Abstract
The genus Corynebacterium is commonly isolated from camel uteri, yet it is rarely identified to the species level. During our routine clinical examination of she-camels brought to the hospital with history of reproductive and systemic health issues, four isolates from the uterus and one isolate from blood could not be assigned to any valid Corynebacterium species. Therefore, we aim to identify these isolates, determine any potential virulence factors, and describe how gene turnover contributed to the evolution of these species. Genome-based and phenotypic identification, along with resistome, mobilome, virulome and phylogenomics analysis, was used to characterize the isolates. The isolates were Gram stain-positive, catalase-positive, and rod-shaped. The isolates were assigned to the genus Corynebacterium based on 16S rRNA gene sequence similarity and phylogenetic analysis. The isolates 3274 and ayman were classified as two new Corynebacterium species based on the average nucleotide identity (ANI) values of 78.46% and 68.88% and digital DNA–DNA hybridization (dDDH) values of 20.9% and 22.4%. The isolates 2581A, 2583C, and 4168A constitute a single Corynebacterium species based on their pairwise ANI value of 99% and dDDH value of more than 90%. In addition, isolates 2581A, 2583C, and 4168A showed ANI values of 75.99%, 75.86%, and 76.04% and dDDH values of 23.1%, 23%, and 22.5% with closely related species, and were designated as single new Corynebacterium species. Genes for mycolic acid and menaquinone biosynthesis were detected in all isolates. The isolates were susceptible to ceftiofur, linezolid, penicillin, erythromycin, and tetracycline. All isolates harbored the antiseptic resistance gene qacA. Moreover, virulence factors involved in cell adhesion and iron acquisition were detected. The evolution of these species is dominated by gene gain rather than gene loss. The majority of these genes are acquired through horizontal gene transfer, mediated by prophages and genomic islands. In summary, we characterized three new Corynebacterium species, expanding the number of new Corynebacterium species from animals. Moreover, we described the mechanism underlying the genome evolution of these new species. The clinical findings and detection of virulence genes highlight the significance of these isolates as possible pathogens, contributing to the development of endometritis in camels.
1. Introduction
The genus Corynebacterium currently comprises 166 officially recognized species, as documented by the List of Prokaryotic names with Standing in Nomenclature (https://lpsn.dsmz.de/genus/Corynebacterium accessed on 14 July 2025) [1]. They have mycolic acid-containing cell walls and are Gram-positive straight or slightly curved bacteria. The environmental presence of Corynebacterium species is widespread and includes soil, water, and human and animal skin and mucous membranes [2,3,4,5]. The pathogenicity spectrum of Corynebacterium includes diseases such as animal mastitis, cattle pyelonephritis, sheep caseous lymphadenitis, and human diphtheria [6,7,8,9,10]. Additionally, Corynebacterium was found in the uterus of various animal species [11,12,13,14,15,16,17]. However, few studies have investigated their virulence factors or species-level classification.
Corynebacterium species were traditionally classified using biochemical profile and chemotaxonomic investigations, as well as genotypic procedures such as DNA-DNA hybridization and 16S rRNA gene sequencing [18]. With the advent of genome sequencing technologies and phylogenomic analysis tools, the classification and description of new Corynebacterium species increasingly rely on whole-genome sequence analysis [19]. A key method in this process is the average nucleotide identity (ANI) approach and digital DNA–DNA hybridization approach (dDDH), which measure the overall similarity between genome sequences [20,21]. At present, an ANI value in the range of 95–96% is widely accepted as the threshold for delineating bacterial species. Values below this cutoff typically indicate that the organisms belong to new species [22,23,24].
Recently, during our regular identification of bacteria from disease camels, we isolated four Corynebacterium species from the uterus of camels with a history of conception failure, as well as one Corynebacterium isolate from the blood. Notably, the five isolates listed above were not assigned to a valid Corynebacterium based on the current 95–96% ANI cutoff or 70% dDDH cutoff. From an epidemiological standpoint, classification at the species level is critical. Thus, the primary goals of the present study are to (1) utilize genome-based analysis and the biochemical profile to classify the isolated Corynebacterium species from camel uterus and blood and (2) clarify the role of gene acquisition and loss on the genome diversity of the isolated Corynebacterium species from camel uterus and blood.
2. Materials and Methods
2.1. Bacterial Isolates
Permission for this study was granted by King Faisal University’s Research Ethics Committee (HAPO-05-HS-003). The study was conducted in the Al-Hasa region of Saudi Arabia at the Camel Research Centre of King Faisal University. During the regular clinical inspection of she-camels brought to the Veterinary Teaching Hospital at King Faisal University, a range of samples were collected and analyzed to investigate potential reproductive and systemic health disease. One particular isolate, designated as ayman, was obtained from the blood of a she-camel suffering from fever. Four isolates designated as 2581C, 2583C, 4168A, and 3274 were isolated from camel uteri with a history of repeated failure to conceive despite multiple breeding attempts. For sample collection, the camel’s skin was disinfected with 70% ethanol before collecting the blood sample from the jugular vein. To ensure aseptic conditions, uterine samples were collected and handled using sterile surgical gloves. The uterine lumen was then sampled with a double-guarded sterile swab (Kruuse, Langeskov, Denmark). The presence or absence of pus in the uterus was further confirmed using transrectal ultrasonography with a 7.5 MHz linear transducer probe (Aloka, Co., Ltd., Tokyo, Japan).
2.2. Isolates Genome Sequence Analysis
To extract genomic DNA and form the blood and uterine isolates, the wizard genomic DNA purification kit (Promega Biotech AB, Madison, WI, USA) was utilized following the guidelines provided by the manufacturer. Utilizing the TruSeq Nano DNA Library Preparation Kits (Illumina, San Diego, CA, USA), DNA libraries were constructed for genome sequencing. Using the NovaSeq technology (Illumina) at Macrogen Inc. in Seoul, Republic of Korea, the produced library samples were pooled and put through high-throughput sequencing, yielding paired-end reads of 151 base pairs. After genome sequencing, Fastqc 0.11.8 was used to evaluate the quality of the paired-end reads [25], and Fastp software version 0.20.0 was used to trim low-quality bases [26]. The paired-end reads were de novo assembled using SPAdes 3.15.4 [27]. Then, only scaffolds having a size more than 500 base pairs and more than 10% coverage were kept for analysis.
Prokka 1.14.6 was used for open reading frame prediction, and the Kyoto Encyclopaedia of Genes and Genomes (KEGG) website was used for functional annotation of metabolic KEGG modules, Phosphotransferase System (PTS), and ATP-Binding Cassette (ABC) transporters [28]. In order to identify putative virulence genes, we used the BLAST 2.16.0 program to perform homology searches against the sequence of Corynebacterium virulence factors that we retrieved from the virulence factor database (VFDB) [29].
Normally, genomic islands (GIs) contain foreign DNA pieces acquired through horizontal transfer. The IslandViewer 4 web server, which is currently the most accurate method for GI prediction, was used to detect GIs [30]. IslandPick, IslandPath-DIMOB, SIGI-HMM, and Islander are the four genomic island prediction techniques that are integrated into the IslandViewer 4 program. We utilized IslandViewer 4 using default settings. It was generally accepted that the genes identified in the GI were horizontally transmitted genes. Finally, PHASTEST (Phage Search Tool Enhanced Release) (https://phastest.ca, accessed on 10 July 2025) was used to locate prophages in the genomes [31].
2.3. Identification of Isolates
Samples were sent to the laboratory and cultured aerobically on 5% sheep blood agar at 37 °C for 48 h. Then, isolates were first screened based on Gram stain. The commercial test API® Coryne system (BioMérieux, Craponne, France) was used to determine the biochemical profile of isolates. The isolates were initially identified as Corynebacterium based on a 16S rRNA sequence identity threshold of 98.7% for species delineation, with sequence BLAST comparisons against Corynebacterium type species [24]. Finally, the genetic similarity between the blood and uterine isolates and Corynebacterium type species was measured using OrthoANI. All pairwise ANI values of the studied species were estimated using the default settings of the command-line version of OrthoANI (OAT v. 1.40 utilizing blastn 2.13.0+) [20]. If two isolates had an OrthoANI value between 95% and 96%, they were regarded as belonging to the same species [24]. The dDDH values were determined using the web tool (http://ggdc.dsmz.de/distcalc2.php, accessed on 20 August 2025) Genome-to-Genome Distance Calculator 3.0 and formula 2 for distance calculations. Isolates were considered a new species if their dDDH values were less than 70% and their OrthoANI values were less than 95–96% [24].
2.4. Phylogenomic Analysis
A total of 161 genomes from Corynebacterium species with validly published names were downloaded from the GenBank database (https://www.ncbi.nlm.nih.gov/datasets/genome/, accessed on 24 June 2025). All of the downloaded genomes were annotated using PROKKA v1.14.6 in order to retain the same annotation. The GET_HOMOLOGUES scripts v22082022 were used to identify core genes with query coverage of ≥70% and sequence similarity of ≥30% [32]. GET_HOMOLOGUES script analysis yielded 173 single-copy core genes. MAFFT v7.490 was used to align each core gene sequence separately using the default parameters [33]. The software raxmlGUI v 2.0.10, which is a graphical user interface for RaxML v2.0 [34], was used to concatenate the individual MAFFT alignments. For tree reconstruction, the concatenated alignments were used as input for IQ-TREE v3.0.0 [35]. Tree branch support was calculated by 1000 replicates. The resulting tree was visualized using iTOL v7.2.1 [36].
2.5. Estimating the Gain and Loss of Genes
Gene family turnover, including gain and loss events, was estimated using BadiRate v1.35, focusing on genomes belonging to the same phylogenetic clades as the Corynebacterium isolates. The Gain and Death (GD) model was used to perform BadiRate analysis [37]. A branch-free rates model (GD-FR-ML) was used to calculate the GD rates of gene families under the assumption that each branch has a unique turnover rate. BadiRate analysis requires an ultrametric tree of the taxa studied. We used the ete v 3.1.3 Python library to transform our phylogenomic ML tree into an ultrametric tree [38]. BadiRate analysis additionally requires gene presence and absence data from the GET_HOMOLOGUES script. The resulting tree was visualized using FigTree v1.4.4.
2.6. Testing for Antibiotic Susceptibility
Susceptibility to antibiotics was assessed on Mueller–Hinton agar following the Kirby–Bauer disk diffusion method and the European Committee on Antimicrobial Susceptibility Testing, Clinical breakpoints. The sensitivity of isolates to linezolid (30 μg), penicillin (10 μg), erythromycin (15 μg), tetracycline (30 μg), and ceftiofur (30 μg) was examined. The command-line AMRFinderPlus 4.0.3 software [39] was used to detect the existence of resistance determinants. The online Comprehensive Antibiotic Resistance Database (https://card.mcmaster.ca/, accessed on 25 August 2025) was used to detect resistance determinants [40].
3. Results
3.1. Clinical Data Associated with Isolates
During the regular clinical inspection of she-camels brought to the Veterinary Teaching Hospital at King Faisal University, a range of samples were collected and analyzed to investigate potential reproductive and systemic health issues. One particular isolate, designated as ayman, was obtained from the blood of a she-camel suffering from fever. The blood was free from known blood parasites. The isolates named 2581A, 2583C, 4168A, and 3274 were collected from the uteri of she-camels that presented with vaginal discharge and had a history of repeated failure to conceive despite multiple breeding attempts, and were therefore diagnosed with clinical endometritis.
3.2. Phenotypic Classification Characteristics
The five Corynebacterium isolates were Gram-positive, rod-shaped bacteria that grew at 37 °C under aerobic conditions on blood agar. The biochemical features of isolates based on API were compared to their closed reference strains, as indicated in Table 1.

Table 1.
Differential features of isolates 2581A, 2581A, and 4168A and the type strain of the closest related species C. endometrii. The isolate ayman and the type strain of the closest related species C. pseudodiphtheriticum. d stands for variable.
3.3. Identification Based on 16S rRNA Gene Sequence
The 16S rRNA gene sequences of isolates 4168A, 2581A, and 2583C were 100% similar, whereas isolates 3274 and ayman were 92–94% similar. The isolates 4168A, 2581A, 2583C, 3274, and ayman were identified as Corynebacterium species based on the 16S rRNA gene sequence similarities (Supplementary Table S1). Isolate 3274 clustered with the type species C. renale NCTC7448, as shown in the 16S rRNA phylogeny tree (Figure 1), showing the highest 16S rRNA gene sequence similarity, 98.95%, with this species. Isolates 4168A, 2581A, and 2583C clustered together with C. endometrii LMM-1653, with sequence similarities of 97.41%, 97.50%, and 97.50% (Figure 1). Isolate ayman clustered with the C. pseudodiphtheriticum DSM 44287 and C. propinquum FDAARGOS 1112 clade but showed the highest 16S rRNA gene sequence similarity, 95.27%, with C. humireducens NBRC 106098 (Supplementary Table S1). The sequence similarity of isolates 4168A, 2581A, 2583C, and ayman was less than the 98.65% cutoff value used to delineate new species. Thus, four putative new species of Corynebacterium could be proposed according to the 16S rRNA gene sequence.
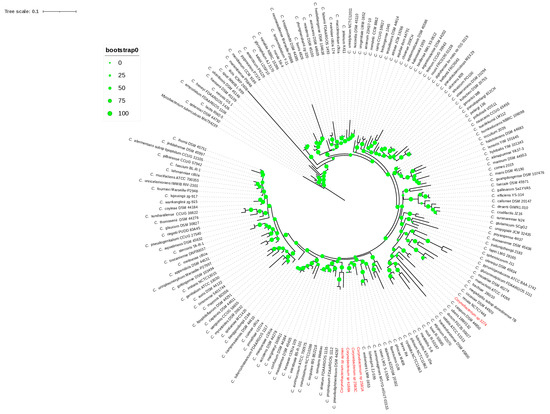
Figure 1.
The 16S rRNA phylogenetic tree. The green circles above the branches are bootstrap support values. Clinical isolates 2581A, 2583C, 4168A, ayman, and 3274 are highlighted in red color.
3.4. Classification via Genome Sequence
We calculated ANI values for each clinical isolate against all Corynebacterium type species to determine the nearest type species (Supplementary Table S2). The ANI values between the type species C. endometrii LMM-1653 and isolates 2581A, 2583C, and 4168A were 75.99%, 75.86%, and 76.04%, while the dDDH values were 23.1%, 23%, and 22.5%, respectively, which were below the cutoff values 95–96% and 70% used to define new species (Figure 2). Thus, they were not classified as C. endometrii, but rather as a new Corynebacterium species. The pairwise ANI values for isolates 2581A, 2583C, and 4168A were greater than 99%. Thus, they are above the cutoff value 95–96% used to define new species. Also, their dDDH values were greater than 90%, which is above the cutoff 70%, indicating that they belong to the same Corynebacterium species. The ANI and dDDH values for isolate 3274 and the type species C. renale NCTC7448 were 78.46% and 20.9%, respectively. The ANI and dDDH values of the isolate ayman with the nearest type species C. pseudodiphtheriticum DSM 44287 were 68.88% and 22.4%, respectively. The five isolates 2581A, 2583C, 4168A, 3274, and ayman exhibited ANI values lower than the threshold value 95–96% used to define new species, indicating that they represent three distinct Corynebacterium species at the genomic level (Figure 2).
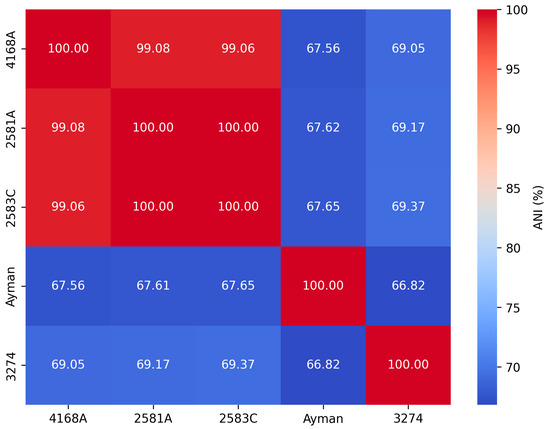
Figure 2.
Pairwise ANI values among clinical isolates of Corynebacterium.
Core-genome phylogeny inference was the second genome-level method used to group the blood and uterine isolates. A high bootstrap value validated a phylogenomic tree based on 173 single-copy core genes that grouped each isolate to its nearest type species (Figure 3). Remarkably, the phylogenomic topology resembles the 16S rRNA phylogeny tree of the isolates mentioned above. Finally, genes for mycolic acid and menaquinone biosynthesis were detected in all isolates. Phylogenetic and genomic taxonomy analyses showed these isolates as new Corynebacterium species, for which we propose the name Corynebacterium cameli sp. nov for the isolates 2581A, 2583C, and 4168A, the name Corynebacterium utericameli sp. nov for the isolate 3274, and Corynebacterium hassacameli sp. nov for the isolate ayman.
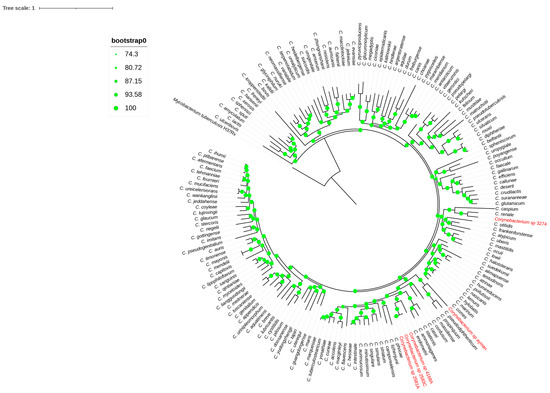
Figure 3.
Phylogenomic tree reconstructed from core genes. The green circles above the branches are bootstrap support. Clinical isolates 2581A, 2583C, 4168A, ayman, and 3274 are highlighted in red color.
3.5. Genome Characteristics and Gene Dynamics
The main genome characteristics of the blood and uterine isolates are presented in (Table 2). In summary, the coding densities for isolates 2581A, 2583C, 4168A, ayman, and 3274 were 87.23, 87.33, 87.54% 88.51%, and 90.03%, respectively. Isolates 2581A, 2583C, 4168A, ayman, and 3274 all exhibited similar GC contents of 59.68%, 59.71%, 59.68%, 51.67%, and 58.78%, respectively. Isolates 2581A, 2583C, and 4168A, which belong to same Corynebacterium species, share a core genome of 2399 genes. Within this group, the number of strain-specific genes was 120, 13, and 392 for isolates 2581A, 2583C, and 4168A, respectively. In contrast, isolates ayman and 3274, which represent different Corynebacterium species, showed much lower gene content similarity with others. Across all five isolates, only 1042 core genes were shared. We detected several different mobile genetic elements, comprising prophages and genomic islands (Table 3). In brief, one intact prophage region with the best match to PHAGE_Coryne_Juicebox NC_048070 was detected across the genome of isolates 2483C, 2581A, 4168A, and 3274. Furthermore, isolate 4168A and 3274 carried one intact prophage region with the best match to PHAGE_Gordon_Nyceirae_NC_031004 and PHAGE_Rhodoc_Hiro_NC_048669, respectively.
Table 2.
General genomic characteristics of Corynebacterium clinical isolates.
Table 3.
Types and features of mobile genetic elements detected in the genomes of the studied isolates.
Additionally, we found that 12.9%, 9.05%, and 12.58% of isolate genes exist within a pool of genomic islands in isolates 2581A, 2583C, and 4168A, respectively. In the case of isolate 3274 and ayman, we found that 6.18% and 6.81% of isolate genes exist within a pool of genomic islands. These genes are involved in metabolism, genetic information processing, environmental information processing, and cellular processes.
Lastly, using the BadiRate v1.35 software, we looked into gene gain/loss (turnover) events for every Corynebacterium isolate through the phylogeny in order to further analyze the dynamics of gene content variation (Figure 4). The results revealed that the number of genes gained outnumbers the number of genes lost in all isolates. Gene gain events were highest in the clade that included isolates 2581A, 2583C, and 4168A. In contrast, the clade containing isolates 3274 had the fewest gene gain events. Furthermore, the turnover rate for gene gain and loss is comparable between isolate 3274 and its nearest type species C. renale.
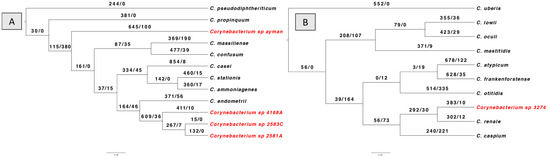
Figure 4.
BadiRate analysis results according to branch-free rates model (GD-FR-ML). The numbers on the branches indicate gains gain/gene loss number. The clinical Corynebacterium isolates are highlighted in red color. (A) shows gene turnover for isolates 2581A, 2583C, 4168A, and ayman. (B) shows the gene turnover for isolate 3274.
3.6. Analysis of Functional Characteristics
To examine the functional profile of Corynebacterium isolates, the transporters and metabolic modules were assembled for every genome using the KEGG database. A detailed examination of metabolic potentials reveals that uterine isolates 2581A, 2583C, and 4168A encode for 63, 63, and 62 complete KEGG metabolic modules and 20, 20, and 19 transporters, respectively. As for the difference from its nearest type species C. endometrii from the cow uterus, it encodes 58 complete KEGG metabolic modules and 18 transporters (Figure 5).
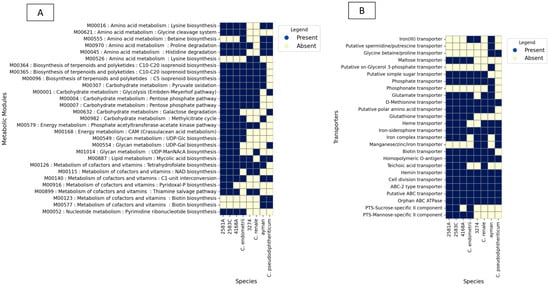
Figure 5.
KEGG BLAST analysis results for isolates 2581A, 2583C, 4168A, 3274, and ayman and their type species. (A) shows metabolic modules. (B) shows ABC and PTS transporters.
In the case of the isolate 3274, examination of metabolic potentials reveals that it encodes 52 complete KEGG metabolic modules and 15 transporters. As for the difference from it its nearest type species C. renale (cow isolate), it encodes 51 complete KEGG metabolic modules and 17 transporters (Figure 5).
In the case of the blood isolate ayman, examination of metabolic potentials reveals that it encodes 52 complete KEGG metabolic modules and 18 transporters. As for the difference from it its nearest type species C. pseudodiphtheriticum, it encodes 49 complete KEGG metabolic modules and 11 transporters (Figure 5).
3.7. Virulence Genes
Analysis of the five Corynebacterium genomes against the VFDB revealed genes involved in adherence. These include genes for the SpaA and SpaD of pili and DIP_RS14950 (Supplementary Table S3). The diphtheria toxins and pld genes were not detected in the five Corynebacterium isolates, but the dtxR regulator gene was present in the five Corynebacterium isolates. Notably, the post-translational modification gene DIP_RS20575, encoding an MdbA thioredoxin-like protein, was present in all isolates. Iron transporters were widely distributed in the isolates, including the heme transporter encoded by hmuTUV genes, the iron (III) transporter encoded by AFuABC genes, and the iron siderophore ABC transporter encoded by FepBDGC and CeuABCD genes (Supplementary Table S3). Genome mining revealed that all isolates shared three out of four two-component systems (TCS): (1) SenX3-RegX3, which has roles in the phosphate starvation response, (2) MprAB, which is essential in the maintenance of persistent infection, and (3) MtrAB, which has roles in the osmotic stress response. The fourth TCS, DesK-DesR, which regulates membrane lipid fluidity, is only found in isolates 4168A and ayman.
3.8. Antibiotic Susceptibility Profile
The uterine isolates and blood isolates showed similar phenotypic antibiotic susceptibility as determined by the disk diffusion method. All the isolates were sensitive to linezolid, penicillin, erythromycin, tetracycline, and ceftiofur. Then, we analyzed the resistomes of uterine isolates and blood isolates. No resistance determinants were found using the command-line AMRFinderPlus 4.0.3 software and by blast against the Comprehensive Antibiotic Resistance Database. KEGG annotation revealed the existence of the multidrug efflux pump encoded by the qacA gene in all isolates, and this was further confirmed by blast against the Comprehensive Antibiotic Resistance Database.
4. Discussion
Here, during our routine clinical examination of the uteri of she-camels with a history of infertility, we found three distinct novel Corynebacterium species. Currently, species identification follows a three-step approach: initial classification based on 16S rRNA gene sequence similarity, followed by phylogeny inference and calculation of ANI or dDDH value [24]. The 16S rRNA gene has been a rapid and accurate genetic method for bacterial identification for many years [41]. Nevertheless, because of its low resolution, this marker is unable to accurately classify some bacteria to the species level [42,43]. The pairwise similarity values of the 16S rRNA gene for isolate 3274 versus validly published Corynebacterium species in this study demonstrated the limits of this gene analysis. According to BLAST results, the similarity was higher than the usual threshold of 98.7% for species delineation [44]. Consequently, isolate 3274 was assigned to C. renale. In contrast, isolate 3274 was considered a new Corynebacterium species, as its dDDH value was less than 70% and its OrthoANI value was less than 95–96% [23]. Thus, this indicates the possibility of incorrect classification when depending exclusively on 16S rRNA gene sequences. Therefore, all isolates were subjected to further genetic testing, including dDDH, OrthoANI, and phylogeny, to achieve correct classification. Finally, to elucidate the evolutionary relationships, taxonomists currently mostly rely on phylogenetic tree inference [45]. Notably, single-gene phylogenetic inference usually provides less resolution than phylogenomic methods, which combine information from across the genome. Consequently, a phylogenomic tree was inferred based on 173 single-copy core genes. Each isolate clustered with its closest type strain, providing further support for their taxonomic classification of the isolates into three distinct novel Corynebacterium species.
Second, for each isolate, we mapped gene gain and loss events in the Corynebacterium phylogeny to gain a better understanding of the dynamics of gene content variations. We observed a dynamic pattern of genome evolution characterized by a higher rate of gene gain than loss on the terminal branch. Our results align with previous research on the human pathogen Acinetobacter baumannii, in which gene gain was a feature of evolution in the terminal branch [46]. Therefore, gene turnover contributed to the overall diversity of gene repertoires of blood and uterine isolates. The predominance of gene gain over gene loss might indicate that these species are expanding or altering their niche. We found various types of mobile genetic elements, consisting of prophages and genomic islands, implying that genetic transfer occurs frequently in Corynebacterium isolates from blood and uterine samples. Therefore, the notable gene gain found in the clinical isolates can be attributed to the horizontal gene transfer mediated by mobile genetic elements. The functional implications of these horizontally transferred genes contributed to genetic diversity among the isolates. In addition, this resulted in enhanced metabolism by gaining genes involved in carbohydrate and amino acid metabolism, as well as transporters. The uterine isolates showed different metabolic profiles although they shared the same niche. Their metabolic profile likely represents their previous niche.
The identification of the MprAB, SenX3-RegX3, and MtrAB two-component systems, associated with osmotic stress response and persistent infection in the human pathogen Mycobacterium tuberculosis [47,48], suggests that our clinical isolates have a gene set supporting their survival in the animal host. Additionally, iron acquisition system detection, which allows sequestered iron to be extracted from the host, suggests adaptation to the iron-limited conditions commonly found in host habitats. In addition, the metabolic modules identified in our clinical isolates support core survival functions and basic metabolism rather than pathways associated with the degradation of environmental contaminants such as aromatic compounds and benzoate derivatives. Clinical findings, along with the above observations, suggest that our isolates are better adapted to survival in the animal host environment and are therefore more likely to represent potential animal pathogens rather than environmental contaminant bacteria.
The detection of Corynebacterium diphtheriae SpaA- and SpaD-type pili in our clinical isolates, which is implicated in host cell attachment, suggests that our clinical isolates have the capacity for colonization [49]. This is further supported by the presence of virulence factor DIP0733, which has been implicated in both the adhesion and invasion of host cells. Moreover, the blood isolate ayman harbored two complete biotin biosynthesis modules (M00123 and M00577), suggesting a self-sufficient metabolic capability that may enhance survival in biotin-limited environments, an attribute linked to increased virulence in pathogens such as Mycobacterium tuberculosis [50]. In contrast, the uterine isolates lacked these biosynthetic modules but contained a biotin uptake transporter system, implying a greater dependence on the host-derived biotin. Collectively, these genomic features, including adhesion and invasion factors and metabolic adaptations homologous to those in Corynebacterium diphtheriae, suggest that the isolates have pathogenic potential and may pose a risk for human infection. Further functional studies are warranted to clarify their role in disease and assess their clinical significance.
The resistome profile and the antimicrobial phenotypic results agreed, and all isolates showed sensitivity to the tested drugs. These findings hold significant practical implications for the treatment of endometritis, where ceftiofur is commonly used as the primary therapeutic agent [51]. However, the effectiveness of standard sanitization methods in veterinary reproductive control may be hampered by the identification of antiseptic resistance genes qacA, which have been shown to mediate antiseptic resistance in Staphylococcus aureus [52]. As a result, resistant bacteria may persist in agricultural or clinical settings, which may pose a risk for zoonotic infection for humans in close contact with camels.
In conclusion, through genome-based taxonomy, we identified three new Corynebacterium species out of five isolates, increasing the total number of Corynebacterium species isolated from animals. Species evolution is dominated by gene gain over gene loss. The majority of the gained genes were acquired through horizontal gene transfer assisted by genomic islands and prophages. The existence of several virulence genes in these species and their isolation from camel with endometritis and fever may suggest that these species are pathogenic. Nevertheless, more proof is required to demonstrate their pathogenicity.
Supplementary Materials
The following supporting information can be downloaded at https://www.mdpi.com/article/10.3390/microorganisms13092090/s1, Table S1: Values for 16S rRNA, from pairwise comparison of Corynebacterium type species against clinical Corynebacterium isolates. Table S2: Values for OrthoANI, from pairwise comparison of Corynebacterium type species against clinical Corynebacterium isolates; Table S3: Virulence factors genes of Corynebacterium isolates from camel uterus and blood.
Funding
This research was funded by the Deanship of Scientific Research, King Faisal University, funding number KFU253128.
Institutional Review Board Statement
The study was approved by the Research Ethics Committee of King Faisal University (HAPO-05-HS-003), approved on 20 September 2020.
Informed Consent Statement
Not applicable. The camels belong to the university. Informed consent was therefore not necessary.
Data Availability Statement
Data are available on a publicly accessible GenBank website, and the accession number is mentioned in Table 2. Further inquiries can be directed to the corresponding author.
Acknowledgments
We are grateful to the Camel Research Centre Farm members of King Faisal University for their assistance.
Conflicts of Interest
The author declares no conflicts of interest.
References
- Parte, A.C.; Sardà Carbasse, J.; Meier-Kolthoff, J.P.; Reimer, L.C.; Göker, M. List of Prokaryotic names with Standing in Nomenclature (LPSN) moves to the DSMZ. Int. J. Syst. Evol. Microbiol. 2020, 70, 5607–5612. [Google Scholar] [CrossRef]
- Yassin, A.F. Corynebacterium ulceribovis sp. nov.; isolated from the skin of the udder of a cow with a profound ulceration. Int. J. Syst. Evol. Microbiol. 2009, 59, 34–37. [Google Scholar] [CrossRef] [PubMed]
- Amao, H.; Akimoto, T.; Komukai, Y.; Sawada, T.; Saito, M.; Takahashi, K.W. Detection of Corynebacterium kutscheri from the oral cavity of rats. Exp. Anim. 2002, 5, 99–102. [Google Scholar] [CrossRef] [PubMed]
- Woudstra, S.; Lücken, A.; Wente, N.; Zhang, Y.; Leimbach, S.; Gussmann, M.K.; Kirkeby, C.; Krömker, V. Reservoirs of Corynebacterium spp. in the Environment of Dairy Cows. Pathogens 2023, 12, 139. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Fang, J.; Xu, Y.; Zhao, W.; Cao, J. Corynebacterium hadale sp. nov. isolated from hadopelagic water of the New Britain Trench. Int. J. Syst. Evol. Microbiol. 2018, 68, 1474–1478. [Google Scholar] [CrossRef]
- Mohd Khalid, M.K.; Ahmad, N.; Hii, S.Y.; Abd Wahab, M.A.; Hashim, R.; Liow, Y.L. Molecular characterization of Corynebacterium diphtheriae isolates in Malaysia between 1981 and 2016. J. Med. Microbiol. 2019, 68, 105–110. [Google Scholar] [CrossRef]
- Dorella, F.A.; Pacheco, L.G.C.; Oliveira, S.C.; Miyoshi, A.; Azevedo, V. Corynebacterium pseudotuberculosis: Microbiology, biochemical properties, pathogenesis and molecular studies of virulence. Vet. Res. 2006, 37, 201–2018. [Google Scholar] [CrossRef]
- Smith, J.S.; Krull, A.C.; Schleining, J.A.; Derscheid, R.J.; Kreuder, A.J. Clinical presentations and antimicrobial susceptibilities of Corynebacterium cystitidis associated with renal disease in four beef cattle. J. Vet. Intern. Med. 2020, 34, 2169–2174. [Google Scholar] [CrossRef]
- Ballas, P.; Rückert, C.; Wagener, K.; Drillich, M.; Kämpfer, P.; Busse, H.J.; Ehling-Schulz, M. Corynebacterium endometrii sp. nov.; isolated from the uterus of a cow with endometritis. Int. J. Syst. Evol. Microbiol. 2020, 70, 146–152. [Google Scholar] [CrossRef]
- Lücken, A.; Wente, N.; Zhang, Y.; Woudstra, S.; Krömker, V. Corynebacteria in Bovine Quarter Milk Samples-Species and Somatic Cell Counts. Pathogens 2021, 10, 831. [Google Scholar] [CrossRef]
- Elshazly, M.O.; El-Rahman, S.S.A.; Hamza, D.A.; Ali, M.E. Pathological and bacteriological studies on reproductive tract abnormalities of she-camels (Camelus dromedarius), emphasizing on zoonotic importance. J. Adv. Vet. Anim. Res. 2020, 7, 633–646. [Google Scholar] [CrossRef] [PubMed]
- Mshelia, G.D.; Okpaje, G.; Voltaire, Y.A.; Egwu, G.O. Comparative studies on genital infections and antimicrobial susceptibility patterns of isolates from camels (Camelus dromedarius) and cows (Bos indicus) in Maiduguri, north-eastern Nigeria. Springerplus 2014, 3, 91. [Google Scholar] [CrossRef] [PubMed]
- Elbir, H.; Almathen, F.; Almuhasen, F.M. Genomic differences among strains of Corynebacterium cystitidis isolated from uterus of camels. J. Infect. Dev. Ctries. 2022, 16, 134–146. [Google Scholar] [CrossRef] [PubMed]
- Frontoso, R.; De Carlo, E.; Pasolini, M.P.; van der Meulen, K.; Pagnini, U.; Iovane, G.; De Martino, L. Retrospective study of bacterial isolates and their antimicrobial susceptibilities in equine uteri during fertility problems. Res. Vet. Sci. 2008, 84, 1–6. [Google Scholar] [CrossRef]
- Getahun, A.M.; Hunderra, G.C.; Gebrezihar, T.G.; Boru, B.G.; Desta, N.T.; Ayana, T.D. Comparative study on lesions of reproductive disorders of cows and female dromedary camels slaughtered at Addis Ababa, Adama and Akaki abattoirs with bacterial isolation and characterization. BMC Vet. Res. 2021, 17, 134. [Google Scholar] [CrossRef]
- Ballas, P.; Reinländer, U.; Schlegl, R.; Ehling-Schulz, M.; Drillich, M.; Wagener, K. Characterization of intrauterine cultivable aerobic microbiota at the time of insemination in dairy cows with and without mild endometritis. Theriogenology 2021, 159, 28–34. [Google Scholar] [CrossRef]
- Schlegl, R.; Drillich, M.; Ballas, P.; Reinländer, U.; Iwersen, M.; Baumgartner, W.; Ehling-Schulz, M.; Wagener, K. Field trial on the post-insemination intrauterine treatment of dairy cows with mild endometritis with cephapirin. Theriogenology 2020, 156, 20–26. [Google Scholar] [CrossRef]
- Negi, V.; Singh, Y.; Schumann, P.; Lal, R. Corynebacterium pollutisoli sp. nov., isolated from hexachlorocyclohexane-contaminated soil. Int. J. Syst. Evol. Microbiol. 2016, 66, 3531–3537. [Google Scholar] [CrossRef]
- Koublová, V.; Sedlář, K.; Sedláček, I.; Musilová, J.; Staňková, E.; Králová, S.; Koudelková, S.; Krsek, D.; Švec, P. Corynebacterium mendelii sp. nov.; a novel bacterium isolated from Adélie penguin oral cavity. Int. J. Syst. Evol. Microbiol. 2024, 74, 006244. [Google Scholar] [CrossRef]
- Lee, I.; Ouk Kim, Y.; Park, S.C.; Chun, J. OrthoANI: An improved algorithm and software for calculating average nucleotide identity. Int. J. Syst. Evol. Microbiol. 2016, 66, 1100–1103. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Göker, M. TYGS is an automated high-throughput platform for state-of-the-art genome-based taxonomy. Nat. Commun. 2019, 10, 2182. [Google Scholar] [CrossRef] [PubMed]
- Richter, M.; Rosselló-Móra, R. Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. USA 2009, 106, 19126–19131. [Google Scholar] [CrossRef] [PubMed]
- Konstantinidis, K.T.; Tiedje, J.M. Genomic insights that advance the species definition for prokaryotes. Proc. Natl. Acad. Sci. USA 2005, 102, 2567–2572. [Google Scholar] [CrossRef]
- Chun, J.; Oren, A.; Ventosa, A.; Christensen, H.; Arahal, D.R.; da Costa, M.S.; Rooney, A.P.; Yi, H.; Xu, X.W.; De Meyer, S.; et al. Proposed minimal standards for the use of genome data for the taxonomy of prokaryotes. Int. J. Syst. Evol. Microbiol. 2018, 68, 461–466. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 22 September 2022).
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Morishima, K. BlastKOALA and GhostKOALA: KEGG Tools for Functional Characterization of Genome and Metagenome Sequences. J. Mol. Biol. 2016, 428, 726–731. [Google Scholar] [CrossRef]
- Liu, B.; Zheng, D.; Jin, Q.; Chen, L.; Yang, J. VFDB 2019: A comparative pathogenomic platform with an interactive web interface. Nucleic Acids Res. 2019, 47, D687–D692. [Google Scholar] [CrossRef]
- Bertelli, C.; Laird, M.R.; Williams, K.P.; Lau, B.Y.; Hoad, G.; Winsor, G.L.; Brinkman, F.S. IslandViewer 4: Expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 2017, 45, W30–W35. [Google Scholar] [CrossRef]
- Wishart, D.S.; Han, S.; Saha, S.; Oler, E.; Peters, H.; Grant, J.R.; Stothard, P.; Gautam, V. PHASTEST: Faster than PHASTER, better than PHAST. Nucleic Acids Res. 2023, 51, W443–W450. [Google Scholar] [CrossRef]
- Contreras-Moreira, B.; Vinuesa, P. GET_HOMOLOGUES, a versatile software package for scalable and robust microbial pangenome analysis. Appl. Environ. Microbiol. 2013, 79, 7696–7701. [Google Scholar] [CrossRef]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef]
- Silvestro, D.; Michalak, I. raxmlGUI: A graphical front-end for RAxML. Org. Divers. Evol. 2012, 12, 335–337. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v6: Recent updates to the phylogenetic tree display and annotation tool. Nucleic Acids Res. 2024, 52, W78–W82. [Google Scholar] [CrossRef] [PubMed]
- Librado, P.; Vieira, F.G.; Rozas, J. BadiRate: Estimating family turnover rates by likelihood-based methods. Bioinformatics 2012, 28, 279–281. [Google Scholar] [CrossRef]
- Huerta-Cepas, J.; Serra, F.; Bork, P. ETE 3: Reconstruction, analysis, and visualization of phylogenomic data. Mol. Biol. Evol. 2016, 33, 1635–1638. [Google Scholar] [CrossRef]
- Feldgarden, M.; Brover, V.; Gonzalez-Escalona, N.; Frye, J.G.; Haendiges, J.; Haft, D.H.; Hoffmann, M.; Pettengill, J.B.; Prasad, A.B.; Tillman, G.E.; et al. AMRFinderPlus and the Reference Gene Catalog Facilitate Examination of the Genomic Links Among Antimicrobial Resistance, Stress Response, and Virulence. Sci. Rep. 2021, 11, 12728. [Google Scholar] [CrossRef]
- Alcock, B.P.; Huynh, W.; Chalil, R.; Smith, K.W.; Raphenya, A.R.; Wlodarski, M.A.; Edalatmand, A.; Petkau, A.; Syed, S.A.; Tsang, K.K.; et al. CARD 2023: Expanded curation, support for machine learning, and resistome prediction at the Comprehensive Antibiotic Resistance Database. Nucleic Acids Res. 2023, 51, D690–D699. [Google Scholar] [CrossRef]
- Pascual, C.; Lawson, P.A.; Farrow, J.A.; Gimenez, M.N.; Collins, M.D. Phylogenetic analysis of the genus Corynebacterium based on the 16S rRNA gene sequences. Int. J. Syst. Bacteriol. 1995, 45, 724–728. [Google Scholar] [CrossRef]
- Rajendhran, J.; Gunasekaran, P. Microbial phylogeny and diversity: Small subunit ribosomal RNA sequence analysis and beyond. Microbiol. Res. 2011, 166, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Lai, Q.; Dong, C.; Sun, F.; Wang, L.; Li, G.; Shao, Z. Phylogenetic diversity of the Bacillus pumilus group and the marine ecotype revealed by multilocus sequence analysis. PLoS ONE 2013, 8, e80097. [Google Scholar] [CrossRef] [PubMed]
- Stackebrandt, E.; Ebers, J. Taxonomic parameters revisited: Tarnished gold standards. Microbiol. Today 2006, 33, 152–155. [Google Scholar]
- Baek, I.; Kim, M.; Lee, I.; Na, S.I.; Goodfellow, M.; Chun, J. Phylogeny Trumps Chemotaxonomy: A Case Study Involving Turicella otitidis. Front. Microbiol. 2018, 9, 834. [Google Scholar] [CrossRef]
- Graña-Miraglia, L.; Lozano, L.F.; Velázquez, C.; Volkow-Fernández, P.; Pérez-Oseguera, Á.; Cevallos, M.A.; Castillo-Ramírez, S. Rapid gene turnover as a significant source of genetic variation in a recently seeded population of a healthcare-associated pathogen. Front. Microbiol. 2017, 8, 1817. [Google Scholar] [CrossRef]
- Zahrt, T.C.; Wozniak, C.; Jones, D.; Trevett, A. Functional analysis of the Mycobacterium tuberculosis MprAB two-component signal transduction system. Infect. Immun. 2003, 71, 6962–6970. [Google Scholar] [CrossRef]
- Parish, T.; Smith, D.A.; Roberts, G.; Betts, J.; Stoker, N.G. The senX3-regX3 two-component regulatory system of Mycobacterium tuberculosis is required for virulence. Microbiology 2003, 149, 1423–1435. [Google Scholar] [CrossRef]
- Mandlik, A.; Swierczynski, A.; Das, A.; Ton-That, H. Corynebacterium diphtheriae employs specific minor pilins to target human pharyngeal epithelial cells. Mol. Microbiol. 2007, 1, 111–124. [Google Scholar] [CrossRef]
- Woong Park, S.; Klotzsche, M.; Wilson, D.J.; Boshoff, H.I.; Eoh, H.; Manjunatha, U.; Blumenthal, A.; Rhee, K.; Barry, C.E., III; Aldrich, C.C.; et al. Evaluating the sensitivity of Mycobacterium tuberculosis to biotin deprivation using regulated gene expression. PLoS Pathog. 2011, 7, e1002264. [Google Scholar] [CrossRef]
- Zaher, H.A.; Al-Fares, A.F.; Mesalam, A. Efficacy of different treatment protocols for endometritis in Camelus dromedarius. Front. Vet. Sci. 2023, 20, 1136823. [Google Scholar] [CrossRef]
- Azrad, M.; Shmuel, C.; Leshem, T.; Hamo, Z.; Baum, M.; Rokney, A.; Agay-Shay, K.; Peretz, A. Reduced Susceptibility to Chlorhexidine among Staphylococcus aureus Isolates in Israel: Phenotypic and Genotypic Tolerance. Antibiotics 2021, 10, 342. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).