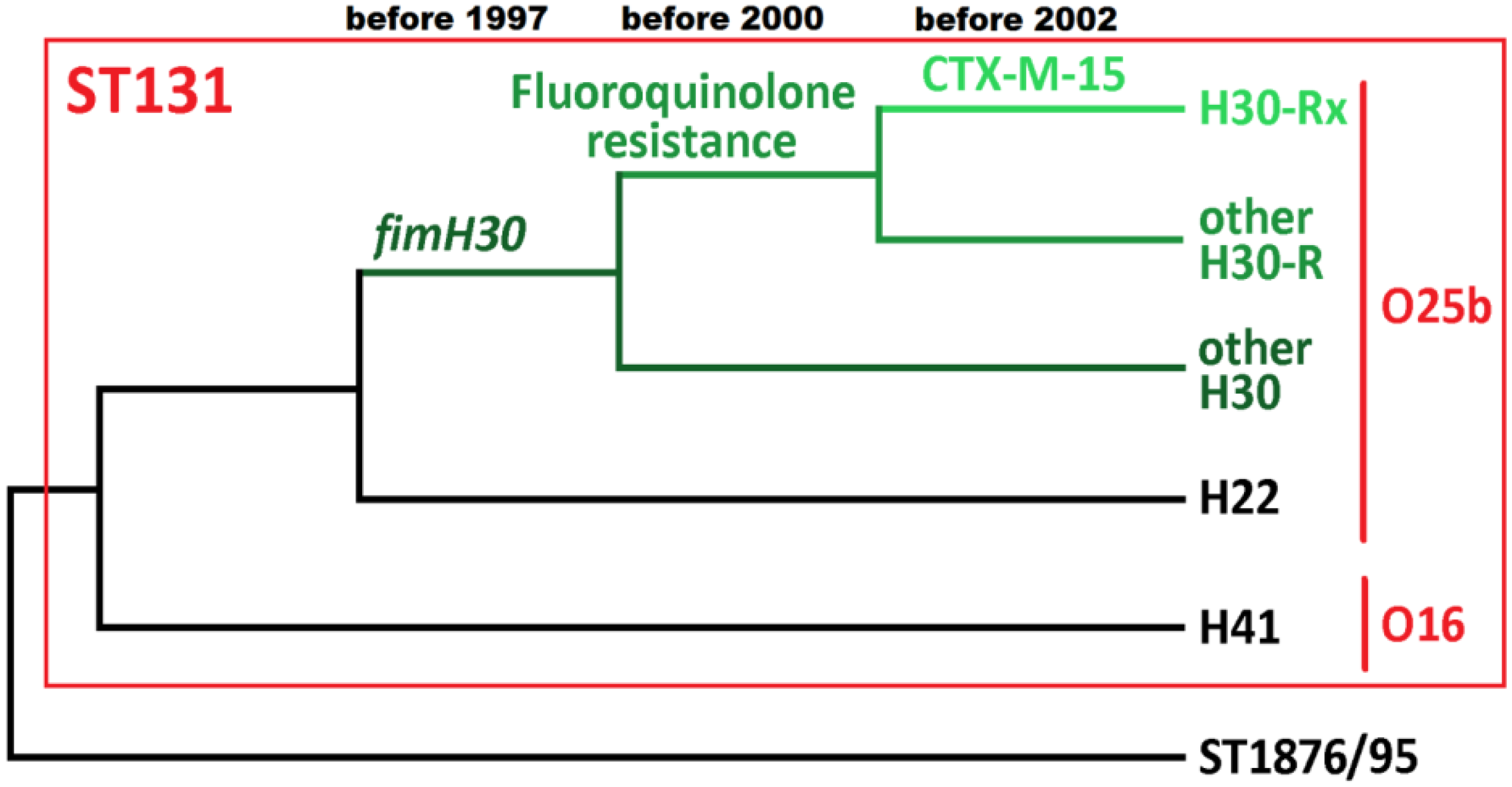
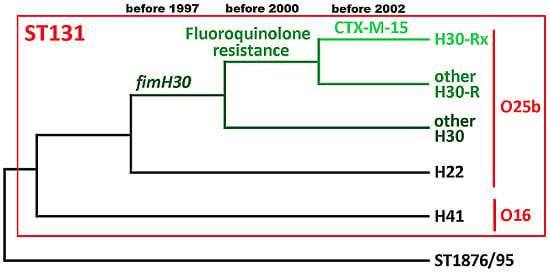
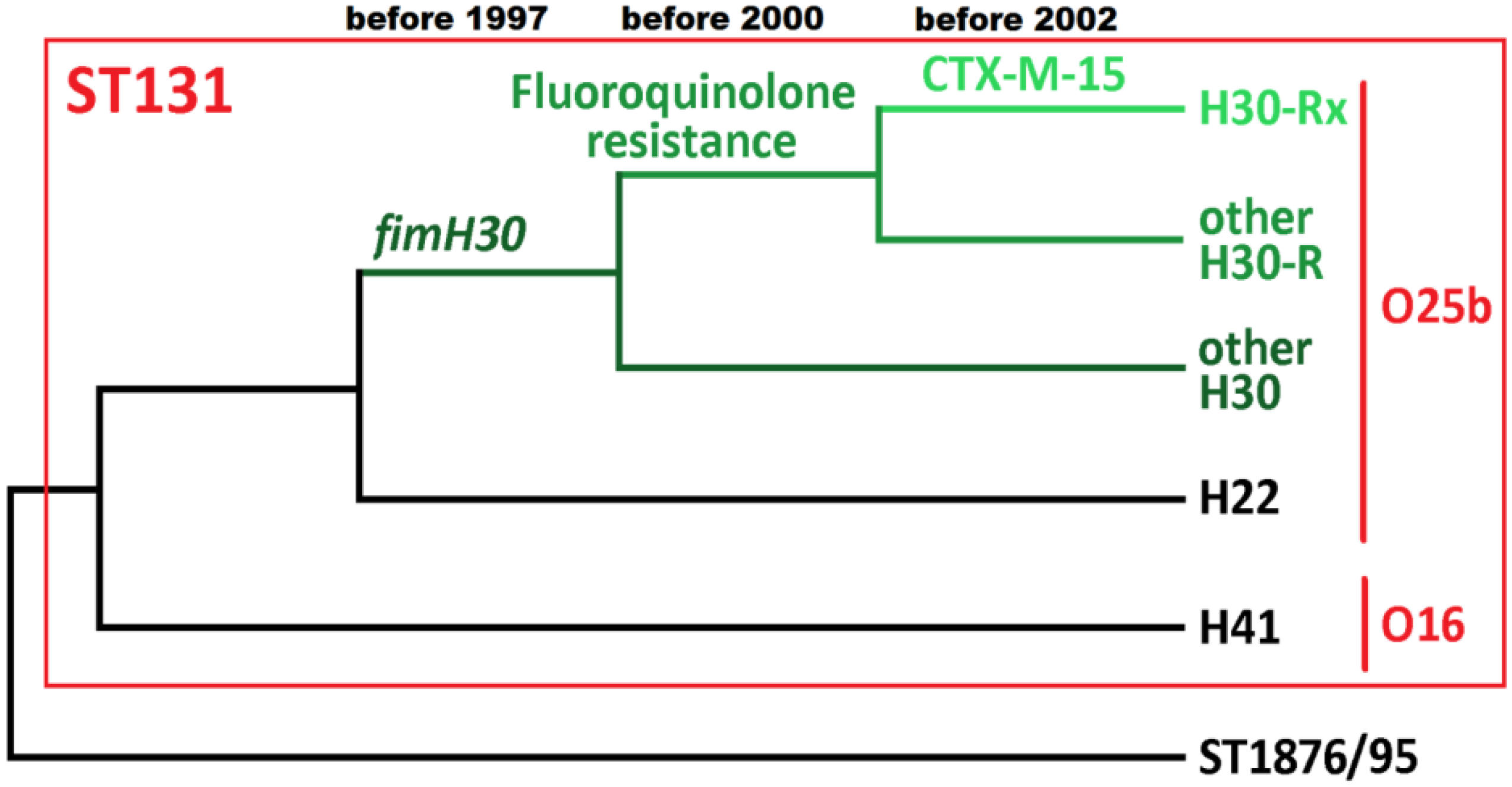
5. The Key Antimicrobial Resistance Elements in ST131
6. How Did ST131 Adapt to Be so Successful?
6.1. Extensive Recombination and Horizontal Gene Transfer
6.2. Maintenance of a Broad Resistome
6.3. Regulatory Fine-Tuning of Gene Expression
6.4. Cross-Antimicrobial Resistance and Compensatory Mutations
6.5. Resistance through Cell Growth Arrest
7. Stopping ST131
7.1. Evolutionary Phylogenomics and Fitness Measurement
7.2. Modelling Historical and Future Epidemiology
7.3. Assessing Cell Growth Arrest
7.4. Avoiding Partial Treatments and Cell Population Heterogeneity
7.5. Dissecting Measurably Evolving Infections
8. Conclusion: Future Avenues for ST131 Infection Genomics
Acknowledgments
Conflicts of Interest
References
- Centers for Disease Control and Prevention. In Antibiotic resistance threats in the United States, 2013; US Department of Health and Human Services, 2013. Available online: http://www.cdc.gov/drugresistance/threat-report-2013/pdf/ar-threats-2013–508.pdf (accessed on 9 September 2014).
- Smith, K.F.; Goldberg, M.; Rosenthal, S.; Carlson, L.; Chen, J.; Chen, C.; Ramachandran, S. Global rise in human infectious disease outbreaks. J. R Soc. Interf. 2014, 11. [Google Scholar] [CrossRef]
- WHO. The Evolving Threat of Antimicrobial Resistance—Options for Action. Available online: http://whqlibdoc.who.int/publications/2012/9789241503181_eng.pdf (accessed on 29 August 2014).
- WHO. Antimicrobial Resistance: Global Report on Surveillance 2014. Available online: http://apps.who.int/iris/bitstream/10665/112642/1/9789241564748_eng.pdf (accessed on 2 September 2014).
- Reuter, S.; Ellington, M.J.; Cartwright, E.J.; Köser, C.U.; Török, M.E.; Gouliouris, T.; Harris, S.R.; Brown, N.M.; Holden, M.T.; Quail, M.; et al. Rapid bacterial whole-genome sequencing to enhance diagnostic and public health microbiology. JAMA Intern. Med. 2013, 173, 1397–1404. [Google Scholar] [CrossRef] [PubMed]
- Achtman, M. Insights from genomic comparisons of genetically monomorphic bacterial pathogens. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2012, 367, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Palmer, A.C.; Kishony, R. Understanding, predicting and manipulating the genotypic evolution of antibiotic resistance. Nat. Rev. Genet 2013, 14, 243–248. [Google Scholar] [CrossRef] [PubMed]
- Noller, A.C.; McEllistrem, M.C.; Pacheco, A.G.; Boxrud, D.J.; Harrison, L.H. Multilocus variable-number tandem repeat analysis distinguishes outbreak and sporadic Escherichia coli O157:H7 isolates. J. Clin. Microbiol. 2003, 41, 5389–5397. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.R.; Nicolas-Chanoine, M.H.; Debroy, C.; Castanheira, M.; Robicsek, A.; Hansen, G.; Weissman, S.; Urban, C.; Platell, J.; Trott, D.; et al. MASTER Investigators. Comparison of Escherichia coli ST131 pulsotypes, by epidemiologic traits, 1967–2009. Emerg. Infect. Dis. 2012, 18, 598–607. [Google Scholar] [CrossRef] [PubMed]
- Mathers, A.J.; Peirano, G.; Pitout, J.D. Escherichia coli ST131: The Quintessential Example of an International Multiresistant High-Risk Clone. Adv. Appl. Microbiol. 2015, 90, 109–154. [Google Scholar] [PubMed]
- Sullivan, C.B.; Diggle, M.A.; Clarke, S.C. Multilocus sequence typing: Data analysis in clinical microbiology and public health. Mol. Biotechnol. 2005, 29, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Coughlan, S.; Barreira, S.; Seoighe, C.; Downing, T. Chapter 3: Genome-wide variant discovery using sequence assembly, mapping and population-wide analysis. In Bioinformatics and Data Analysis in Microbiology, 1st ed.; Bishop, O.T., Ed.; Caister Academic Press: Norfolk, UK, 2014; pp. 51–80. [Google Scholar]
- Escherichia coli MLST Database. Available online: http://mlst.warwick.ac.uk/mlst/dbs/Ecoli (accessed on 23 August 2014).
- Bryant, J.; Chewapreecha, C.; Bentley, S.D. Developing insights into the mechanisms of evolution of bacterial pathogens from whole-genome sequences. Future Microbiol. 2012, 7, 1283–1296. [Google Scholar] [CrossRef] [PubMed]
- Aarestrup, F.M.; Brown, E.W.; Detter, C.; Gerner-Smidt, P.; Gilmour, M.W.; Harmsen, D.; Hendriksen, R.S.; Hewson, R.; Heymann, D.L.; Johansson, K.; et al. Integrating genome-based informatics to modernize global disease monitoring, information sharing, and response. Emerg. Infect. Dis. 2012, 18. [Google Scholar] [CrossRef]
- Gault, G.; Weill, F.X.; Mariani-Kurkdjian, P.; Jourdan-da Silva, N.; King, L.; Aldabe, B.; Charron, M.; Ong, N.; Castor, C.; Mace, M.; et al. Outbreak of haemolytic uraemic syndrome and bloody diarrhoea due to Escherichia coli O104:H4, south-west France, June 2011. Euro Surveill. 2011, 16. pii: 19905. [Google Scholar] [PubMed]
- Frank, C.; Werber, D.; Cramer, J.P.; Askar, M.; Faber, M.; an der Heiden, M.; Bernard, H.; Fruth, A.; Prager, R.; Spode, A.; et al. Epidemic profile of Shiga-toxin-producing Escherichia coli O104:H4 outbreak in Germany. N. Engl. J. Med. 2011, 365, 1771–1780. [Google Scholar] [CrossRef] [PubMed]
- Mellmann, A.; Harmsen, D.; Cummings, C.A.; Zentz, E.B.; Leopold, S.R.; Rico, A.; Prior, K.; Szczepanowski, R.; Ji, Y.; Zhang, W.; et al. Prospective genomic characterization of the German enterohemorrhagic Escherichia coli O104:H4 outbreak by rapid next generation sequencing technology. PLoS ONE 2011, 6, e22751. [Google Scholar] [CrossRef] [PubMed]
- Rasko, D.A.; Webster, D.R.; Sahl, J.W.; Bashir, A.; Boisen, N.; Scheutz, F.; Paxinos, E.E.; Sebra, R.; Chin, C.S.; Iliopoulos, D.; et al. Origins of the E. coli strain causing an outbreak of hemolytic-uremic syndrome in Germany. N. Engl. J. Med. 2011, 365, 709–717. [Google Scholar] [CrossRef] [PubMed]
- Sherry, N.L.; Porter, J.L.; Seemann, T.; Watkins, A.; Stinear, T.P.; Howden, B.P. Outbreak investigation using high-throughput genome sequencing within a diagnostic microbiology laboratory. J. Clin. Microbiol. 2013, 51, 1396–1401. [Google Scholar] [CrossRef] [PubMed]
- Underwood, A.P.; Dallman, T.; Thomson, N.R.; Williams, M.; Harker, K.; Perry, N.; Adak, B.; Willshaw, G.; Cheasty, T.; Green, J.; et al. Public health value of next-generation DNA sequencing of enterohemorrhagic Escherichia coli isolates from an outbreak. J. Clin. Microbiol. 2013, 51, 232–237. [Google Scholar] [CrossRef] [PubMed]
- Eyre, D.W.; Golubchik, T.; Gordon, N.C.; Bowden, R.; Piazza, P.; Batty, E.M.; Ip, C.L.; Wilson, D.J.; Didelot, X.; O’Connor, L.; et al. A pilot study of rapid benchtop sequencing of Staphylococcus aureus and Clostridium difficile for outbreak detection and surveillance. BMJ Open 2012, 2, e001124. [Google Scholar] [CrossRef] [PubMed]
- Parkinson, N.J.; Maslau, S.; Ferneyhough, B.; Zhang, G.; Gregory, L.; Buck, D.; Ragoussis, J.; Ponting, C.P.; Fischer, M.D. Preparation of high-quality next-generation sequencing libraries from picogram quantities of target DNA. Genome Res. 2012, 22, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Koser, C.U.; Fraser, L.J.; Ioannou, A.; Becq, J.; Ellington, M.J.; Holden, M.T.; Reuter, S.; Török, M.E.; Bentley, S.D.; Parkhill, J.; et al. Rapid single-colony whole-genome sequencing of bacterial pathogens. J. Antimicrob. Chemother. 2014, 69, 1275–1281. [Google Scholar] [CrossRef] [PubMed]
- Quail, M.A.; Smith, M.; Jackson, D.; Leonard, S.; Skelly, T.; Swerdlow, H.P.; Gu, Y.; Ellis, P. SASI-Seq: Sample assurance Spike-Ins, and highly differentiating 384 barcoding for Illumina sequencing. BMC Genomics 2014, 15. [Google Scholar] [CrossRef] [PubMed]
- Lasken, R.S.; McLean, J.S. Recent advances in genomic DNA sequencing of microbial species from single cells. Nat. Rev. Genet 2014, 15, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Gole, J.; Gore, A.; Richards, A.; Chiu, Y.J.; Fung, H.L.; Bushman, D.; Chiang, H.I.; Chun, J.; Lo, Y.H.; Zhang, K. Massively parallel polymerase cloning and genome sequencing of single cells using nanoliter microwells. Nat. Biotechnol. 2013, 31, 1126–1132. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.E.; Peluso, P.; Baybayan, P.; Yeadon, P.J.; Yu, C.; Fisher, W.; Chin, C.S.; Rapicavoli, N.A.; Rank, D.R.; Li, J.; et al. Long-read, whole genome shotgun sequence data from five model organisms. Nat. Sci. Data 2014. [Google Scholar] [CrossRef]
- Chin, C.S.; Alexander, D.H.; Marks, P.; Klammer, A.A.; Drake, J.; Heiner, C.; Clum, A.; Copeland, A.; Huddleston, J.; Eichler, E.E.; et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods 2013, 10, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Conlan, S.; Thomas, P.J.; Deming, C.; Park, M.; Lau, A.F.; Dekker, J.P.; Snitkin, E.S.; Clark, T.A.; Luong, K.; Song, Y.; et al. Single-molecule sequencing to track plasmid diversity of hospital-associated carbapenemase-producing Enterobacteriaceae. Sci. Transl. Med. 2014, 6. [Google Scholar] [CrossRef]
- Beatson, S.A.; Walker, M.J. Microbiology: Tracking antibiotic resistance. Science 2014, 345, 1454–1455. [Google Scholar] [CrossRef] [PubMed]
- Latif, H.; Li, H.J.; Charusanti, P.; Palsson, B.Ø.; Aziz, R.K. A Gapless, Unambiguous Genome Sequence of the Enterohemorrhagic Escherichia coli O157:H7 Strain EDL933. Genome Announc. 2014, 2, e00821-14. [Google Scholar] [CrossRef] [PubMed]
- Quick, J.; Quinlan, A.R.; Loman, N.J. A reference bacterial genome dataset generated on the MinION™ portable single-molecule nanopore sequencer. Gigascience 2014, 3, 22. [Google Scholar] [CrossRef] [PubMed]
- Trecarichi, E.M.; Cauda, R.; Tumbarello, M. Detecting risk and predicting patient mortality in patients with extended-spectrum beta-lactamase-producing Enterobacteriaceae bloodstream infections. Future Microbiol. 2012, 7, 1173–1189. [Google Scholar] [CrossRef] [PubMed]
- Salipante, S.J.; Roach, D.J.; Kitzman, J.O.; Snyder, M.W.; Stackhouse, B.; Butler-Wu, S.M.; Lee, C.; Cookson, B.T.; Shendure, J. Large-scale genomic sequencing of extraintestinal pathogenic Escherichia coli strains. Genome Res. 2015, 25, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Russo, T.A.; Johnson, J.R. Proposal for a new inclusive designation for extraintestinal pathogenic isolates of Escherichia coli: ExPEC. J. Infect. Dis. 2000, 181, 1753–1754. [Google Scholar] [CrossRef] [PubMed]
- Foxman, B. The epidemiology of urinary tract infection. Nat. Rev. Urol. 2010, 7, 653–660. [Google Scholar] [CrossRef] [PubMed]
- Didelot, X.; Meric, G.; Falush, D.; Darling, A.E. Impact of homologous and non-homologous recombination in the genomic evolution of Escherichia coli. BMC Genomics 2012, 13, 256. [Google Scholar] [CrossRef] [PubMed]
- Wiles, T.J.; Kulesus, R.R.; Mulvey, M.A. Origins and virulence mechanisms of uropathogenic Escherichia coli. Exp. Mol. Pathol. 2008, 85, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Day, M.J.; Mafura, M.T.; Nunez-Garcia, J.; Fenner, J.J.; Sharma, M.; Rodríguez, I.; van Essen-Zandbergen, A.; Dierikx, C.; Kadlec, K.; et al. Comparative analysis of ESBL-positive Escherichia coli isolates from animals and humans from the UK, The Netherlands and Germany. PLoS ONE 2013, 8, e75392. [Google Scholar] [CrossRef]
- Tango, C.N.; Mansur, A.R.; Oh, D.H. Fumaric Acid and Slightly Acidic Electrolyzed Water Inactivate Gram Positive and Gram Negative Foodborne Pathogens. Microorganisms 2015, 3, 34–46. [Google Scholar] [CrossRef]
- Vincent, C.; Boerlin, P.; Daignault, D.; Dozois, C.M.; Dutil, L.; Galanakis, C.; Reid-Smith, R.J.; Tellier, P.P.; Tellis, P.A.; Ziebell, K.; et al. Food reservoir for Escherichia coli causing urinary tract infections. Emerg. Infect. Dis. 2010, 16, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Lupindu, A.M.; Olsen, J.E.; Ngowi, H.A.; Msoffe, P.L.; Mtambo, M.M.; Scheutz, F.; Dalsgaard, A. Occurrence and characterization of Shiga toxin-producing Escherichia coli O157:H7 and other non-sorbitol-fermenting E. coli in cattle and humans in urban areas of Morogoro, Tanzania. Vector Borne Zoonotic Dis. 2014, 14, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Ewers, C.; Grobbel, M.; Stamm, I.; Kopp, P.A.; Diehl, I.; Semmler, T.; Fruth, A.; Beutlich, J.; Guerra, B.; Wieler, L.H.; et al. Emergence of human pandemic O25:H4-ST131 CTX-M-15 extended-spectrum-beta-lactamase-producing Escherichia coli among companion animals. J. Antimicrob. Chemother. 2010, 65, 651–660. [Google Scholar] [CrossRef] [PubMed]
- Nebbia, P.; Tramuta, C.; Odore, R.; Nucera, D.; Zanatta, R.; Robino, P. Genetic and phenotypic characterisation of Escherichia coli producing cefotaximase-type extended-spectrum beta-lactamases: First evidence of the ST131 clone in cats with urinary infections in Italy. J. Feline Med. Surg. 2014, 16, 966–971. [Google Scholar] [CrossRef] [PubMed]
- Köhler, C.D.; Dobrindt, U. What defines extraintestinal pathogenic Escherichia coli? Int. J. Med. Microbiol. 2011, 301, 642–647. [Google Scholar] [CrossRef] [PubMed]
- Madigan, T.; Johnson, J.R.; Clabots, C.; Johnston, B.D.; Porter, S.B.; Slater, B.S.; Banerjee, R. Extensive household outbreak of urinary tract infection and intestinal colonization due to extended-spectrum beta lactamase (ESBL)-producing Escherichia coli sequence type 131 (ST131). Clin. Infect. Dis. 2015. [Google Scholar] [CrossRef]
- Dahbi, G.; Mora, A.; López, C.; Alonso, M.P.; Mamani, R.; Marzoa, J.; Coira, A.; García-Garrote, F.; Pita, J.M.; Velasco, D.; et al. Emergence of new variants of ST131 clonal group among extraintestinal pathogenic Escherichia coli producing extended-spectrum beta-lactamases. Int. J. Antimicrob. Agents 2013, 42, 347–351. [Google Scholar] [CrossRef] [PubMed]
- Izdebski, R.; Baraniak, A.; Fiett, J.; Adler, A.; Kazma, M.; Salomon, J.; Lawrence, C.; Rossini, A.; Salvia, A.; Vidal Samso, J.; et al. Clonal structure, extended-spectrum beta-lactamases, and acquired AmpC-type cephalosporinases of Escherichia coli populations colonizing patients in rehabilitation centers in four countries. Antimicrob. Agents Chemother. 2013, 57, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, Y.; Yamamoto, M.; Higuchi, T.; Komori, T.; Tsuboi, F.; Hayashi, A.; Sugimoto, Y.; Hotta, G.; Matsushima, A.; Nagao, M.; et al. Prevalence of plasmid-mediated AmpC beta-lactamase-producing Escherichia coli and spread of the ST131 clone among extended-spectrum beta-lactamase-producing E. coli in Japan. Int. J. Antimicrob. Agents 2012, 40, 158–162. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.R.; Urban, C.; Weissman, S.J.; Jorgensen, J.H.; Lewis, J.S.; Hansen, G.; Edelstein, P.H.; Robicsek, A.; Cleary, T.; Adachi, J.; et al. Molecular epidemiological analysis of Escherichia coli sequence type ST131 (O25, H4) and blaCTX-M-15 among extended-spectrum-beta-lactamase-producing E. coli from the United States, 2000 to 2009. Antimicrob. Agents Chemother. 2012, 56, 2364–2370. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.F.; Freeman, J.T.; Nicholson, B.; Keiger, A.; Lancaster, S.; Joyce, M.; Woods, C.W.; Cook, E.; Adcock, L.; Louis, S.; et al. Widespread dissemination of CTX-M-15 genotype extended-spectrum-beta-lactamase-producing enterobacteriaceae among patients presenting to community hospitals in the southeastern United States. Antimicrob. Agents Chemother. 2014, 58, 1200–1202. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.R.; Menard, M.; Johnston, B.; Kuskowski, M.A.; Nichol, K.; Zhanel, G.G. Epidemic clonal groups of Escherichia coli as a cause of antimicrobial-resistant urinary tract infections in Canada, 2002 to 2004. Antimicrob. Agents Chemother. 2009, 53, 2733–2739. [Google Scholar] [CrossRef] [PubMed]
- Fennell, J.; Vellinga, A.; Hanahoe, B.; Morris, D.; Boyle, F.; Higgins, F.; Lyons, M.; O’Connell, K.; Keady, D.; Cormican, M. Increasing prevalence of ESBL production among Irish clinical Enterobacteriaceae from 2004 to 2008: An observational study. BMC Infect. Dis. 2012, 12, 116. [Google Scholar] [CrossRef] [PubMed]
- Clark, G.; Paszkiewicz, K.; Hale, J.; Weston, V.; Constantinidou, C.; Penn, C.; Achtman, M.; McNally, A. Genomic analysis uncovers a phenotypically diverse but genetically homogeneous Escherichia coli ST131 clone circulating in unrelated urinary tract infections. J. Antimicrob. Chemother. 2012, 67, 868–877. [Google Scholar] [CrossRef] [PubMed]
- Rogers, B.A.; Sidjabat, H.E.; Paterson, D.L. Escherichia coli O25b-ST131: A pandemic, multiresistant, community-associated strain. J. Antimicrob. Chemother. 2011, 66, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Woodford, N.; Ward, M.E.; Kaufmann, M.E.; Turton, J.; Fagan, E.J.; James, D.; Johnson, A.P.; Pike, R.; Warner, M.; Cheasty, T.; et al. Community and hospital spread of Escherichia coli producing CTX-M extended-spectrum beta-lactamases in the UK. J. Antimicrob. Chemother. 2004, 54, 735–743. [Google Scholar] [CrossRef] [PubMed]
- Escobar-Paramo, P.; Grenet, K.; Le Menac’h, A.; Rode, L.; Salgado, E.; Amorin, C.; Gouriou, S.; Picard, B.; Rahimy, M.C.; Andremont, A.; et al. Large-scale population structure of human com- mensal Escherichia coli isolates. Appl. Environ. Microbiol. 2004, 70, 5698–5700. [Google Scholar] [CrossRef] [PubMed]
- Escobar-Paramo, P.; Le Menac’h, A.; Le Gall, T.; Amorin, C.; Gouriou, S.; Picard, B.; Skurnik, D.; Denamur, E. Identification of forces shaping the commensal Escherichia coli genetic structure by comparing animal and human isolates. Environ. Microbiol. Rep. 2006, 8, 1975–1984. [Google Scholar] [CrossRef]
- Le Gall, T.; Clermont, O.; Gouriou, S.; Picard, B.; Nassif, X.; Denamur, E.; Tenaillon, O. Extraintestinal virulence is a coincidental by-product of commensalism in B2 phylogenetic group Escherichia coli strains. Mol. Biol. Evol. 2007, 24, 2373–2384. [Google Scholar] [CrossRef] [PubMed]
- Platell, J.L.; Johnson, J.R.; Cobbold, R.N.; Trott, D.J. Multidrug-resistant extraintestinal pathogenic Escherichia coli of sequence type ST131 in animals and foods. Vet. Microbiol. 2011, 153, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Zong, Z.; Yu, R. blaCTX-M-carrying Escherichia coli of the O25b ST131 clonal group have emerged in China. Diagn. Microbiol. Infect. Dis. 2011, 69, 228–231. [Google Scholar] [CrossRef] [PubMed]
- Olesen, B.; Hansen, D.S.; Nilsson, F.; Frimodt-Møller, J.; Leihof, R.F.; Struve, C.; Scheutz, F.; Johnston, B.; Krogfelt, K.A.; Johnson, J.R. Prevalence and characteristics of the epidemic multiresistant Escherichia coli ST131 clonal group among extended-spectrum beta-lactamase-producing E. coli isolates in Copenhagen, Denmark. J. Clin. Microbiol. 2013, 51, 1779–1785. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, Y.; Yamamoto, M.; Nagao, M.; Hotta, G.; Matsushima, A.; Ito, Y.; Takakura, S.; Ichiyama, S. Emergence and spread of B2-ST131-O25b, B2-ST131-O16 and D-ST405 clonal groups among extended-spectrum-beta-lactamase-producing Escherichia coli in Japan. J. Antimicrob. Chemother. 2012, 67, 2612–2620. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.R.; Johnston, B.; Clabots, C.; Kuskowski, M.A.; Castanheira, M. Escherichia coli sequence type ST131 as the major cause of serious multidrug-resistant E. coli infections in the United States 2007. Clin. Infect. Dis. 2010, 51, 286–294. [Google Scholar] [CrossRef] [PubMed]
- Lau, S.H.; Kaufmann, M.E.; Livermore, D.M.; Woodford, N.; Willshaw, G.A.; Cheasty, T.; Stamper, K.; Reddy, S.; Cheesbrough, J.; Bolton, F.J.; et al. UK epidemic Escherichia coli strains A-E, with CTX-M-15 beta-lactamase, all belong to the international O25:H4-ST131 clone. J. Antimicrob. Chemother. 2008, 62, 1241–1244. [Google Scholar] [CrossRef] [PubMed]
- Alobwede, I.; M’Zali, F.H.; Livermore, D.M.; Heritage, J.; Todd, N.; Hawkey, P.M. CTX-M extended-spectrum beta-lactamase arrives in the UK. J. Antimicrob. Chemother. 2003, 51, 470–471. [Google Scholar] [CrossRef] [PubMed]
- Olesen, B.; Frimodt-Møller, J.; Leihof, R.F.; Struve, C.; Johnston, B.; Hansen, D.S.; Scheutz, F.; Krogfelt, K.A.; Kuskowski, M.A.; Clabots, C.; et al. Temporal trends in antimicrobial resistance and virulence-associated traits within the Escherichia coli sequence type 131 clonal group and its H30 and H30-Rx subclones, 1968 to 2012. Antimicrob. Agents Chemother. 2014, 58, 6886–6895. [Google Scholar] [CrossRef] [PubMed]
- Huttner, A.; Harbarth, S.; Carlet, J.; Cosgrove, S.; Goossens, H.; Holmes, A.; Jarlier, V.; Voss, A.; Pittet, D. Antimicrobial resistance: A global view from the 2013 World Healthcare-Associated Infections Forum. Antimicrob. Resist. Infect. Control. 2013, 2, 31. [Google Scholar] [CrossRef] [PubMed]
- Gupta, K.; Scholes, D.; Stamm, W.E. Increasing prevalence of antimicrobial resistance among uropathogens causing acute uncomplicated cystitis in women. JAMA 1999, 281, 736–738. [Google Scholar] [CrossRef] [PubMed]
- Andersson, D.I.; Hughes, D. Evolution of antibiotic resistance at non-lethal drug concentrations. Drug Resist. Updat. 2012, 15, 162–172. [Google Scholar] [CrossRef] [PubMed]
- Oz, T.; Guvenek, A.; Yildiz, S.; Karaboga, E.; Tamer, Y.T.; Mumcuyan, N.; Ozan, V.B.; Senturk, G.H.; Cokol, M.; Yeh, P.; et al. Strength of selection pressure is an important parameter contributing to the complexity of antibiotic resistance evolution. Mol. Biol. Evol. 2014, 31, 2387–2401. [Google Scholar] [CrossRef] [PubMed]
- Harris, S.; Morris, C.; Morris, D.; Cormican, M.; Cummins, E. The effect of hospital effluent on antimicrobial resistant E. coli within a municipal wastewater system. Environ. Sci. Process. Impacts 2013, 15, 617–622. [Google Scholar] [CrossRef] [PubMed]
- Galvin, S.; Boyle, F.; Hickey, P.; Vellinga, A.; Morris, D.; Cormican, M. Enumeration and characterization of antimicrobial-resistant Escherichia coli bacteria in effluent from municipal, hospital, and secondary treatment facility sources. Appl. Environ. Microbiol. 2010, 76, 4772–4779. [Google Scholar] [CrossRef] [PubMed]
- Udikovic-Kolic, N.; Wichmann, F.; Broderick, N.A.; Handelsman, J. Bloom of resident antibiotic-resistant bacteria in soil following manure fertilization. Proc. Natl. Acad. Sci. USA 2014, 111, 15202–15207. [Google Scholar] [CrossRef] [PubMed]
- Currie, C.J.; Berni, E.; Jenkins-Jones, S.; Poole, C.D.; Ouwens, M.; Driessen, S.; de Voogd, H.; Butler, C.C.; Morgan, C.L. Antibiotic treatment failure in four common infections in UK primary care 1991–2012: Longitudinal analysis. BMJ 2014, 349, g5493. [Google Scholar] [CrossRef] [PubMed]
- Morris, D.; Boyle, F.; Buckley, V.; Xu, L.; Hanahoe, B.; Hawkey, P.; Cormican, M. CTX-M enzymes are the predominant extended-spectrum beta-lactamases produced by Enterobacteriaceae in Ireland. J. Antimicrob. Chemother. 2009, 64, 864–866. [Google Scholar] [CrossRef] [PubMed]
- Morris, D.; McGarry, E.; Cotter, M.; Passet, V.; Lynch, M.; Ludden, C.; Hannan, M.M.; Brisse, S.; Cormican, M. Detection of OXA-48 carbapenemase in the pandemic clone Escherichia coli O25b:H4-ST131 in the course of investigation of an outbreak of OXA-48-producing Klebsiella pneumoniae. Antimicrob. Agents Chemother. 2012, 56, 4030–4031. [Google Scholar] [CrossRef] [PubMed]
- Agabou, A.; Pantel, A.; Ouchenane, Z.; Lezzar, N.; Khemissi, S.; Satta, D.; Sotto, A.; Lavigne, J.P. First description of OXA-48-producing Escherichia coli and the pandemic clone ST131 from patients hospitalised at a military hospital in Algeria. Eur. J. Clin. Microbiol. Infect. Dis. 2014, 33, 1641–1646. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.C.; Zhang, R.; Hu, Y.Y.; Zhou, H.W.; Chen, G.X. Emergence of Escherichia coli sequence type 131 isolates producing KPC-2 carbapenemase in China. Antimicrob. Agents Chemother. 2014, 58, 1146–1152. [Google Scholar] [CrossRef] [PubMed]
- Morris, D.; Boyle, F.; Ludden, C.; Condon, I.; Hale, J.; O’Connell, N.; Power, L.; Boo, T.W.; Dhanji, H.; Lavallee, C.; et al. Production of KPC-2 carbapenemase by an Escherichia coli clinical isolate belonging to the international ST131 clone. Antimicrob. Agents Chemother. 2011, 55, 4935–4936. [Google Scholar] [CrossRef] [PubMed]
- Riley, L.W. Pandemic lineages of extraintestinal pathogenic Escherichia coli. Clin. Microbiol. Infect. 2014, 20, 380–390. [Google Scholar] [CrossRef] [PubMed]
- Blanco, J.; Mora, A.; Mamani, R.; Lopez, C.; Blanco, M.; Dahbi, G.; Herrera, A.; Marzoa, J.; Fernandez, V.; de la Cruz, F.; et al. Four main virotypes among extended-spectrum-beta-lactamase-producing isolates of Escherichia coli O25b:H4-B2-ST131: Bacterial, epidemiological, and clinical characteristics. J. Clin. Microbiol. 2013, 51, 3358–3367. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, R.; Johnston, B.; Lohse, C.; Porter, S.B.; Clabots, C.; Johnson, J.R. Escherichia coli sequence type 131 is a dominant, antimicrobial-resistant clonal group associated with healthcare and elderly hosts. Infect. Control. Hosp. Epidemiol. 2013, 34, 361–369. [Google Scholar] [CrossRef] [PubMed]
- Livermore, D.M.; Woodford, N. The beta-lactamase threat in Enterobacteriaceae, Pseudomonas and Acinetobacter. Trends Microbiol. 2006, 14, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Pelly, H.; O’Connell, E.; Hanahoe, B.; Chambers, C.; Biernacka, K.; Gray, S.; Cormican, M. Outbreak of extended spectrum beta-lactamase producing E. coli in a nursing home in Ireland, May 2006. Euro Surveill. 2006, 11, E060831.1. [Google Scholar] [PubMed]
- Burke, L.; Humphreys, H.; Fitzgerald-Hughes, D. The revolving door between hospital and community: Extended-spectrum beta-lactamase-producing Escherichia coli in Dublin. J. Hosp. Infect. 2012, 81, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Pitout, J.D.; Gregson, D.B.; Campbell, L.; Laupland, K.B. Molecular characteristics of extended-spectrum-beta-lactamase-producing Escherichia coli isolates causing bacteremia in the Calgary Health Region from 2000 to 2007: Emergence of clone ST131 as a cause of community-acquired infections. Antimicrob. Agents Chemother. 2009, 53, 2846–2851. [Google Scholar] [CrossRef] [PubMed]
- Karami, N.; Helldal, L.; Welinder-Olsson, C.; Ahrén, C.; Moore, E.R. Sub-typing of extended-spectrum-beta-lactamase-producing isolates from a nosocomial outbreak: Application of a 10-loci generic Escherichia coli multi-locus variable number tandem repeat analysis. PLoS ONE 2013, 8, e83030. [Google Scholar] [CrossRef] [PubMed]
- Shakir, S.M.; Goldbeck, J.M.; Robison, D.; Eckerd, A.M.; Chavez-Bueno, S. Genotypic and Phenotypic Characterization of Invasive Neonatal Escherichia coli Clinical Isolates. Am. J. Perinatol. 2014, 31, 975–982. [Google Scholar] [CrossRef] [PubMed]
- Ludden, C.; Cormican, M.; Vellinga, A.; Johnson, J.R.; Austin, B.; Morris, D. Colonisation with ESBL-producing and carbapenemase-producing Enterobacteriaceae, vancomycin-resistant enterococci, and meticillin-resistant Staphylococcus aureus in a long-term care facility over one year. BMC Infect. Dis. 2015, 15, 168. [Google Scholar] [CrossRef] [PubMed]
- Suetens, C. Healthcare-associated infections in European long-term care facilities: How big is the challenge? Euro Surveill. 2012, 17. pii. 20259. [Google Scholar] [PubMed]
- Peirano, G.; Pitout, J.D.D. Molecular epidemiology of Escherichia coli producing CTX-M β-lactamases: The worldwide emergence of clone ST131 O25:H4. Int. J. Antimicrob. Agents 2010, 35, 316–321. [Google Scholar] [CrossRef] [PubMed]
- Van Tonder, A.J.; Mistry, S.; Bray, J.E.; Hill, D.M.; Cody, A.J.; Farmer, C.L.; Klugman, K.P.; von Gottberg, A.; Bentley, S.D.; Parkhill, J.; et al. Defining the estimated core genome of bacterial populations using a Bayesian decision model. PLoS Comput. Biol. 2014, 10, e1003788. [Google Scholar] [CrossRef] [PubMed]
- Hooper, S.D.; Berg, O.G. Duplication is more common among laterally transferred genes than among indigenous genes. Genome Biol. 2003, 4, R48. [Google Scholar] [CrossRef] [PubMed]
- McNally, A.; Cheng, L.; Harris, S.R.; Corander, J. The evolutionary path to extraintestinal pathogenic, drug-resistant Escherichia coli is marked by drastic reduction in detectable recombination within the core genome. Genome Biol. Evol. 2013, 5, 699–710. [Google Scholar] [CrossRef] [PubMed]
- Rasko, D.A.; Rosovitz, M.J.; Myers, G.S.; Mongodin, E.F.; Fricke, W.F.; Gajer, P.; Crabtree, J.; Sebaihia, M.; Thomson, N.R.; Chaudhuri, R.; et al. The pangenome structure of Escherichia coli: comparative genomic analysis of Escherichia coli commensal and pathogenic isolates. J. Bacteriol. 2008, 190, 6881–6893. [Google Scholar] [CrossRef] [PubMed]
- Price, L.B.; Johnson, J.R.; Aziz, M.; Clabots, C.; Johnston, B.; Tchesnokova, V.; Nordstrom, L.; Billig, M.; Chattopadhyay, S.; Stegger, M.; et al. The epidemic of extended-spectrum-beta-lactamase-producing Escherichia coli ST131 is driven by a single highly pathogenic subclone, H30-Rx. mBio 2013, 4, e00377-13. [Google Scholar] [PubMed]
- Treangen, T.J.; Rocha, E.P. Horizontal transfer, not duplication, drives the expansion of protein families in prokaryotes. PLoS Genet 2011, 7, e1001284. [Google Scholar] [CrossRef] [PubMed]
- Bobay, L.M.; Touchon, M.; Rocha, E.P. Pervasive domestication of defective prophages by bacteria. Proc. Natl. Acad. Sci. USA 2014, 111, 12127–12132. [Google Scholar] [CrossRef] [PubMed]
- Näsvall, J.; Sun, L.; Roth, J.R.; Andersson, D.I. Real-time evolution of new genes by innovation, amplification, and divergence. Science 2012, 338, 384–387. [Google Scholar] [CrossRef] [PubMed]
- Bobay, L.M.; Rocha, E.P.; Touchon, M. The adaptation of temperate bacteriophages to their host genomes. Mol. Biol. Evol. 2013, 30, 737–751. [Google Scholar] [CrossRef] [PubMed]
- Waldor, M.K.; Friedman, D.I. Phage regulatory circuits and virulence gene expression. Curr. Opin. Microbiol. 2005, 8, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Edlin, G.; Lin, L.; Bitner, R. Reproductive fitness of P1, P2, and Mu lysogens of Escherichia coli. J. Virol. 1977, 21, 560–564. [Google Scholar] [PubMed]
- Rabinovich, L.; Sigal, N.; Borovok, I.; Nir-Paz, R.; Herskovits, A.A. Prophage excision activates Listeria competence genes that promote phagosomal escape and virulence. Cell 2012, 150, 792–802. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Kim, Y.; Ma, Q.; Hong, S.H.; Pokusaeva, K.; Sturino, J.M.; Wood, T.K. Cryptic prophages help bacteria cope with adverse environments. Nat. Commun. 2010, 1. [Google Scholar] [CrossRef]
- Gödeke, J.; Paul, K.; Lassak, J.; Thormann, K.M. Phage-induced lysis enhances biofilm formation in Shewanella oneidensis MR-1. ISME J. 2011, 5, 613–626. [Google Scholar] [CrossRef] [PubMed]
- Wozniak, R.A.; Fouts, D.E.; Spagnoletti, M.; Colombo, M.M.; Ceccarelli, D.; Garriss, G.; Déry, C.; Burrus, V.; Waldor, M.K. Comparative ICE genomics: insights into the evolution of the SXT/R391 family of ICEs. PLoS Genet 2009, 5, e1000786. [Google Scholar] [CrossRef] [PubMed]
- D’Agata, E.M.; Dupont-Rouzeyrol, M.; Magal, P.; Olivier, D.; Ruan, S. The impact of different antibiotic regimens on the emergence of antimicrobial-resistant bacteria. PLoS ONE 2008, 3, e4036. [Google Scholar] [CrossRef] [PubMed]
- Palmer, A.C.; Kishony, R. Opposing effects of target overexpression reveal drug mechanisms. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Avasthi, T.S.; Kumar, N.; Baddam, R.; Hussain, A.; Nandanwar, N.; Jadhav, S.; Ahmed, N. Genome of multidrug-resistant uropathogenic Escherichia coli strain NA114 from India. J. Bacteriol. 2011, 193, 4272–4273. [Google Scholar] [CrossRef] [PubMed]
- Andersen, P.S.; Stegger, M.; Aziz, M.; Contente-Cuomo, T.; Gibbons, H.S.; Keim, P.; Sokurenko, E.V.; Johnson, J.R.; Price, L.B. Complete Genome Sequence of the Epidemic and Highly Virulent CTX-M-15-Producing H30-Rx Subclone of Escherichia coli ST131. Genome Announc 2013, 1, e00988-13. [Google Scholar] [CrossRef] [PubMed]
- Forde, B.M.; Zakour, B.; Stanton-Cook, M.; Phan, M.D.; Totsika, M.; Peters, K.M.; Chan, K.G.; Schembri, M.A.; Upton, M.; Beatson, S.A. The Complete Genome Sequence of Escherichia coli EC958: A High Quality Reference Sequence for the Globally Disseminated Multidrug Resistant E. coli O25b:H4-ST131 Clone. PLoS ONE 2014, 9, e104400. [Google Scholar] [CrossRef] [PubMed]
- Johnson, T.J.; Hargreaves, M.; Shaw, K.; Snippes, P.; Lynfield, R.; Aziz, M.; Price, L.B. Complete Genome Sequence of a Carbapenem-Resistant Extraintestinal Pathogenic Escherichia coli Strain Belonging to the Sequence Type 131 H30R Subclade. Genome Announc. 2015, 3. [Google Scholar] [CrossRef]
- Totsika, M.; Beatson, S.A.; Sarkar, S.; Phan, M.D.; Petty, N.K.; Bachmann, N.; Szubert, M.; Sidjabat, H.E.; Paterson, D.L.; Upton, M.; et al. Insights into a multidrug resistant Escherichia coli pathogen of the globally disseminated ST131 lineage: Genome analysis and virulence mechanisms. PLoS ONE 2011, 6, e26578. [Google Scholar] [CrossRef] [PubMed]
- Clermont, O.; Lavollay, M.; Vimont, S.; Deschamps, C.; Forestier, C.; Branger, C.; Denamur, E.; Arlet, G. The CTX-M-15-producing Escherichia coli diffusing clone belongs to a highly virulent B2 phylogenetic subgroup. J. Antimicrob. Chemother. 2008, 61, 1024–1028. [Google Scholar] [CrossRef] [PubMed]
- Petty, N.K.; Zakour, B.; Stanton-Cook, M.; Skippington, E.; Totsika, M.; Forde, B.M.; Phan, M.D.; Moriel, D.G.; Peters, K.M.; Davies, M.; et al. Global dissemination of a multidrug resistant Escherichia coli clone. Proc. Natl. Acad. Sci. USA 2014, 111, 5694–5699. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.R.; Tchesnokova, V.; Johnston, B.; Clabots, C.; Roberts, P.L.; Billig, M.; Riddell, K.; Rogers, P.; Qin, X.; Butler-Wu, S.; et al. Abrupt emergence of a single dominant multi-drug-resistant strain of Escherichia coli. J. Infect. Dis. 2013, 207, 919–928. [Google Scholar] [CrossRef] [PubMed]
- Peirano, G.; van der Bij, A.K.; Freeman, J.L.; Poirel, L.; Nordmann, P.; Costello, M.; Pitout, J.D.; Tchesnokova, V.L. Characteristics of Escherichia coli sequence type 131 isolates that produce extended-spectrum beta-lactamases: Global distribution of the H30-Rx sublineage. Antimicrob. Agents Chemother. 2014, 58, 3762–3767. [Google Scholar] [CrossRef] [PubMed]
- Peirano, G.; Pitout, J.D. Fluoroquinolone-resistant Escherichia coli sequence type 131 isolates causing bloodstream infections in a Canadian region with a centralized laboratory system: Rapid emergence of the H30-Rx sublineage. Antimicrob. Agents Chemother. 2014, 58, 2699–2703. [Google Scholar] [CrossRef] [PubMed]
- Cantón, R.; Coque, T.M. The CTX-M beta-lactamase pandemic. Curr. Opin. Microbiol. 2006, 9, 466–475. [Google Scholar] [CrossRef] [PubMed]
- Rooney, P.J.; O’Leary, M.C.; Loughrey, A.C.; McCalmont, M.; Smyth, B.; Donaghy, P.; Badri, M.; Woodford, N.; Karisik, E.; Livermore, D.M. Nursing homes as a reservoir of extended-spectrum beta-lactamase (ESBL)-producing ciprofloxacin-resistant Escherichia coli. J. Antimicrob. Chemother. 2009, 64, 635–641. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, R.; Johnson, J.R. Escherichia coli ST131: Variations on a theme of clonal expansion. Enferm Infecc Microbiol. Clin. 2013, 31, 355–356. [Google Scholar] [CrossRef] [PubMed]
- Gullberg, E.; Cao, S.; Berg, O.G.; Ilbäck, C.; Sandegren, L.; Hughes, D.; Andersson, D.I. Selection of resistant bacteria at very low antibiotic concentrations. PLoS Pathog. 2011, 7, e1002158. [Google Scholar] [CrossRef] [PubMed]
- Adams-Sapper, S.; Diep, B.A.; Perdreau-Remington, F.; Riley, L.W. Clonal composition and community clustering of drug-susceptible and -resistant Escherichia coli isolates from bloodstream infections. Antimicrob. Agents Chemother. 2013, 57, 490–497. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.R.; Murray, A.C.; Gajewski, A.; Sullivan, M.; Snippes, P.; Kuskowski, M.A.; Smith, K.E. Isolation and molecular characterization of nalidixic acid-resistant extraintestinal pathogenic Escherichia coli from retail chicken products. Antimicrob. Agents Chemother. 2003, 47, 2161–2168. [Google Scholar] [CrossRef] [PubMed]
- Pitout, J.D. Extraintestinal Pathogenic Escherichia coli: A Combination of Virulence with Antibiotic Resistance. Front Microbiol. 2012, 3, 9. [Google Scholar] [CrossRef] [PubMed]
- Nicolas-Chanoine, M.H.; Bertrand, X.; Madec, J.Y. Escherichia coli ST131, an intriguing clonal group. Clin. Microbiol. Rev. 2014, 27, 543–574. [Google Scholar] [CrossRef] [PubMed]
- Dhanji, H.; Doumith, M.; Rooney, P.J.; O’Leary, M.C.; Loughrey, A.C.; Hope, R.; Woodford, N.; Livermore, D.M. Molecular epidemiology of fluoroquinolone-resistant ST131 Escherichia coli producing CTX-M extended-spectrum beta-lactamases in nursing homes in Belfast, UK. J. Antimicrob. Chemother. 2011, 66, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Bush, K.; Jacoby, G.A. Updated functional classification of beta-lactamases. Antimicrob. Agents Chemother. 2010, 54, 969–976. [Google Scholar] [CrossRef] [PubMed]
- Jensen, L.B.; Hasman, H.; Agersø, Y.; Emborg, H.-D.; Aarestrup, F.M. First description of an oxyiminocephalosporin-resistant, ESBL-carrying Escherichia coli isolated from meat sold in Denmark. J. Antimicrob. Chemother. 2006, 57, 793–794. [Google Scholar] [CrossRef] [PubMed]
- Yong, D.; Toleman, M.A.; Giske, C.G.; Cho, H.S.; Sundman, K.; Lee, K.; Walsh, T.R. Characterization of a new metallo-beta-lactamase gene, bla(NDM-1), and a novel erythromycin esterase gene carried on a unique genetic structure in Klebsiella pneumoniae sequence type 14 from India. Antimicrob. Agents Chemother. 2009, 53, 5046–5054. [Google Scholar] [CrossRef] [PubMed]
- Naas, T.; Cuzon, G.; Gaillot, O.; Courcol, R.; Nordmann, P. When carbapenem-hydrolyzing beta-lactamase KPC meets Escherichia coli ST131 in France. Antimicrob. Agents Chemother. 2011, 55, 4933–4934. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.A.; Qureshi, Z.A.; Adams-Haduch, J.M.; Park, Y.S.; Shutt, K.A.; Doi, Y. Features of infections due to Klebsiella pneumoniae carbapenemase-producing Escherichia coli: Emergence of sequence type 131. Clin. Infect Dis. 2012, 55, 224–231. [Google Scholar] [CrossRef] [PubMed]
- Mantengoli, E.; Luzzaro, F.; Pecile, P.; Cecconi, D.; Cavallo, A.; Attala, L.; Bartoloni, A.; Rossolini, G.M. Escherichia coli ST131 producing extended-spectrum beta-lactamases plus VIM-1 carbapenemase: Further narrowing of treatment options. Clin. Infect. Dis. 2011, 52, 690–691. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.-J.; Tsai, L.-H.; Wu, J.-J. Emergence of the IMP-8 metallo-beta-lactamase in the Escherichia coli ST131 clone in Taiwan. Int. J. Antimicrob. Agents 2012, 40, 281–282. [Google Scholar] [CrossRef] [PubMed]
- Peirano, G.; Schreckenberger, P.C.; Pitout, J.D. Characteristics of NDM-1-producing Escherichia coli isolates that belong to the successful and virulent clone ST131. Antimicrob. Agents Chemother. 2011, 55, 2986–2988. [Google Scholar] [CrossRef] [PubMed]
- Seema, K.; Ranjan Sen, M.; Upadhyay, S.; Bhattacharjee, A. Dissemination of the New Delhi metallo-beta-lactamase-1 (NDM-1) among Enterobacteriaceae in a tertiary referral hospital in north India. J. Antimicrob. Chemother. 2011, 66, 1646–1647. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, P.; Rodrigues, C.; Shetty, A.; Kapadia, F.; Hedge, A.; Soman, R. New Delhi Metallo-beta lactamase (NDM-1) in Enterobacteriaceae: Treatment options with carbapenems compromised. J. Assoc. Physicians India 2010, 58, 147–149. [Google Scholar] [PubMed]
- Perry, J.D.; Naqvi, S.H.; Mirza, I.A.; Alizai, S.A.; Hussain, A.; Ghirardi, S.; Orenga, S.; Wilkinson, K.; Woodford, N.; Zhang, J.; et al. Prevalence of faecal carriage of Enterobacteriaceae with NDM-1 carbapenemase at military hospitals in Pakistan, and evaluation of two chromogenic media. J. Antimicrob. Chemother. 2011, 66, 2288–2294. [Google Scholar] [CrossRef] [PubMed]
- Bonnin, R.A.; Poirel, L.; Carattoli, A.; Nordmann, P. Characterization of an IncFII plasmid encoding NDM-1 from Escherichia coli ST131. PLoS ONE 2012, 7, e34752. [Google Scholar] [CrossRef] [PubMed]
- Gaillot, O.; Clement, C.; Simonet, M.; Philippon, A. Novel transferable beta-lactam resistance with cephalosporinase characteristics in Salmonella enteritidis. J. Antimicrob. Chemother. 1997, 39, 85–87. [Google Scholar] [CrossRef] [PubMed]
- Sousa, A.; Magalhães, S.; Gordo, I. Cost of antibiotic resistance and the geometry of adaptation. Mol. Biol. Evol. 2012, 29, 1417–1428. [Google Scholar] [CrossRef] [PubMed]
- LeClerc, J.E.; Li, B.G.; Payne, W.L.; Cebula, T.A. High mutation frequencies among Escherichia coli and Salmonella pathogens. Science 1996, 274, 1208–1211. [Google Scholar] [CrossRef] [PubMed]
- Olofsson, S.K.; Geli, P.; Andersson, D.I.; Cars, O. Pharmacodynamic model to describe the concentration-dependent selection of cefotaxime-resistant Escherichia coli. Antimicrob. Agents Chemother. 2005, 49, 5081–5091. [Google Scholar] [CrossRef] [PubMed]
- Silva, R.F.; Mendonça, S.C.; Carvalho, L.M.; Reis, A.M.; Gordo, I.; Trindade, S.; Dionisio, F. Pervasive sign epistasis between conjugative plasmids and drug-resistance chromosomal mutations. PLoS Genet 2011, 7, e1002181. [Google Scholar] [CrossRef] [PubMed]
- Trindade, S.; Sousa, A.; Xavier, K.B.; Dionisio, F.; Ferreira, M.G.; Gordo, I. Positive epistasis drives the acquisition of multidrug resistance. PLoS Genet 2009, 5, e1000578. [Google Scholar] [CrossRef] [PubMed]
- Morgand, M.; Vimont, S.; Bleibtreu, A.; Boyd, A.; Thien, H.V.; Zahar, J.R.; Denamur, E.; Arlet, G. Extended-spectrum beta-lactamase-producing Escherichia coli infections in children: Are community-acquired strains different from nosocomial strains? Int. J. Med. Microbiol. 2014, 304, 970–976. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Beltrán, J.; Tourret, J.; Tenaillon, O.; López, E.; Bourdelier, E.; Costas, C.; Matic, I.; Denamur, E.; Blázquez, J. High Recombinant Frequency in Extraintestinal Pathogenic Escherichia coli Strains. Mol. Biol. Evol. 2015. [Google Scholar] [CrossRef]
- Leflon-Guibout, V.; Blanco, J.; Amaqdouf, K.; Mora, A.; Guize, L.; Nicolas-Chanoine, M.H. Absence of CTX-M enzymes but high prevalence of clones, including clone ST131, among fecal Escherichia coli isolates from healthy subjects living in the area of Paris, France. J. Clin. Microbiol. 2008, 46, 3900–3905. [Google Scholar] [CrossRef] [PubMed]
- Angst, D.C.; Hall, A.R. The cost of antibiotic resistance depends on evolutionary history in Escherichia coli. BMC Evol. Biol. 2013, 13, 163. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, K.; Goto, K.; Nakane, K.; Arakawa, Y. Molecular epidemiology of extended-spectrum beta-lactamases and Escherichia coli isolated from retail foods including chicken meat in Japan. Foodborne Pathog. Dis. 2014, 11, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Randall, L.P.; Clouting, C.; Horton, R.A.; Coldham, N.G.; Wu, G.; Clifton-Hadley, F.A.; Davies, R.H.; Teale, C.J. Prevalence of Escherichia coli carrying extended-spectrum beta-lactamases (CTX-M and TEM-52) from broiler chickens and turkeys in Great Britain between 2006 and 2009. J. Antimicrob. Chemother. 2011, 66, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Kutumbaka, K.K.; Han, S.; Mategko, J.; Nadala, C.; Buser, G.L.; Cassidy, M.P.; Beldavs, Z.G.; Weissman, S.J.; Morey, K.E.; Vega, R.; et al. Draft Genome Sequence of blaNDM-1-Positive Escherichia coli O25b-ST131 Clone Isolated from an Environmental Sample. Genome Announc. 2014, 2. [Google Scholar] [CrossRef] [PubMed]
- Morar, M.; Wright, G.D. The genomic enzymology of antibiotic resistance. Annu. Rev. Genet 2010, 44, 25–51. [Google Scholar] [CrossRef] [PubMed]
- Soo, V.W.; Hanson-Manful, P.; Patrick, W.M. Artificial gene amplification reveals an abundance of promiscuous resistance determinants in Escherichia coli. Proc. Natl. Acad. Sci. USA 2011, 108, 1484–1489. [Google Scholar] [CrossRef] [PubMed]
- Rodrigo, G.; Baji´c, D.; Elola, I.; Poyatos, J.F. Dual autogenous control of the multiple antibiotic resistance phenotype in Escherichia coli. Available online: http://biorxiv.org/content/early/2014/08/19/008169 (accessed on 3 September 2014).
- Ravva, S.V.; Cooley, M.B.; Sarreal, C.Z.; Mandrell, R.E. Fitness of Outbreak and Environmental Strains of Escherichia coli O157:H7 in Aerosolizable Soil and Association of Clonal Variation in Stress Gene Regulation. Pathogens 2014, 3, 528–548. [Google Scholar] [CrossRef] [PubMed]
- Imming, P.; Sinning, C.; Meyer, A. Drugs, their targets and the nature and number of drug targets. Nat. Rev. Drug Discov. 2006, 5, 821–834. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, D.J. Stochastic modelling for quantitative description of heterogeneous biological systems. Nat. Rev. Genet 2009, 10, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, M.L.; le Coq, D.; Jules, M.; Aymerich, S.; Radulescu, O.; Declerck, N.; Royer, C.A. Reconciling molecular regulatory mechanisms with noise patterns of bacterial metabolic promoters in induced and repressed states. Proc. Natl. Acad. Sci. USA 2012, 109, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Wolf, L.; Silander, O.K.; van Nimwegen, E.J. Expression noise facilitates the evolution of gene regulation. Available online: http://biorxiv.org/content/early/2014/07/18/007237 (accessed on 25 July 2014).
- Cheng, A.A.; Dinge, H.; Lu, T.K. Enhanced killing of antibiotic-resistant bacteria enabled by massively parallel combinatorial genetics. Proc. Natl. Acad. Sci. USA 2014, 111, 12462–12467. [Google Scholar] [CrossRef] [PubMed]
- Silander, O.K.; Nikolic, N.; Zaslaver, A.; Bren, A.; Kikoin, I.; Alon, U.; Ackermann, M. A genome-wide analysis of promoter-mediated phenotypic noise in Escherichia coli. PLoS Genet 2012, 8, e1002443. [Google Scholar] [CrossRef] [PubMed]
- Bollenbach, T.; Kishony, R. Resolution of gene regulatory conflicts caused by combinations of antibiotics. Mol. Cell 2011, 42, 413–425. [Google Scholar] [CrossRef] [PubMed]
- Caroff, N.; Espaze, E.; Gautreau, D.; Richet, H.; Reynaud, A. Analysis of the effects of −42 and −32 ampC promoter mutations in clinical isolates of Escherichia coli hyperproducing ampC. J. Antimicrob. Chemother. 2000, 45, 783–788. [Google Scholar] [CrossRef] [PubMed]
- Teichmann, S.A.; Babu, M.M. Gene regulatory network growth by duplication. Nat. Genet 2004, 36, 492–496. [Google Scholar] [CrossRef] [PubMed]
- Toprak, E.; Veres, A.; Michel, J.B.; Chait, R.; Hartl, D.L.; Kishony, R. Evolutionary paths to antibiotic resistance under dynamically sustained drug selection. Nat. Genet 2011, 44, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Thomas, R. Laws for the dynamics of regulatory networks. Int. J. Dev. Biol. 1998, 42, 479–485. [Google Scholar] [PubMed]
- Kim, S.; Lieberman, T.D.; Kishony, R. Alternating antibiotic treatments constrain evolutionary paths to multidrug resistance. Proc. Natl. Acad. Sci. USA 2014, 111, 14494–14499. [Google Scholar] [CrossRef] [PubMed]
- Adler, M.; Anjum, M.; Andersson, D.I.; Sandegren, L. Influence of acquired beta-lactamases on the evolution of spontaneous carbapenem resistance in Escherichia coli. J. Antimicrob. Chemother. 2013, 68, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Raeside, C.; Gaffé, J.; Deatherage, D.E.; Tenaillon, O.; Briska, A.M.; Ptashkin, R.N.; Cruveiller, S.; Médigue, C.; Lenski, R.E.; Barrick, J.E.; et al. Large chromosomal rearrangements during a long-term evolution experiment with Escherichia coli. mBio 2014, 5, e01377-14. [Google Scholar] [CrossRef] [PubMed]
- Aldridge, B.B.; Fernandez-Suarez, M.; Heller, D.; Ambravaneswaran, V.; Irimia, D.; Toner, M.; Fortune, S.M. Asymmetry and aging of mycobacterial cells lead to variable growth and antibiotic susceptibility. Science 2012, 335, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Klumpp, S.; Zhang, Z.; Hwa, T. Growth rate-dependent global effects on gene expression in bacteria. Cell 2009, 139, 1366–1375. [Google Scholar] [CrossRef] [PubMed]
- Elowitz, M.B.; Levine, A.J.; Siggia, E.D.; Swain, P.S. Stochastic gene expression in a single cell. Science 2002, 297, 1183–1186. [Google Scholar] [CrossRef] [PubMed]
- Balaban, N.Q.; Merrin, J.; Chait, R.; Kowalik, L.; Leibler, S. Bacterial persistence as a phenotypic switch. Science 2004, 305, 1622–1625. [Google Scholar] [CrossRef] [PubMed]
- Fasani, R.A.; Savageau, M.A. Molecular mechanisms of multiple toxin-antitoxin systems are coordinated to govern the persister phenotype. Proc. Natl. Acad. Sci. USA 2013, 110, E2528–E2537. [Google Scholar] [CrossRef] [PubMed]
- Fridman, O.; Goldberg, A.; Ronin, I.; Shoresh, N.; Balaban, N.Q. Optimization of lag time underlies antibiotic tolerance in evolved bacterial populations. Nature 2014, 513, 418–421. [Google Scholar] [CrossRef] [PubMed]
- Lou, C.; Li, Z.; Ouyang, Q. A molecular model for persister in E. coli. J. Theor. Biol. 2008, 255, 205–209. [Google Scholar] [CrossRef] [PubMed]
- Grundmann, H. Towards a global antibiotic resistance surveillance system: A primer for a roadmap. Upsala J. Med. Sci. 2014, 119, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Li, L.M.; Grassly, N.C.; Fraser, C. Genomic analysis of emerging pathogens: Methods, application and future trends. Genome Biol. 2014, 15, 541. [Google Scholar] [CrossRef] [PubMed]
- Webber, M.A.; Ricci, V.; Whitehead, R.; Patel, M.; Fookes, M.; Ivens, A.; Piddock, L.J. Clinically relevant mutant DNA gyrase alters supercoiling, changes the transcriptome, and confers multidrug resistance. mBio 2013, 4, e00273-13. [Google Scholar] [CrossRef] [PubMed]
- Joensen, K.G.; Scheutz, F.; Lund, O.; Hasman, H.; Kaas, R.S.; Nielsen, E.M.; Aarestrup, F.M. Real-time whole-genome sequencing for routine typing, surveillance, and outbreak detection of verotoxigenic Escherichia coli. J. Clin. Microbiol. 2014, 52, 1501–1510. [Google Scholar] [CrossRef] [PubMed]
- Caboche, S.; Audebert, C.; Hot, D. High-Throughput Sequencing, a Versatile Weapon to Support Genome-Based Diagnosis in Infectious Diseases: Applications to Clinical Bacteriology. Pathogens 2014, 3, 258–279. [Google Scholar] [CrossRef] [PubMed]
- Alqasim, A.; Scheutz, F.; Zong, Z.; McNally, A. Comparative genome analysis identifies few traits unique to the Escherichia coli ST131 H30Rx clade and extensive mosaicism at the capsule locus. BMC Genomics 2014, 15, 830. [Google Scholar] [CrossRef] [PubMed]
- Yahara, K.; Furuta, Y.; Oshima, K.; Yoshida, M.; Azuma, T.; Hattori, M.; Uchiyama, I.; Kobayashi, I. Chromosome painting in silico in a bacterial species reveals fine population structure. Mol. Biol. Evol. 2013, 30, 1454–1464. [Google Scholar] [CrossRef] [PubMed]
- Yahara, K.; Didelot, X.; Ansari, M.A.; Sheppard, S.K.; Falush, D. Efficient inference of recombination hot regions in bacterial genomes. Mol. Biol. Evol. 2014, 31, 1593–1605. [Google Scholar] [CrossRef] [PubMed]
- Shimodaira, H. An approximately unbiased test of phylogenetic tree selection. Syst. Biol. 2002, 51, 492–508. [Google Scholar] [CrossRef] [PubMed]
- Schierup, M.H. Consequences of recombination on traditional phylogenetic analysis. Genetics 2000, 156, 879–891. [Google Scholar] [PubMed]
- Barrick, J.E.; Kauth, M.R.; Strelioff, C.C.; Lenski, R.E. Escherichia coli rpoB mutants have increased evolvability in proportion to their fitness defects. Mol. Biol. Evol. 2010, 27, 1338–1347. [Google Scholar] [CrossRef] [PubMed]
- Pybus, O.G.; Rambaut, A.; Belshaw, R.; Freckleton, R.P.; Drummond, A.J.; Holmes, E.C. Phylogenetic evidence for deleterious mutation load in RNA viruses and its contribution to viral evolution. Mol. Biol. Evol. 2007, 24, 845–852. [Google Scholar] [CrossRef] [PubMed]
- Neher, R.A.; Russell, C.A.; Shraiman, B.I. Predicting evolution from the shape of genealogical trees. Elife 2014, 3. [Google Scholar] [CrossRef]
- Dobrindt, U. (Patho-)Genomics of Escherichia coli. Int. J. Med. Microbiol. 2005, 295, 357–371. [Google Scholar] [CrossRef] [PubMed]
- Freeman, J.T.; McBride, S.J.; Heffernan, H.; Bathgate, T.; Pope, C.; Ellis-Pegler, R.B. Community-onset genitourinary tract infection due to CTX-M-15-Producing Escherichia coli among travelers to the Indian subcontinent in New Zealand. Clin. Infect. Dis. 2008, 47, 689–692. [Google Scholar] [CrossRef] [PubMed]
- Laupland, K.B.; Church, D.L.; Vidakovich, J.; Mucenski, M.; Pitout, J.D. Community-onset extended-spectrum beta-lactamase (ESBL) producing Escherichia coli: Importance of international travel. J. Infect. 2008, 57, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Pitout, J.D.; Campbell, L.; Church, D.L.; Wang, P.W.; Guttman, D.S.; Gregson, D.B. Using a commercial DiversiLab semiautomated repetitive sequence-based PCR typing technique for identification of Escherichia coli clone ST131 producing CTX-M-15. J. Clin. Microbiol. 2009, 47, 1212–1215. [Google Scholar] [CrossRef] [PubMed]
- Van der Bij, A.K.; Pitout, J.D. The role of international travel in the worldwide spread of multiresistant Enterobacteriaceae. J. Antimicrob. Chemother. 2012, 67, 2090–2100. [Google Scholar] [CrossRef] [PubMed]
- Haigh, J. The accumulation of deleterious genes in a population—Muller’s Ratchet. Theor. Popul. Biol. 1978, 14, 251–267. [Google Scholar] [CrossRef] [PubMed]
- Landis, M.J.; Bedford, T. Phylowood: Interactive web-based animations of biogeographic and phylogeographic histories. Bioinformatics 2014, 30, 123–124. [Google Scholar] [CrossRef] [PubMed]
- Illingworth, C.J.; Mustonen, V. A method to infer positive selection from marker dynamics in an asexual population. Bioinformatics 2012, 6, 831–837. [Google Scholar] [CrossRef]
- Skoglund, P.; Sjödin, P.; Skoglund, T.; Lascoux, M.; Jakobsson, M. Investigating Population History Using Temporal Genetic Differentiation. Mol. Biol. Evol. 2014, 31, 2516–2527. [Google Scholar] [CrossRef] [PubMed]
- Foll, M.; Poh, Y.P.; Renzette, N.; Ferrer-Admetlla, A.; Bank, C.; Shim, H.; Malaspinas, A.S.; Ewing, G.; Liu, P.; Wegmann, D.; et al. Influenza virus drug resistance: a time-sampled population genetics perspective. PLoS Genet 2014, 10, e1004185. [Google Scholar] [CrossRef] [PubMed]
- Luksza, M.; Lässig, M. A predictive fitness model for influenza. Nature 2014, 507, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, D.A.; Volz, E.M.; Koelle, K. Phylodynamic inference for structured epidemiological models. PLoS Comput. Biol. 2014, 10, e1003570. [Google Scholar] [CrossRef] [PubMed]
- Török, M.E.; Reuter, S.; Bryant, J.; Köser, C.U.; Stinchcombe, S.V.; Nazareth, B.; Ellington, M.J.; Bentley, S.D.; Smith, G.P.; Parkhill, J.; et al. Rapid whole-genome sequencing for investigation of a suspected tuberculosis outbreak. J. Clin. Microbiol. 2013, 51, 611–614. [Google Scholar] [CrossRef] [PubMed]
- Harris, S.R.; Cartwright, E.J.; Török, M.E.; Holden, M.T.; Brown, N.M.; Ogilvy-Stuart, A.L.; Ellington, M.J.; Quail, M.A.; Bentley, S.D.; Parkhill, J.; et al. Whole-genome sequencing for analysis of an outbreak of meticillin-resistant Staphylococcus aureus: A descriptive study. Lancet Infect. Dis. 2013, 13, 130–136. [Google Scholar] [CrossRef] [PubMed]
- Gardy, J.L.; Johnston, J.C.; Ho Sui, S.J.; Cook, V.J.; Shah, L.; Brodkin, E.; Rempel, S.; Moore, R.; Zhao, Y.; Holt, R.; et al. Whole-genome sequencing and social-network analysis of a tuberculosis outbreak. N. Engl. J. Med. 2011, 364, 730–739. [Google Scholar] [CrossRef] [PubMed]
- Machuca, J.; Briales, A.; Labrador, G.; Díaz-de-Alba, P.; López-Rojas, R.; Docobo-Pérez, F.; Martínez-Martínez, L.; Rodríguez-Baño, J.; Pachón, M.E.; Pascual, A.; et al. Interplay between plasmid-mediated and chromosomal-mediated fluoroquinolone resistance and bacterial fitness in Escherichia coli. J. Antimicrob. Chemother. 2014, 69, 3203–3215. [Google Scholar] [CrossRef] [PubMed]
- Török, M.E.; Harris, S.R.; Cartwright, E.J.; Raven, K.E.; Brown, N.M.; Allison, M.E.; Greaves, D.; Quail, M.A.; Limmathurotsakul, D.; Holden, M.T.; et al. Zero tolerance for healthcare-associated MRSA bacteraemia: Is it realistic? J. Antimicrob. Chemother. 2014, 69, 2238–2245. [Google Scholar] [CrossRef] [PubMed]
- Bull, J.J. Evolution of phenotypic variance. Evolution 1987, 41, 303–315. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Munsky, B.; Neuert, G.; van Oudenaarden, A. Using gene expression noise to understand gene regulation. Science 2012, 336, 183–187. [Google Scholar] [CrossRef] [PubMed]
- Yun, H.S.; Hong, J.; Lim, H.C. Regulation of ribosome synthesis in Escherichia coli: Effects of temperature and dilution rate changes. Biotechnol. Bioeng. 1996, 52, 615–624. [Google Scholar] [CrossRef] [PubMed]
- Zaslaver, A.; Kaplan, S.; Bren, A.; Jinich, A.; Mayo, A.; Dekel, E.; Alon, U.; Itzkovitz, S. Invariant distribution of promoter activities in Escherichia coli. PLoS Comput. Biol. 2009, 5, e1000545. [Google Scholar] [CrossRef] [PubMed]
- Sabarly, V.; Bouvet, O.; Glodt, J.; Clermont, O.; Skurnik, D.; Diancourt, L.; de Vienne, D.; Denamur, E.; Dillmann, C. The decoupling between genetic structure and metabolic phenotypes in Escherichia coli leads to continuous phenotypic diversity. J. Evol. Biol. 2011, 24, 1559–1571. [Google Scholar] [CrossRef] [PubMed]
- Kiviet, D.J.; Nghe, P.; Walker, N.; Boulineau, S.; Sunderlikova, V.; Tans, S.J. Stochasticity of metabolism and growth at the single-cell level. Nature 2014, 514, 376–379. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, M.; Good, T.A.; Wales, M.E.; Hua, J.P.; Wild, J.R. Modeling allosteric regulation of de novo pyrimidine biosynthesis in Escherichia coli. J. Theor. Biol. 2005, 234, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Hart, Y.; Madar, D.; Yuan, J.; Bren, A.; Mayo, A.E.; Rabinowitz, J.D.; Alon, U. Robust control of nitrogen assimilation by a bifunctional enzyme in E. coli. Mol. Cell 2011, 41, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Levine, E.; Hwa, T. Stochastic fluctuations in metabolic pathways. Proc. Natl. Acad. Sci. USA 2007, 104, 9224–9229. [Google Scholar] [CrossRef] [PubMed]
- Wilson, W.A.; Roach, P.J.; Montero, M.; Baroja-Fernández, E.; Muñoz, F.J.; Eydallin, G.; Viale, A.M.; Pozueta-Romero, J. Regulation of glycogen metabolism in yeast and bacteria. FEMS Microbiol. Rev. 2010, 34, 952–985. [Google Scholar] [PubMed]
- El-Mansi, E.M.; Holms, W.H. Control of carbon flux to acetate excretion during growth of Escherichia coli in batch and continuous cultures. J. Gen Microbiol. 1989, 135, 2875–2883. [Google Scholar] [PubMed]
- Bremer, H.; Dennis, P.P. Modulation of chemical composition and other parameters of the cell at different exponential growth rates. In EcoSal—Escherichia coli and Salmonella, Cellular and Molecular Biology; Curtiss, R., III, Ed.; ASM Press: Washington, DC, U.S.A., 2008. [Google Scholar]
- Baguley, D.; Lim, E.; Bevan, A.; Pallet, A.; Faust, S.N. Prescribing for children—Taste and palatability affect adherence to antibiotics: A review. Arch. Dis. Childhood 2012, 97, 293–297. [Google Scholar] [CrossRef]
- Ambrose, P.G.; Bhavnani, S.M.; Ellis-Grosse, E.J.; Drusano, G.L. Pharmacokinetic-pharmacodynamic considerations in the design of hospital-acquired or ventilator-associated bacterial pneumonia studies: look before you leap! Clin. Infect. Dis. 2010, 51, S103–S110. [Google Scholar] [CrossRef] [PubMed]
- Baquero, F.; Negri, M.C. Selective compartments for resistant microorganisms in antibiotic gradients. Bioessays 1997, 19, 731–736. [Google Scholar] [CrossRef] [PubMed]
- Hermsen, R.; Deris, J.B.; Hwa, T. On the rapidity of antibiotic resistance evolution facilitated by a concentration gradient. Proc. Natl. Acad. Sci. USA 2012, 109, 10775–10780. [Google Scholar] [CrossRef]
- Andersson, D.I.; Hughes, D. Persistence of antibiotic resistance in bacterial populations. FEMS Microbiol. Rev. 2011, 35, 901–911. [Google Scholar] [CrossRef] [PubMed]
- Greulich, P.; Waclaw, B.; Allen, R.J. Mutational pathway determines whether drug gradients accelerate evolution of drug-resistant cells. Phys. Rev. Lett. 2012, 109. [Google Scholar] [CrossRef] [PubMed]
- Levin, B.R.; Concepcion-Acevedo, J.; Udekwu, K. Persistence: A copacetic and parsimonious hypothesis for the existence of non-inherited resistance to antibiotics. Curr. Opin. Microbiol. 2014, 21, 18–21. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Gamez, S.; Hill, A.L.; Rosenbloom, D.I.S.; Petrov, D.A.; Nowak, M.A.; Pennings, P. Imperfect drug penetration leads to spatial monotherapy and rapid evolution of multi-drug resistance. Available online: http://biorxiv.org/content/early/2014/12/19/013003 (accessed on 9 January 2015).
- Kussell, E.; Leibler, S. Phenotypic diversity, population growth, and information in fluctuating environments. Science 2005, 309, 2075–2078. [Google Scholar] [CrossRef] [PubMed]
- Deris, J.B.; Kim, M.; Zhang, Z.; Okano, H.; Hermsen, R.; Groisman, A.; Hwa, T. The innate growth bistability and fitness landscapes of antibiotic-resistant bacteria. Science 2013, 342. [Google Scholar] [CrossRef]
- Moyed, H.S.; Bertrand, K.P. hipA, a newly recognized gene of Escherichia coli K-12 that affects frequency of persistence after inhibition of murein synthesis. J. Bacteriol. 1983, 155, 768–775. [Google Scholar] [PubMed]
- Young, B.C.; Golubchik, T.; Batty, E.M.; Fung, R.; Larner-Svensson, H.; Votintseva, A.A.; Miller, R.R.; Godwin, H.; Knox, K.; Everitt, R.G.; et al. Evolutionary dynamics of Staphylococcus aureus during progression from carriage to disease. Proc. Natl. Acad. Sci. USA 2012, 109, 4550–4555. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, T.D.; Flett, K.B.; Yelin, I.; Martin, T.R.; McAdam, A.J.; Priebe, G.P.; Kishony, R. Genetic variation of a bacterial pathogen within individuals with cystic fibrosis provides a record of selective pressures. Nat. Genet 2013, 46, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.C.; Becq, J.; Fraser, L.; Schulz-Trieglaff, O.; Bond, N.J.; Foweraker, J.; Bruce, K.D.; Smith, G.P.; Welch, M. Genomic variation among contemporary Pseudomonas aeruginosa isolates from chronically infected cystic fibrosis patients. J. Bacteriol. 2012, 194, 4857–4866. [Google Scholar] [CrossRef] [PubMed]
- Folkvardsen, D.B.; Svensson, E.; Thomsen, V.Ø.; Rasmussen, E.M.; Bang, D.; Werngren, J.; Hoffner, S.; Hillemann, D.; Rigouts, L. Can molecular methods detect 1% isoniazid resistance in Mycobacterium tuberculosis? J. Clin. Microbiol. 2013, 51, 1596–1599. [Google Scholar] [CrossRef] [PubMed]
- Korolev, K.S.; Müller, M.J.; Karahan, N.; Murray, A.W.; Hallatschek, O.; Nelson, D.R. Selective sweeps in growing microbial colonies. Phys. Biol. 2012, 9. [Google Scholar] [CrossRef]
- Coward, C.; Restif, O.; Dybowski, R.; Grant, A.J.; Maskell, D.J.; Mastroeni, P. The effects of vaccination and immunity on bacterial infection dynamics in vivo. PLoS Pathog. 2014, 10, e1004359. [Google Scholar] [CrossRef] [PubMed]
- Webb, G.F.; D’Agata, E.M.; Magal, P.; Ruan, S. A model of antibiotic-resistant bacterial epidemics in hospitals. Proc. Natl. Acad. Sci. USA 2005, 102, 13343–13348. [Google Scholar] [CrossRef] [PubMed]
- Ankomah, P.; Levin, B.R. Exploring the collaboration between antibiotics and the immune response in the treatment of acute, self-limiting infections. Proc. Natl. Acad. Sci. USA 2014, 111, 8331–8338. [Google Scholar] [CrossRef] [PubMed]
- Stoesser, N.; Batty, E.M.; Eyre, D.W.; Morgan, M.; Wyllie, D.H.; Del Ojo Elias, C.; Johnson, J.R.; Walker, A.S.; Peto, T.E.; Crook, D.W. Predicting antimicrobial susceptibilities for Escherichia coli and Klebsiella pneumoniae isolates using whole genomic sequence data. J. Antimicrob. Chemother. 2013, 68, 2234–2244. [Google Scholar] [PubMed]
- Köser, C.U.; Ellington, M.J.; Peacock, S.J. Whole-genome sequencing to control antimicrobial resistance. Trends Genet 2014, 30, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Raoult, D. Editorial: emerging clones of bacterial epidemics in the genomic area. Clin. Microbiol. Infect. 2014, 20, 371–372. [Google Scholar] [CrossRef] [PubMed]
- Grad, Y.H.; Lipsitch, M. Epidemiologic data and pathogen genome sequences: A powerful synergy for public health. Genome Biol. 2014, 15, 538. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, Y.; Choi, P.J.; Li, G.W.; Chen, H.; Babu, M.; Hearn, J.; Emili, A.; Xie, X.S. Quantifying E. coli proteome and transcriptome with single-molecule sensitivity in single cells. Science 2010, 329, 533–538. [Google Scholar] [CrossRef] [PubMed]
- Lalaouna, D.; Eyraud, A.; Chabelskaya, S.; Felden, B.; Massé, E. Regulatory RNAs involved in bacterial antibiotic resistance. PLoS Pathog. 2014, 10, e1004299. [Google Scholar] [CrossRef] [PubMed]
- Beaulaurier, J.; Zhu, S.; Sebra, R.; Zhang, X.S.; Rosenbluh, C.; Deikus, G.; Shen, N.; Munera, D.; Waldor, M.K.; Blaser, M.; et al. Single molecule-level detection and long read-based phasing of epigenetic variations in bacterial methylomes. Available online: http://biorxiv.org/content/early/2014/09/16/007823 (accessed on 25 September 2014).
- Fang, G.; Munera, D.; Friedman, D.I.; Mandlik, A.; Chao, M.C.; Banerjee, O.; Feng, Z.; Losic, B.; Mahajan, M.C.; Jabado, O.J.; et al. Genome-wide mapping of methylated adenine residues in pathogenic Escherichia coli using single-molecule real-time sequencing. Nat. Biotechnol. 2012, 30, 1232–1239. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Horinouchi, T.; Furusawa, C. Prediction of antibiotic resistance by gene expression profiles. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Downing, T. Tackling Drug Resistant Infection Outbreaks of Global Pandemic Escherichia coli ST131 Using Evolutionary and Epidemiological Genomics. Microorganisms 2015, 3, 236-267. https://doi.org/10.3390/microorganisms3020236
Downing T. Tackling Drug Resistant Infection Outbreaks of Global Pandemic Escherichia coli ST131 Using Evolutionary and Epidemiological Genomics. Microorganisms. 2015; 3(2):236-267. https://doi.org/10.3390/microorganisms3020236
Chicago/Turabian StyleDowning, Tim. 2015. "Tackling Drug Resistant Infection Outbreaks of Global Pandemic Escherichia coli ST131 Using Evolutionary and Epidemiological Genomics" Microorganisms 3, no. 2: 236-267. https://doi.org/10.3390/microorganisms3020236
APA StyleDowning, T. (2015). Tackling Drug Resistant Infection Outbreaks of Global Pandemic Escherichia coli ST131 Using Evolutionary and Epidemiological Genomics. Microorganisms, 3(2), 236-267. https://doi.org/10.3390/microorganisms3020236