Abstract
Growth stage is a key factor influencing the composition and richness of the porcine gut microbiota. The stage-specific alterations in gut microbiota of indigenous Chinese pig breeds and cultivated breeds remain to be elucidated. This study conducted 16S rRNA sequencing analysis on fecal microbiota from Dahe black pigs across distinct growth stages. Samples included lactating sows, suckling piglets, weaned piglets, pigs weighing 50–100 kg, pigs weighing 120–150 kg, and pigs weighing > 200 kg. The results indicated that Escherichia shigella (12.4% vs. 16.2%), Lactobacillus (5.9% vs. 6.3%), and Rikenellaceae RC9 gut group (3.9% vs. 4.2%) were dominant genera shared between lactating sows and suckling piglets. The relative abundance of Eubacterium brachy group was significantly higher in lactating sows, whereas Flavonifractor was significantly lower compared to suckling piglets (p < 0.05). Compared to pigs weighing > 120 kg, lactating sows exhibited 22 differentially abundant genera, including Escherichia shigella, Cloacibacillus, Fusobacterium, Faecalibacterium, and Prevotella (p < 0.05). In suckling piglets, Firmicutes and Bacteroidota constituted 47.4% and 27.3% of the microbiota, respectively. Their relative abundance increased with body weight, reaching 52.6% and 33.3% in pigs weighing > 200 kg. Proteobacteria decreased from 17.3% in suckling piglets to 2.0% in >200 kg pigs. Spirochaetota declined from 2.5% in suckling piglets to 0.9% in weaned piglets and then increased to 6.9% in >200 kg pigs. Lactobacillus peaked at 15.7% in weaned piglets, while Escherichia shigella reached its maximum (16.2%) in suckling piglets, both gradually declining thereafter. Streptococcus abundance remained relatively stable (1.1% in suckling piglets; 4.5% in weaned piglets). Prevotellaceae NK3B31 group registered 2.9% in suckling piglets, increased to 7.1% in weaned piglets, and then declined to 2.6% in >200 kg pigs. Mitsuokella, Bilophila, Succinivibrio, Romboutsia, and Desulfovibrio were identified as the top five genera discriminating suckling and weaned piglets. Similarly, Lachnospiraceae XPB1014 group, Clostridium sensu stricto 1, Turicibacter, Quinella, and p 1088 a5 gut group were key discriminators between weaned piglets and 50–100 kg pigs. These identified microbial taxa represent potential candidate targets for modulating the developmental timing of growth phases in pigs, offering possibilities for either advancing or delaying specific physiological timepoints.
1. Introduction
The composition of the gut microbiota varies substantially across individuals and developmental stages, with the bacterial domain constituting the predominant component in swine [1]. Early gut colonizers in pigs are critical for establishing a permanent microbial community architecture that exerts enduring influences on porcine health and growth performance [2]. Unlike the relatively stable gut microbiota in adults, the neonatal microbiota dramatically fluctuates, exhibiting high adaptability and plasticity while being continually influenced by maternal factors through direct skin contact and breastfeeding after birth [3]. A meta-analysis of 3313 fecal samples across 60 timepoints (0–183 days) spanning six countries and four continents identified “study” and growth stage as the primary drivers of swine gut microbiota variation, outweighing age, hypervariable region, sequencing platform, and country of origin [4]. Host age was the dominant factor in shaping the gut microbiota of piglets after weaning, which follows a highly structured developmental program characterized by post-weaning changes [5,6]. Gut microbial diversity undergoes continuous age-dependent shifts, with aged pigs typically exhibiting reduced beneficial taxa and increased abundance of Bacteroides [7].
During porcine development, microbial succession drives dynamic compositional changes in the gut microbiome, which exhibits clear spatial variation along the gastrointestinal tract and is dominated by Firmicutes and Bacteroidetes [8]. Dominating the porcine gut from birth to 2 days, Escherichia, Clostridium, Fusobacterium, Streptococcus, and Enterococcus genera establish initial colonization, while Clostridiaceae (predominant at 6 h postnatal) decline to ≤1% abundance by day 20, and Enterobacteriaceae surge by post-weaning day 5 before significantly reducing after post-weaning day 11 [9]. Compositional shifts across porcine growth stages (suckling, weaning, nursery, grower, finisher) reveal 19 core genera (dominated by Bacteroides, Prevotella, and Lactobacillus) with both these “core” bacteria and enterotypes invariably transitioning to an adult-like profile with age [10]. Distinct developmental stages in pigs exhibit stage-specific gut microbial profiles.
Through prolonged artificial selection under diverse natural environments, Chinese indigenous pig breeds have undergone significant alterations in the relative abundance of their gut microbiota. In Wuzhishan pigs, a Chinese indigenous miniature breed, the abundance of Firmicutes and fecal microbial diversity increased progressively across four developmental stages: pre-weaning piglet, weaning piglet, growing pig, and sow, while the proportions of Bacteroidetes declined [11]. Fresh fecal samples from six Tunchang pig stages (pre-weaning/weaning piglets, growing pigs, suckling/empty sows, adult males) showed increased Firmicutes and Spirochaetes with decreased Bacteroidetes and Proteobacteria over development [12]. However, among China’s 83 indigenous pig breeds and 38 cultivated breeds, the gut microbial developmental characteristics of numerous porcine genetic resources remain systematically uncharacterized.
Machine learning has gained increasing prevalence across diverse fields, including microbiome data analysis. Distinct microbial genera were employed as predictors for the prevalence of foodborne pathogens during key broiler growth phases: the START phase (weeks 2–4), MID phase (weeks 5–7), and END phase (weeks 8–11) [13]. Machine learning integrated with Shapley Additive Explanation (SHAP) methodology delivers interpretable insights into models that identify signature microbiota based on milk urea nitrogen concentrations [14]. Current research on the developmental stages of the gut microbiota in Dahe black pigs remains limited. Machine learning offers a viable approach to identifying pivotal microbial taxa characteristic of distinct growth phases, thereby laying the groundwork for subsequent strain selection and validation. The present study selected Dahe black pigs—a cultivated breed derived from Dahe pigs and Duroc genetics—as the experimental model. We comprehensively profiled the intestinal microbial features using 16S rRNA sequencing across distinct developmental stages in this breed. By integrating machine learning approaches, stage-specific core microorganisms were identified. This study identifies potential microbial candidates for screening that may temporally modulate developmental stages through subsequent probiotic supplementation.
2. Materials and Methods
2.1. Animals and Sample Collection
All the animal procedures were approved by the Animal Care and Use Committee of Yunnan Agricultural University (approval code 202403081). Fecal samples were collected from pigs at an ecological farm in Fuyuan County, Qujing City, Yunnan Province, China. Environmental variables were strictly controlled throughout the study. All 323 fecal samples were collected within a single month (April 2024) to eliminate seasonal effects. Sows were individually housed and received feed devoid of probiotics, antibiotics, or veterinary medications. Piglets followed standardized developmental protocols: creep feeding initiated at postnatal day 14, weaning at day 28 with immediate transfer to nursery pens, and entry into the fattening phase at day 70. Consistent hygiene practices, feeding regimes, and environmental conditions were maintained within each production phase according to the farm’s standard operating procedures (SOPs). These samples included lactating sows (SE, n = 10), lactating piglets (SP, n = 55), weaned piglets (WP, n = 54), pigs weighing 50–100 kg (P50_100, n = 52), pigs weighing 120–150 kg (P120_150, n = 101), and pigs weighing more than 200 kg (P200, n = 51). Only the internal portion of the feces was collected. All animals received stage-specific antibiotic-free diets meeting or exceeding nutritional requirements recommended by NRC (2012).
2.2. Microbial DNA Extraction and Sequencing
DNA was extracted from fecal samples using the HiPure Stool DNA Kit (Magen, Guangzhou, China) according to the manufacturer’s protocol. The V3-V4 region of bacterial 16S rRNA genes was amplified with primers 341F (5′-CCTACGGGNGGCWGCAG-3′) and 806R (5′-GGACTACHVGGGTATCTAAT-3′). PCR products were purified with AMPure XP Beads (Beckman Coulter, Brea, CA, USA), quantified using a Qubit 3.0 Fluorometer, and constructed into sequencing libraries with the Illumina DNA Prep Kit (Illumina, San Diego, CA, USA). Library quality was assessed on an ABI StepOnePlus Real-Time PCR System (Life Technologies, Foster City, CA, USA). Qualified libraries were sequenced on the Illumina NovaSeq 6000 platform with PE250 configuration.
2.3. Sequencing Data Analysis
Sequencing data were processed using FASTP (v0.18.0) to remove reads containing (1) ≥10% unidentified nucleotides (N), (2) >50% bases with Phred quality scores ≤20, or (3) adapter sequences [15]. Clean reads were assembled into tags via FLASH (v1.2.11) with a minimum overlap of 10 bp and a maximum mismatch rate of 2% [16]. Raw tags were truncated at the first base position where consecutive low-quality bases (Q ≤ 3) reached a length threshold of 3. Post-truncation tags were further filtered by discarding those with consecutive high-quality base lengths < 75% of total tag length [17]. Clean tags were clustered into operational taxonomic units (OTUs) at a 97% similarity threshold using the UPARSE algorithm in USEARCH (v11.0.667) [18]. Chimeric sequences were detected and removed via UCHIME, with subsequent analyses performed on effective tags [19].
OTU sequences were taxonomically annotated against the SILVA database (v138.2) [20]. Species composition was visualized using stacked bar plots generated with the R package ggplot2 (v2.2.1) [21]. Venn diagrams were constructed with the R package VennDiagram [22]. Differential microbial features were identified via LEfSe using thresholds of LDA score ≥ 2.0 and p < 0.05 [23]. Alpha diversity indices (Shannon/Simpson) and beta diversity (PCoA based on Bray–Curtis distance) were computed using the vegan package in R for both pairwise and multi-group comparisons [24].
2.4. Machine Learning Based on the Fecal Microbiome
The K-medoids algorithm (also termed PAM, Partitioning Around Medoids) is a standard clustering method for microbial community typing, grouping samples with analogous species composition into distinct clusters. For each cluster, a medoid is computed. Through iterative optimization, the algorithm minimizes the total sum of dissimilarity distances between all samples. The optimal K value (number of clusters) is determined by maximizing the average silhouette width, which quantitatively evaluates cluster cohesion and separation.
The SHAP algorithm treats each feature as a “contributor” to the prediction outcome, explaining its importance in specific predictions made by ML and random forest models. SHAP quantifies each feature’s contribution to the model output by assigning a Shapley value, which indicates both the direction and magnitude of the feature’s influence [25]. For a specific prediction, the cumulative Shapley values across all features plus the average prediction value yield the model’s final output, thereby revealing individual feature contributions. Feature contributions derived from 16S rRNA data are determined by the sum of the cumulative SHAP values for a specific prediction and the average prediction value. Global interpretation computes the mean SHAP value for each feature across all samples, highlighting overall feature importance, whereas local interpretation assesses the impact of each feature on an individual prediction [26].
2.5. Statistical Analysis
Machine learning analysis was performed using Python (version 3.10.2). Prior to model construction, the dataset was randomly split into training (70%) and testing (30%) sets. Significance analyses were conducted using R software (version 4.1.1) and SPSS 22.0. The Mann–Whitney U test was employed for two-group comparisons, while the Kruskal–Wallis test was applied for multi-group comparisons. A threshold of p < 0.05 was considered statistically significant. Permutational multivariate analysis of variance (PERMANOVA) was performed using the adonis function (vegan package) to assess significant differences between groups. The graphical elements in the abstract were sourced from BioRender (https://www.biorender.com/) and created using draw.io software (v22.0.2).
3. Results
3.1. Vertical Transmission of Gut Microbiota from Sows to Offspring
SE and SP shared 961 OTUs, with 238 OTUs unique to SE and 136 OTUs unique to SP (Figure 1A). Principal coordinates analysis (PCoA) based on Bray–Curtis distance revealed distinct clustering of SE and SP samples (Figure 1B). No significant differences were observed in Shannon or Simpson indices (p > 0.05; Figure 1C,D). Both SE and SP exhibited dominant phyla Firmicutes (46.3% vs. 47.4%), Bacteroidota (26.7% vs. 27.3%), and Proteobacteria (13.1% vs. 17.3%) (Figure 1E). The relative abundance of Synergistota was higher in SE (3.6%) than in SP (0.5%). Dominant genera shared by SE and SP included Escherichia shigella (12.4%, 16.2%), Lactobacillus (5.9%, 6.3%), and Rikenellaceae RC9 gut group (3.9%, 4.2%) (Figure 1F). Cloacibacillus (in phyla Synergistota) demonstrated greater abundance in SE versus SP. Using LDA > 2 and p < 0.05 as thresholds, differential microbial analysis revealed significantly elevated abundance of Eubacterium brachy group in SE compared to SP, whereas Flavonifractor showed reduced abundance in SE (Figure 1G, Table S1). Overall, the microbiota composition of the SE and SP groups was similar, with observed differences mainly due to changes in the relative abundance of specific bacterial genera.
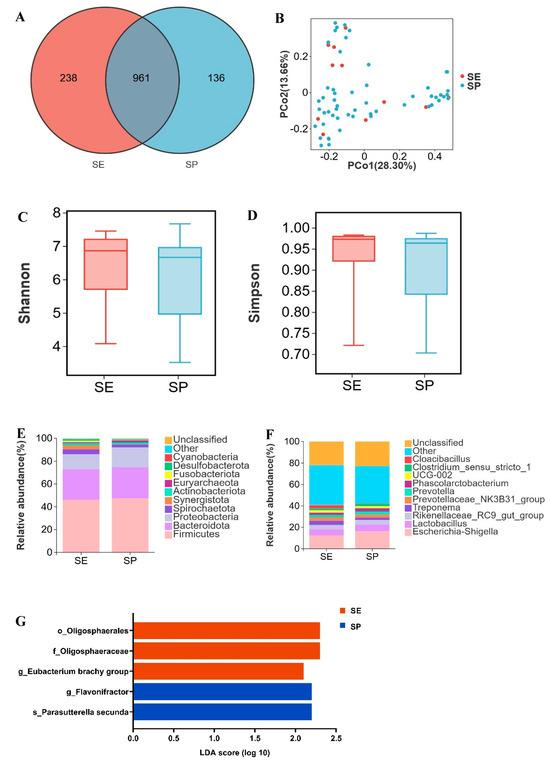
Figure 1.
Vertical transmission of microbiota from Dahe sows to their offspring. (A) Venn diagram of OTUs between lactating sows (SE) and suckling piglets (SP), (B) two-dimensional PCoA plot of all SE and SP samples, (C) Shannon index comparison (Mann–Whitney U test, p = 0.3), (D) Simpson index comparison (Mann−Whitney U test, p = 0.4), (E) top 10 phyla by relative abundance in SE and SP, (F) top 10 genera by relative abundance in SE and SP (unclassified = unannotated microorganisms, other = unlisted taxa), (G) LEfSe analysis (“o_” = order, “f_” = family, “g_” = genus, “s_” = species).
3.2. Differential Gut Microbial Abundance Between Lactating Sows and Adult Swine
Piglet gut microbiota primarily derives from sows, with significant compositional differences observed between lactating sows (SE; body weight: 150–200 kg) and market-weight pigs. Integration of gut microbiota from all pigs ≥ 120 kg (designated PNE) enabled analysis of microbial alterations in lactating sows. Although 822 OTUs were shared between SE and PNE, 377 OTUs were unique to SE and 460 to PNE (Figure 2A). Principal coordinates analysis demonstrated distinct clustering patterns separating SE and PNE (Figure 2B). A significant difference was indeed observed between the SE and PNE groups (PERMANOVA, p = 0.001, R2 = 0.0878). The Shannon and Simpson indices were significantly lower in SE compared to PNE (p < 0.05) (Figure 2C,D).
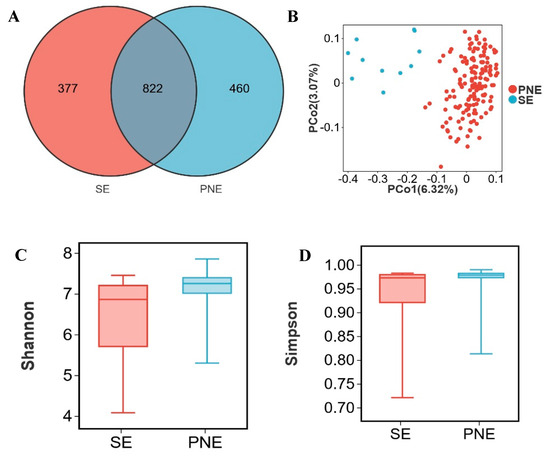
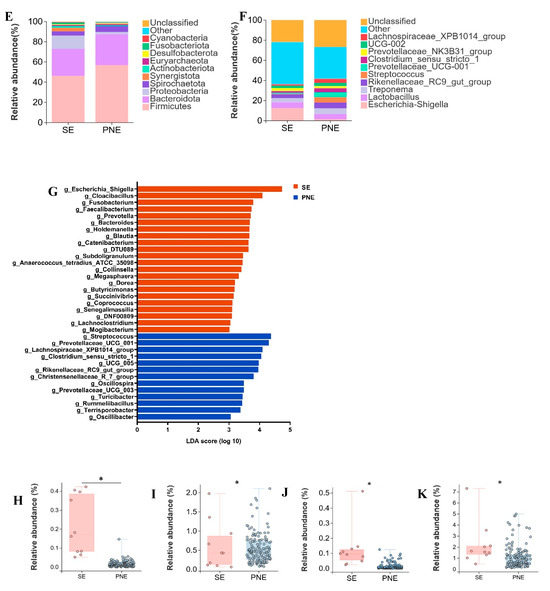
Figure 2.
Distinct characteristic of microbial relative abundance during the lactation period. (A) Venn diagram of OTUs between lactating sows (SE) and pigs weighing > 120 kg (PNE), (B) two- dimensional PCoA plot of all SE and PNE samples, (C) Shannon index comparison (Mann–Whitney U test, p = 0.01), (D) Simpson index comparison (Mann–Whitney U test, p = 0.04), (E) top 10 phyla by relative abundance in SE and PNE, (F) top 10 genera by relative abundance in SE and PNE, (G) analysis of bacterial genera with LDA scores greater than 3 and p < 0.05 using LEfSe, “g_” = genus level, (H) the relative abundance of Lachnoclostridium, (I) the relative abundance of Ruminococcus, (J) the relative abundance of Ruminococcus torques group, (K) the relative abundance of Prevotella. * p < 0.05.
Firmicutes (56.8%, 46.3%), Bacteroidota (30.6%, 26.7%), Proteobacteria (2.2%, 13.1%), and Spirochaetota (5.9%, 4.2%) were predominant in the feces of PNE and SE (Figure 2E). The relative abundance of Escherichia shigella in SE (12.4%) was higher than in PNE (1.4%). In contrast, the levels of Streptococcus, Prevotellaceae UCG 001, and Clostridium sensu stricto 1 were lower in SE (0.5%, 0.9%, and 1.7%, respectively) compared to PNE (5.3%, 4.9%, and 4.0%) (Figure 2F). Using LDA > 2.0 and p < 0.05 as thresholds to screen for differential microbes, 59 differential genera were identified in the SE group and 32 differential genera in the PNE group (Supplementary Table S2). Genera with LDA scores > 3.0 were filtered from Supplementary Table S2. The SE group exhibited 22 differential genera, including Escherichia shigella, Cloacibacillus, Fusobacterium, Faecalibacterium, and Prevotella, whereas the PNE group contained 13 differential genera, such as Streptococcus, Prevotellaceae UCG 001, Lachnospiraceae XPB1014 group, Clostridium sensu stricto 1, and UCG-005 (Figure 2G). The relative abundances of Lachnoclostridium, Ruminococcus, Ruminococcus torques group, and Prevotella in lactating sow fecal samples were higher than in other pigs over 120 kg.
3.3. Changes of Gut Microbiota Across Porcine Growth Stages
In different growth stages of Dahe black pigs (including SP, WP, P50_100, P120_150, and P200), 566 OTUs were shared among all groups, while 148, 157, 58, 67, and 54 OTUs were uniquely identified in each respective stage (Figure 3A). During the SP and WP stages, the gut microbiota of Dahe black pigs underwent substantial changes, with significant variations in microbial composition and relative abundance among individuals. However, microbial profiles gradually converged with age, exhibiting increased similarity (Figure 3B).
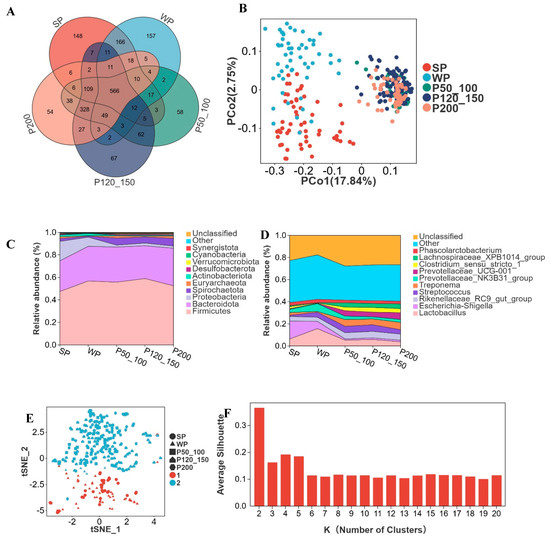
Figure 3.
Advancing age accompanies alterations in microbial composition and relative abundance. (A) Venn diagram of OTUs across developmental stages, suckling piglets (SP), weaned piglets (WP), pigs weighing 50–100 kg (P50_100), 120–150 kg (P120_150), and >200 kg (P200), (B) two-dimensional PCoA plot of all samples, (C) top 10 phyla by relative abundance, (D) top 10 genera by relative abundance, (E) cluster-based regrouping of samples via PCoA, (F) cluster count determination with silhouette width metric, where higher values indicate superior clustering.
Firmicutes and Bacteroidota constituted 47.4% and 27.3%, respectively, at the SP stage, with their relative abundance increasing as body weight grew, reaching 52.6% and 33.3% at P200 (Figure 3C). Proteobacteria accounted for 17.3% at the SP stage but progressively decreased to 2.0% by P200. Spirochaetota represented 2.5% at SP, declined to 0.9% at WP, and then subsequently increased to 6.9% at P200. Lactobacillus peaked at 15.7% in WP, while Escherichia shigella reached its highest level at 16.2% in SP, both gradually decreasing thereafter (Figure 3D). The relative abundance of Streptococcus was 1.1% in SP and 4.5% in WP, remaining relatively stable thereafter. Prevotellaceae NK3B31 group registered 2.9% in SP, increased to 7.1% in WP, and then declined to 2.6% at P200. Treponema, Prevotellaceae UCG 001, Clostridium sensu stricto 1, and Lachnospiraceae XPB1014 group maintained relative abundances below 3% during SP/WP but exceeded 3% in P50_100, P120_150, and P200 stages. The K-medoids algorithm partitioned all genera into two clusters. SP and WP were grouped into Cluster 1, while P50_100, P120_150, and P200 formed Cluster 2 (Figure 3E,F).
3.4. Critical Transition of Microbial Abundance from Suckling to Weaned Piglets
SP and WP shared 811 OTUs, while SP harbored 286 unique OTUs and WP contained 225 distinct OTUs (Figure 4A). Approximately two-thirds of individual samples formed separate clusters in PCoA ordination between SP and WP groups (Figure 4B). No significant differences (p > 0.05) were observed in the Shannon and Simpson indices between the two groups (Figure 4 C, D). At the phylum level, the dominant bacterial phyla in both SP and WP groups were Firmicutes (SP: 47.4%, WP: 56.8%), Bacteroidota (SP: 27.3%, WP: 30.9%), Proteobacteria (SP: 17.3%, WP: 8.1%), Actinobacteriota (SP: 1.6%, WP: 1.9%), and Spirochaetota (SP: 2.5%, WP: 0.9%) (Figure 4E). At the genus level, the relative abundances of Escherichia shigella (16.2%, 6.6%), Lactobacillus (6.3%, 15.7%), Prevotellaceae NK3B31 group (2.9%, 7.1%), Prevotella (3.0%, 5.7%), Streptococcus (1.1%, 4.5%), Parabacteroides (2.0%, 2.3%), Faecalibacterium (0.9%, 3.2%), and Lachnospiraceae NK4A136 group (1.7%, 2.1%) changed between the WP and SP groups (Figure 4F). Using an LDA score > 2 and p < 0.05 as thresholds, 63 differentially abundant genera were identified in the SP group and 46 in the WP group (Supplementary Table S3). Genera with LDA scores >3.0 were filtered from Supplementary Table S3. The SP group comprised 20 differential genera, including Escherichia shigella, Treponema, Clostridium sensu stricto 1, UCG-002, and Methanobrevibacter, while the WP group encompassed 15 differential genera, such as Lactobacillus, Prevotellaceae NK3B31 group, Streptococcus, Prevotella, and Faecalibacterium (Figure 4G). Mitsuokella, Bilophila, Succinivibrio, Romboutsia, and Desulfovibrio were identified as the top five genera contributing most significantly to distinguishing between the SP and WP groups (Figure 4H, Supplementary Table S4). The relative abundance of these genera was significantly higher in SP compared to WP (p < 0.05) (Figure 4I–M).
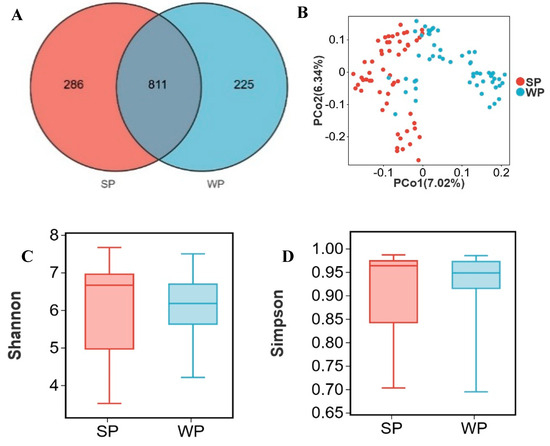
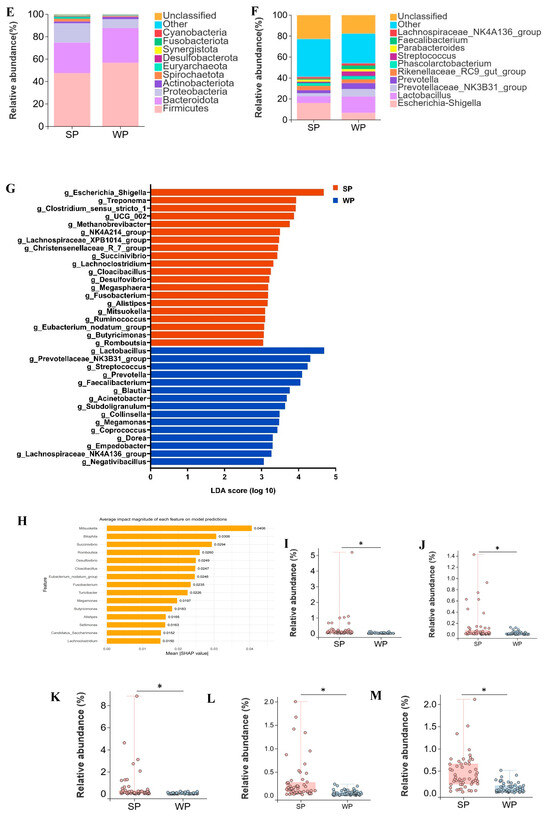
Figure 4.
Alterations in gut microbiota from suckling to weaned piglets. (A) Venn diagram of OTUs between suckling piglets (SP) and weaned piglets (WP), (B) two-dimensional PCoA plot of all SP and WP samples, (C) Shannon index comparison (Mann–Whitney U test, p = 0.4), (D) Simpson index comparison (Mann–Whitney U test, p = 0.7), (E) top 10 phyla by relative abundance in SP and WP, (F) top 10 genera by relative abundance in SP and WP, (G) analysis of bacterial genera with LDA scores greater than 3 and p < 0.05 using LEfSe, (H) SHAP interprets and visualizes machine learning outcomes, (I–M) the relative abundance of Mitsuokella (I), Bilophila (J), Succinivibrio (K), Romboutsia (L), and Desulfovibrio (M). * p < 0.05.
3.5. Changes in Microbial Abundance from Post-Weaning to Finishing Stage (50–100 kg Body Weight)
The WP and P50_100 stages represent the pivotal transition phase where intestinal microbiota succession in Dahe black pigs culminates in stabilization. WP and P50_100 shared 663 OTUs, while WP and P50_100 harbored 373 and 609 unique OTUs, respectively (Figure 5A). PCoA revealed distinct clustering of WP and P50_100 groups, with microbial composition converging as age increased (Figure 5B). Both the Shannon and Simpson indices were significantly higher in the P50_100 group compared to WP (Figure 5C,D). Firmicutes (WP: 56.8%, P50_100: 55.7%) and Bacteroidota (WP: 30.9%, P50_100: 31.2%) dominated the fecal microbiota in both groups (Figure 5E). The relative abundance of Proteobacteria was higher in WP (8.1%) than in P50_100 (1.7%), whereas Spirochaetota abundance was lower in WP (0.9%) versus P50_100 (6.3%). Distinct genus-level community composition was observed between groups (Figure 5F). Specifically, Lactobacillus (15.7% vs. 5.2%), Prevotellaceae NK3B31 group (7.1% vs. 3.1%), Escherichia shigella (6.6% vs. 1.2%), and Prevotella (5.7% vs. 0.9%) were enriched in WP relative to P50_100. Conversely, Rikenellaceae RC9 gut group (3.8% vs. 5.6%), Treponema (0.7% vs. 6.1%), Prevotellaceae UCG 001 (0.2% vs. 4.9%), and UCG-005 (1.5% vs. 3.1%) showed reduced abundance in WP. LEfSe analysis identified 67 and 61 differentially abundant genera in the WP and P50_100 groups, respectively (Supplementary Table S5). Genera with LDA scores > 3.0 were filtered from Supplementary Table S4. The WP group comprised 22 differential genera, including Lactobacillus, Escherichia shigella, Prevotella, Prevotellaceae NK3B31 group, and Faecalibacterium, while the P50_100 group encompassed 22 differential genera, such as Treponema, Prevotellaceae UCG 001, Lachnospiraceae XPB1014 group, Christensenellaceae R7 group, and Clostridium sensu stricto 1 (Figure 5G). The machine learning model selected Lachnospiraceae XPB1014 group, Clostridium sensu stricto 1, Turicibacter, Quinella, and p 1088 a5 gut group as the top five contributing microbes discriminating WP and P50_100 (Figure 5H, Supplementary Table S6). Consistently, the relative abundance of Lachnospiraceae XPB1014 group, Clostridium sensu stricto 1, Turicibacter, Quinella, and p 1088 a5 gut group were significantly elevated in P50_100 compared to WP (p < 0.05) (Figure 5I–M).
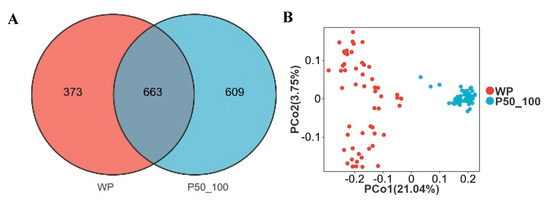
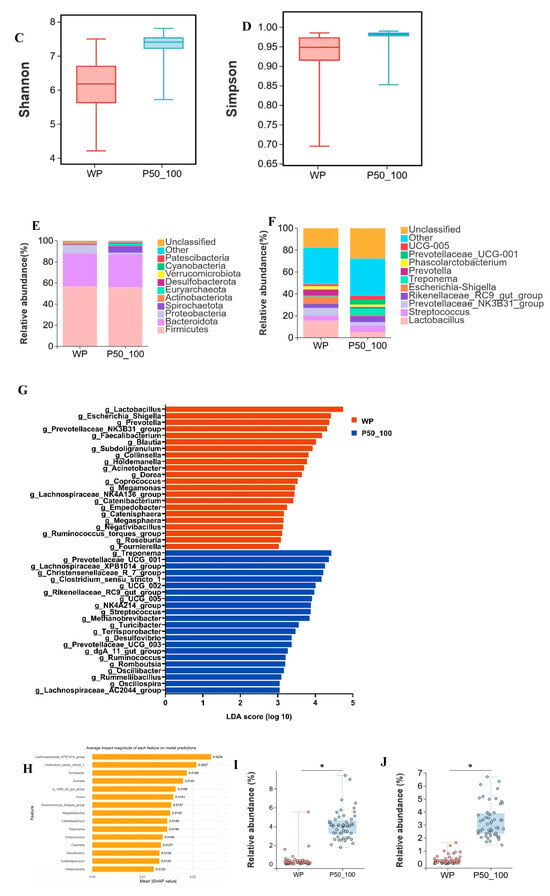
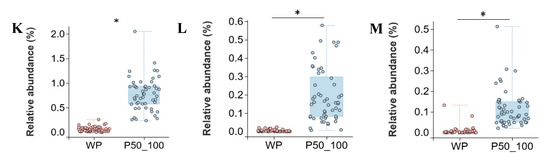
Figure 5.
The period from weaned piglets to the 50–100 kg growth stage constitutes a critical transition point for microbial alterations. (A) Venn diagram of OTUs between weaned piglets (WP) and pigs weighing 50–100 kg (P50_100), (B) two-dimensional PCoA plot of all WP and P50_100 samples, (C) Shannon index comparison (Mann–Whitney U test, p < 0.01), (D) Simpson index comparison (Mann–Whitney U test, p < 0.01), (E) top 10 phyla by relative abundance, (F) top 10 genera by relative abundance, (G) analysis of bacterial genera with LDA scores greater than 3 and p < 0.05 using LEfSe, (H) SHAP interprets and visualizes machine learning outcomes, (I–M) the relative abundance of Lachnospiraceae XPB1014 group (I), Clostridium sensu stricto 1 (J), Turicibacter (K), Quinella (L), and p 1088 a5 gut group (M). * p < 0.05.
4. Discussion
Gut microbiota influences vital body processes, with early-life colonization being crucial for shaping intestinal development [27]. In this study, the fecal microbiota of 323 Dahe black pigs across different growth stages were analyzed to characterize their composition and abundance features using 16S rRNA sequencing. The results indicate that early-life microbial colonization in Dahe black piglets relies heavily on the sow’s microbiota. Weaning represents a critical turning point, after which microbial communities gradually stabilize in composition and richness.
Both Dahe black sows and piglets exhibited dominant phyla Firmicutes (46.3% vs. 47.4%), Bacteroidota (26.7% vs. 27.3%), and Proteobacteria (13.1% vs. 17.3%); the dominant genera included Escherichia shigella (12.4% vs. 16.2%), Lactobacillus (5.9% vs. 6.3%), and Rikenellaceae RC9 gut group (3.9% vs. 4.2%). The fecal microbiota of newborn piglets initially resembled environmental sources (slatted floor, sow’s milk, nipple surfaces) but proved transient, ultimately being succeeded by sow fecal microbiota [28]. Sow milk microbiota shapes piglet gut microbiomes, with Corynebacterium and Streptococcus being dominant in colostrum, while Lactobacillus, Ruminococcaceae, Lachnospiraceae, and Clostridiales are enriched in mature milk [29]. The sow fecal microbiota exerts a persistent influence on the fecal microbiota of offspring piglets until at least 10 days of age [30]. Bacterial and fungal taxa detected in sow feces were also present in piglet gastric and cecal digesta, supporting their role in neonatal gut colonization; Lactobacillus largely increased in piglet intestines from days 3 to 7, while post-weaning, plant-glycan fermenters (e.g., Prevotella-9) appeared to replace milk–glycan fermenting Fusobacterium and Bacteroides [31]. Maternal dietary supplementation with Lactobacillus reuteri exerted profound effects on the early-life overall microbial composition and maturation of the fecal microbiota in offspring piglets [32]. This included an increased relative abundance of beneficial bacteria in the piglets’ meconium, such as Romboutsia, Lactobacillus, Blautia, Butyricicoccus, and Ruminococcus. These findings indicate that the initial gut microbiota composition in piglets is primarily governed by the sow, suggesting that modulating the sow’s gut microbiota represents a feasible pathway for indirectly regulating the piglet microbiome.
Analysis of gut microbiota from 613 healthy individuals identified three distinct enterotypes, an Escherichia shigella-dominant enterotype, a mixed enterotype characterized by Bacteroides and Faecalibacterium, and a Prevotella-dominant enterotype, with age emerging as the primary driver of microbial variation [33]. The core genus in the vaginal secretions and colostrum of sows was Pseudomonas, and in piglets at 1 d of age, Pseudomonas and Escherichia shigella were most abundant [34]. The gut microbiota of sows on day 3 antepartum was characterized by an increased relative abundance of Escherichia shigella, Fusobacterium, and Bacteroides, alongside a decreased relative abundance of Alloprevotella, Prevotellaceae UCG 003, and Ruminococcus 1 [35]. The infant gut microbiota exhibited high heterogeneity, forming three distinct clusters: a Bifidobacterium-enriched cluster, a Bacteroides-enriched cluster, and an Escherichia shigella-enriched cluster [36]. Healthy large white piglets exhibited a continuous decrease in Lactobacillus and Escherichia shigella alongside a gradual increase in Prevotella [37]. This shift in the relationship between Escherichia shigella and Prevotella could potentially explain the piglet diarrhea. In this study, the relative abundance of Escherichia shigella gradually decreased with increasing age in piglets, while there were progressive increases in Lactobacillus and Prevotella. This microbial shift may contribute to the enhanced disease resilience observed in Chinese indigenous pig breeds.
Distinct differences in gut microbiota composition and richness were observed between lactating sows and non-pregnant/non-lactating Dahe black pigs. The intestinal microbiota of Mangalica pigs exhibited resilience to dietary intake variations (limit feeding vs. ad libitum), and PERMANOVA confirmed that diet significantly influenced microbial community composition (p = 0.012, R2 = 0.37182) [38]. Significant differences were indeed observed between lactating sows and non-pregnant/non-lactating Dahe black pigs (PERMANOVA, p = 0.001, R2 = 0.0878); however, the lower R2 value compared to dietary factors suggests that physiological stage may have a limited impact on gut microbiota composition. The relative abundance of Escherichia shigella was significantly higher in lactating sows compared to non-pregnant/non-lactating pigs. Conversely, lactating sows exhibited significantly lower relative abundances of Streptococcus, Prevotellaceae UCG 001, and Clostridium sensu stricto 1. Furthermore, fecal samples from lactating sows showed significantly higher relative abundances of Lachnoclostridium, Ruminococcus, Ruminococcus torques group, and Prevotella than those from non-pregnant/non-lactating pigs. During late gestation in sows, Rothia, Moraxella, and Streptococcus dominated the oral microbiota, while Clostridium sensu stricto 1 prevailed in the rectal microbiota, and Streptococcus emerged as the most abundant genus in both vaginal and colostrum samples [39]. Compared to gestation and empty phases, the lactation stage demonstrated significantly higher relative abundance of bacterial genera, predominantly belonging to Firmicutes (e.g., Lachnospiraceae, Ruminococcus) and Bacteroidetes (e.g., Paraprevotellaceae, Prevotella) [40,41]. Cellulose-degrading microorganisms in the porcine intestine include Bacteroides succinogenes and Ruminococcus flavefaciens [42,43]. In the present study, lactating sows of the Dahe black pig breed exhibited higher dietary fiber intake levels compared to the comparative cohorts, which partially explains the observed higher relative abundance of Ruminococcus in lactating sows.
Swine fecal bacterial composition varied across growth stages, with Bacteroidetes decreasing as weight increased; differential abundance testing between growers and finishers revealed nearly half of the species as shared OTUs, indicating that community differences are primarily driven by abundance variation [44]. Bacteroidetes and Firmicutes were the predominant phyla in the fecal microbiota of healthy pigs at 14, 36, 48, 60, and 70 days of age [45]. Prevotella, Clostridium, Alloprevotella, and Ruminococcus, and the Rikenellaceae RC9 gut group, were found in 99% of all fecal samples [46]. Compared to older pigs (>90% Firmicutes, ~2% Proteobacteria), one-month-old pigs exhibited significantly lower Firmicutes (73%) and higher Proteobacteria (16.3%), while Bacteroidetes showed fluctuations (5.1%→0.9%→4.9% at 1/2/6 months) and Actinobacteria replaced Spirochaetes as the fourth dominant phylum post-weaning before declining to fifth by six months [47]. Longitudinal analysis of 18 pigs across the lactation (d 0, 11, 20), nursery (d 27, 33, 41, 50, 61), growing (d 76, 90, 104, 116), and finishing stages (d 130, 146, 159, 174) revealed 19 gut microbiome phyla (dominated by Firmicutes and Bacteroidetes) with alpha diversity increasing overall [48]. Gut microbiota in Dahe black pigs exhibited distinct characteristics across growth stages, including an age-dependent increase in the relative abundance of Firmicutes and a gradual decline in Proteobacteria.
The early intestinal colonizers belonging to Bacteroides, Escherichia shigella, Clostridium sensu stricto 1, and Fusobacterium, increasing abundances of Prevotella, Butyricimonas, and Christensenellaceae R7 group as the piglets aged, which indicates the adaptation of the piglets to a cereal-based diet rich in oligosaccharides and starch [49]. The predominant genera in suckling piglet microbiota (Fusobacterium, Lactobacillus, Bacteroides, Escherichia shigella, and Megasphaera) were replaced post-weaning by Clostridium sensu stricto 1, Roseburia, Paraprevotella, Clostridium XIVa, and Blautia [50]. During suckling, Lactobacillus and Bacteroides dominated the gut microbiota, with the latter progressively replaced by Clostridium sensu stricto 1 as piglets aged; upon weaning, the microbiota became enriched with fiber-degrading bacteria [51]. Post-weaning gut microbiome changes rapidly, characterized by decreased relative abundance of Escherichia shigella and increased abundance of Copromorpha, Clostridium, Fusicatenibacter saccharivorans, Intestinibacter, Oliverpabstia intestinalis, Phascolarctobacterium, Prevotella, and Ruminococcus [52]. The fecal microbiota of Pietrain × (Large White × Landrace) castrated male and female pigs’ feces microbiota evolved strongly from 52 to 99 days of age, with an increased abundance of Streptococcaceae and a decreased abundance of Lactobacillaceae; during the finishing stage, microbiota kept evolving at a slower rate [53]. Similar to previous studies, the relative abundance of Escherichia shigella in the feces of Dahe black pigs gradually decreased with age, while the relative abundance of fiber-degrading microorganisms (such as Prevotella) increased.
Machine learning was applied to feature selection of porcine gut microbiota. Feature selection using random forest impurity-based importance scores identified a core set of 25 to 35 key microbial genera in the Iberian pig, among which Acetitomaculum, Escherichia shigella, and Lachnospiraceae FCS020 group were consistently identified as the most important features across all scenarios [54]. Mitsuokella, Bilophila, Succinivibrio, Romboutsia, and Desulfovibrio were identified as the top five genera contributing most significantly to distinguishing between the suckling and weaned piglets. Sixty-day-old large white pig populations consistently split into two enterotypes with either Prevotella and Mitsuokella (PM enterotype) or Ruminococcus and Treponema (RT enterotype) as keystone taxa [55]. There were higher abundances of Roseburia, Ruminococcus, Coprococcus, Dorea, and Lachnospira in weaned piglets compared to prior to weaning [56]. The significant post-weaning shift (3–7 days) in the fecal microbiome of all piglets included increased relative abundance of several Prevotella spp. and butyrate-producing species (Butyricicoccus porcorum, Faecalibacterium prausnitzii, Megasphaera elsdenii) immediately following weaning [57]. Members of the Succinivibrio demonstrated significant negative correlations with growth and carcass traits [58]. In the present study, Succinivibrio was significantly enriched in suckling piglets compared to weaned piglets. It is postulated that the early growth and development of Dahe black pigs are linked to the reduced relative abundance of Succinivibrio.
Lachnospiraceae XPB1014 group, Clostridium sensu stricto 1, Turicibacter, Quinella, and p 1088 a5 gut group were the top five contributing microbes discriminating Dahe black weaned pigs and 50–100 kg pigs. Lachnospiracea XPB1014 group is a beneficial bacterium involved in the synthesis of short-chain fatty acids [59]. A negative correlation was observed between the Lachnospiraceae XPB1014 group and LPS-binding protein levels, which were positively correlated with intramuscular fat (IMF) content [60,61]. Christensenellaceae R7 group, Ruminococcaceae NK4A214 group, Lachnospiraceae XPB1014 group, Eubacterium coprostanoligenes group, Rikenellaceae RC9 gut group, Lachnospiraceae AC2044 group, and Prevotellaceae UCG 001 comprised the predominant SCFA-synthesizing bacteria [62]. Lachnospiraceae XPB1014 group exhibited marked enrichment at the genus level within the colonic microbiota of Shaziling pigs in response to reduced dietary protein [63]. Prevotellaceae UCG 001, Alistipes, and Clostridium sensu stricto 1 were highly positively correlated with pig backfat thickness and intramuscular fatness [64]. This indicates that during the ontogenic progression of Dahe black pigs, microbial consortia involved in fiber degradation and lipid metabolism supersede previously dominant taxa to establish themselves as the emergent functional core.
5. Conclusions
In conclusion, the intestinal microbiota of Dahe black pigs exhibits fluctuations during the early stages of development. In contrast, the microbiota of growing pigs and finishing pigs demonstrates relative stability. The initial colonization of the piglet gut microbiome is derived from the sow, resulting in similarity between the microbiota of suckling piglets and their dams. Furthermore, the relative abundance profile of the maternal microbiota during lactation differs from that observed in pigs weighing over 120 kg. Utilizing machine learning approaches, we identified two critical transition points in the developmental trajectory of the Dahe black pig microbiota: the shift from suckling to weaning and the transition from weaning to the 50–100 kg growth phase. Key microbial taxa associated with the first transition period (suckling to weaning) include Mitsuokella, Bilophila, Succinivibrio, Romboutsia, and Desulfovibrio. Key taxa related to the second transition period (weaning to 50–100 kg) comprise Lachnospiraceae XPB1014 group, Clostridium sensu stricto 1, Turicibacter, Quinella, and p 1088 a5 gut group. These identified microbial taxa represent potential candidate targets for modulating the developmental timing of growth phases in pigs, offering possibilities for either advancing or delaying specific physiological timepoints. Although this study sought to characterize key microbial shifts across different stages, technical constraints of 16S rRNA sequencing preclude reliable resolution of species-level differences. Nonetheless, the identified genus-level signatures delineate a focused scope for subsequent microbiological validation. Future research should prioritize longitudinal tracking of individual pigs to map microbial successions from birth (day 0) through market age.
Supplementary Materials
The following supporting information can be downloaded at https://www.mdpi.com/article/10.3390/microorganisms13092038/s1, Table S1: LEfSe analysis of sows and piglets; Table S2: LEfSe analysis in lactating sows and non-lactating pigs; Table S3: LEfSe analysis in suckling and weaned piglets; Table S4: The performance metrics of SP vs WP; Table S5: Lefse analysis in weaned piglets and live weight (50–100 kg) pigs; Table S6: The performance metrics of WP vs. P50-100.
Author Contributions
S.Z., conceived the study. L.Y., formal analysis, software, writing, and editing. W.C., methodology and investigation. G.S. formal analysis. H.J., Y.X. and W.Z., investigation. J.Z., formal analysis. All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded by the National Key R&D Program of China, grant number 2024YFD1800404; the National Natural Science Foundation of China, grant numbers 32360808, 31760645, 31260592, and 31060331; Yunnan Fundamental Research Projects, grant number 202501AS070086; and the Sci-Tech Service Station of Farmer Academician in Fuyuan County.
Institutional Review Board Statement
All the animal procedures were approved by the Animal Care and Use Committee of Yunnan Agricultural University (approval code 202403081).
Informed Consent Statement
Not applicable.
Data Availability Statement
The original contributions presented in this study are included in the article/Supplementary Materials. Further inquiries can be directed to the corresponding authors.
Acknowledgments
Technical support was provided by Gene Denovo Biotechnology Co., Ltd. (Guangzhou, China), including access to sequencing platforms and bioinformatic analysis.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| OTUs | operational taxonomic units |
References
- Upadhaya, S.D.; Kim, I.H. Maintenance of gut microbiome stability for optimum intestinal health in pigs—A review. J. Anim. Sci. Biotechnol. 2022, 13, 140. [Google Scholar] [CrossRef]
- Guevarra, R.B.; Lee, J.H.; Lee, S.H.; Seok, M.J.; Kim, D.W.; Kang, B.N.; Johnson, T.J.; Isaacson, R.E.; Kim, H.B. Piglet gut microbial shifts early in life: Causes and effects. J. Anim. Sci. Biotechnol. 2019, 10, 1. [Google Scholar] [CrossRef] [PubMed]
- Tian, M.; Li, Q.; Zheng, T.; Yang, S.; Chen, F.; Guan, W.; Zhang, S. Maternal microbe-specific modulation of the offspring microbiome and development during pregnancy and lactation. Gut Microbes 2023, 15, 2206505. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.; Ricker, N.; Holman, D.B.; Johnson, T.A. Meta-analysis reveals the predictable dynamic development of the gut microbiota in commercial pigs. Microbiol. Spectr. 2023, 11, e0172223. [Google Scholar] [CrossRef] [PubMed]
- Gaio, D.; DeMaere, M.Z.; Anantanawat, K.; Eamens, G.J.; Falconer, L.; Chapman, T.A.; Djordjevic, S.; Darling, A.E. Phylogenetic diversity analysis of shotgun metagenomic reads describes gut microbiome development and treatment effects in the post-weaned pig. PLoS ONE 2022, 17, e0270372. [Google Scholar] [CrossRef]
- Gaio, D.; DeMaere, M.Z.; Anantanawat, K.; Chapman, T.A.; Djordjevic, S.P.; Darling, A.E. Post-weaning shifts in microbiome composition and metabolism revealed by over 25,000 pig gut metagenome-assembled genomes. Microb. Genom. 2021, 7, 000501. [Google Scholar]
- Lim, M.Y.; Song, E.J.; Kang, K.S.; Nam, Y.D. Age-related compositional and functional changes in micro-pig gut microbiome. Geroscience 2019, 41, 935–944. [Google Scholar] [CrossRef] [PubMed]
- Isaacson, R.; Kim, H.B. The intestinal microbiome of the pig. Anim. Health Res. Rev. 2012, 13, 100–109. [Google Scholar] [CrossRef]
- Patil, Y.; Gooneratne, R.; Ju, X.H. Interactions between host and gut microbiota in domestic pigs: A review. Gut Microbes 2020, 11, 310–334. [Google Scholar] [CrossRef]
- Luo, Y.; Ren, W.; Smidt, H.; Wright, A.G.; Yu, B.; Schyns, G.; McCormack, U.M.; Cowieson, A.J.; Yu, J.; He, J.; et al. Dynamic Distribution of Gut Microbiota in Pigs at Different Growth Stages: Composition and Contribution. Microbiol. Spectr. 2022, 10, e0068821. [Google Scholar] [CrossRef]
- Shao, M.; Wang, Z.; He, Y.; Tan, Z.; Zhang, J. Fecal microbial composition and functional diversity of Wuzhishan pigs at different growth stages. AMB Express 2021, 11, 88. [Google Scholar] [CrossRef]
- Tan, Z.; Li, J.; He, Y.; Wang, F.; Xiao, Q. Characteristics of gut microbiota and metabolomic of Hainan Tunchang pigs at various growth stages. Gene 2024, 900, 148161. [Google Scholar] [CrossRef] [PubMed]
- Ayoola, M.B.; Pillai, N.; Nanduri, B.; Rothrock, M.J., Jr.; Ramkumar, M. Predicting foodborne pathogens and probiotics taxa within poultry-related microbiomes using a machine learning approach. Anim. Microbiome 2023, 5, 57. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Wang, H.; Qin, L.; Wang, T.; Zhang, Y.; Sun, Y. Interpretable machine learning reveals microbiome signatures strongly associated with dairy cow milk urea nitrogen. iScience 2024, 27, 109955. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Magoč, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef]
- Bokulich, N.A.; Subramanian, S.; Faith, J.J.; Gevers, D.; Gordon, J.I.; Knight, R.; Mills, D.A.; Caporaso, J.G. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat. Methods 2013, 10, 57–59. [Google Scholar] [CrossRef]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef]
- Pruesse, E.; Quast, C.; Knittel, K.; Fuchs, B.M.; Ludwig, W.; Peplies, J.; Glöckner, F.O. SILVA: A comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 2007, 35, 7188–7196. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2. Wiley Interdiscip. Rev. Comput. Stat. 2011, 3, 180–185. [Google Scholar] [CrossRef]
- Chen, H.; Boutros, P.C. VennDiagram: A package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinform. 2011, 12, 35. [Google Scholar] [CrossRef]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [PubMed]
- Oksanen, J.; Blanchet, F.G.; Kindt, R.; Legendre, P.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Sólymos, P.; Stevens, M.H.H.; Wagner, H. Package ‘vegan’. In Community Ecology Package; University of Helsinki: Helsinki, Finland, 2010. [Google Scholar]
- Wu, H.; Li, Y.; Jiang, Y.; Li, X.; Wang, S.; Zhao, C.; Yang, X.; Chang, B.; Yang, J.; Qiao, J. Machine learning prediction of obesity-associated gut microbiota: Identifying Bifidobacterium pseudocatenulatum as a potential therapeutic target. Front. Microbiol. 2025, 15, 1488656. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Fang, Y.; Li, S.; Zeng, L.; Chen, S.; Li, Z.; Ji, G.; Yang, X.; Wu, W. Interpretable machine learning algorithms reveal gut microbiome features associated with atopic dermatitis. Front. Immunol. 2025, 16, 1528046. [Google Scholar] [CrossRef]
- Nowland, T.L.; Kirkwood, R.N.; Pluske, J.R. Review: Can early-life establishment of the piglet intestinal microbiota influence production outcomes? Animal 2022, 16 (Suppl. 2), 100368. [Google Scholar] [CrossRef]
- Chen, X.; Xu, J.; Ren, E.; Su, Y.; Zhu, W. Co-occurrence of early gut colonization in neonatal piglets with microbiota in the maternal and surrounding delivery environments. Anaerobe 2018, 49, 30–40. [Google Scholar] [CrossRef]
- Chen, W.; Mi, J.; Lv, N.; Gao, J.; Cheng, J.; Wu, R.; Ma, J.; Lan, T.; Liao, X. Lactation Stage-Dependency of the Sow Milk Microbiota. Front. Microbiol. 2018, 9, 945. [Google Scholar] [CrossRef]
- Nowland, T.L.; Kirkwood, R.N.; Plush, K.J.; Barton, M.D.; Torok, V.A. Exposure to maternal feces in lactation influences piglet enteric microbiota, growth, and survival preweaning. J. Anim. Sci. 2021, 99, skab170. [Google Scholar] [CrossRef]
- Yosi, F.; Lerch, F.; Vötterl, J.C.; Koger, S.; Verhovsek, D.; Metzler-Zebeli, B.U. Lactation-related dynamics of bacterial and fungal microbiomes in feces of sows and gut colonization in suckling and newly weaned piglets. J. Anim. Sci. 2024, 102, skae321. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Wang, X.; Ma, Y.; Cai, S.; Yang, L.; Fan, Y.; Zeng, X.; Qiao, S. Lactobacillus reuteri improves the development and maturation of fecal microbiota in piglets through mother-to-infant microbe and metabolite vertical transmission. Microbiome 2022, 10, 211. [Google Scholar] [CrossRef]
- Wu, J.; Shen, H.; Lv, Y.; He, J.; Xie, X.; Xu, Z.; Yang, P.; Qian, W.; Bai, T.; Hou, X. Age over sex: Evaluating gut microbiota differences in healthy Chinese populations. Front. Microbiol. 2024, 15, 1412991. [Google Scholar] [CrossRef]
- Li, Y.; Liu, Y.; Ma, Y.; Ge, X.; Zhang, X.; Cai, C.; Yang, Y.; Lu, C.; Liang, G.; Guo, X.; et al. Effects of Maternal Factors and Postpartum Environment on Early Colonization of Intestinal Microbiota in Piglets. Front. Vet. Sci. 2022, 9, 815944. [Google Scholar] [CrossRef]
- Gaukroger, C.H.; Edwards, S.A.; Walshaw, J.; Nelson, A.; Adams, I.P.; Stewart, C.J.; Kyriazakis, I. Shifting sows: Longitudinal changes in the periparturient faecal microbiota of primiparous and multiparous sows. Animal 2021, 15, 100135. [Google Scholar] [CrossRef]
- Marrs, T.; Jo, J.H.; Perkin, M.R.; Rivett, D.W.; Witney, A.A.; Bruce, K.D.; Logan, K.; Craven, J.; Radulovic, S.; Versteeg, S.A.; et al. Gut microbiota development during infancy: Impact of introducing allergenic foods. J. Allergy Clin. Immunol. 2021, 147, 613–621.e9. [Google Scholar] [CrossRef]
- Yang, Q.; Huang, X.; Wang, P.; Yan, Z.; Sun, W.; Zhao, S.; Gun, S. Longitudinal development of the gut microbiota in healthy and diarrheic piglets induced by age-related dietary changes. Microbiologyopen 2019, 8, e923. [Google Scholar] [CrossRef] [PubMed]
- Hallowell, H.A.; Higgins, K.V.; Roberts, M.; Johnson, R.M.; Bayne, J.; Maxwell, H.S.; Brandebourg, T.; Hiltbold Schwartz, E. Longitudinal Analysis of the Intestinal Microbiota in the Obese Mangalica Pig Reveals Alterations in Bacteria and Bacteriophage Populations Associated With Changes in Body Composition and Diet. Front. Cell Infect. Microbiol. 2021, 11, 698657. [Google Scholar] [CrossRef] [PubMed]
- Piirainen, V.; König, E.; Husso, A.; Heinonen, M.; Iivanainen, A.; Pessa-Morikawa, T.; Niku, M. Bacterial profiles of the oral, vaginal, and rectal mucosa and colostrum of periparturient sows. PLoS ONE 2025, 20, e0317513. [Google Scholar] [CrossRef]
- Liu, H.; Hou, C.; Li, N.; Zhang, X.; Zhang, G.; Yang, F.; Zeng, X.; Liu, Z.; Qiao, S. Microbial and metabolic alterations in gut microbiota of sows during pregnancy and lactation. FASEB J. 2019, 33, 4490–4501. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.J.; Li, H.; Xie, P.F.; Li, Z.H.; Li, H.W.; Yin, Y.L.; Blachier, F.; Kong, X.F. Stages of pregnancy and weaning influence the gut microbiota diversity and function in sows. J. Appl. Microbiol. 2019, 127, 867–879. [Google Scholar] [CrossRef]
- Varel, V.H.; Fryda, S.J.; Robinson, I.M. Cellulolytic bacteria from pig large intestine. Appl. Environ. Microbiol. 1984, 47, 219–221. [Google Scholar] [CrossRef]
- Varel, V.H. Activity of fiber-degrading microorganisms in the pig large intestine. J. Anim. Sci. 1987, 65, 488–496. [Google Scholar] [CrossRef]
- Kim, J.; Nguyen, S.G.; Guevarra, R.B.; Lee, I.; Unno, T. Analysis of swine fecal microbiota at various growth stages. Arch. Microbiol. 2015, 197, 753–759. [Google Scholar] [CrossRef]
- Mach, N.; Berri, M.; Estellé, J.; Levenez, F.; Lemonnier, G.; Denis, C.; Leplat, J.J.; Chevaleyre, C.; Billon, Y.; Doré, J.; et al. Early-life establishment of the swine gut microbiome and impact on host phenotypes. Environ. Microbiol. Rep. 2015, 7, 554–569. [Google Scholar] [CrossRef]
- Holman, D.B.; Brunelle, B.W.; Trachsel, J.; Allen, H.K. Meta-analysis To Define a Core Microbiota in the Swine Gut. mSystems 2017, 2, e00004-17. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Wang, Y.; Liu, S.; Huang, J.; Zhai, Z.; He, C.; Ding, J.; Wang, J.; Wang, H.; Fan, W.; et al. The dynamic distribution of porcine microbiota across different ages and gastrointestinal tract segments. PLoS ONE 2015, 10, e0117441. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Tsai, T.; Deng, F.; Wei, X.; Chai, J.; Knapp, J.; Apple, J.; Maxwell, C.V.; Lee, J.A.; Li, Y.; et al. Longitudinal investigation of the swine gut microbiome from birth to market reveals stage and growth performance associated bacteria. Microbiome 2019, 7, 109. [Google Scholar] [CrossRef] [PubMed]
- Saladrigas-García, M.; Durán, M.; D’Angelo, M.; Coma, J.; Pérez, J.F.; Martín-Orúe, S.M. An insight into the commercial piglet’s microbial gut colonization: From birth towards weaning. Anim. Microbiome 2022, 4, 68. [Google Scholar] [CrossRef]
- Chen, L.; Xu, Y.; Chen, X.; Fang, C.; Zhao, L.; Chen, F. The Maturing Development of Gut Microbiota in Commercial Piglets during the Weaning Transition. Front. Microbiol. 2017, 8, 1688. [Google Scholar] [CrossRef]
- Mahmud, M.R.; Jian, C.; Uddin, M.K.; Huhtinen, M.; Salonen, A.; Peltoniemi, O.; Venhoranta, H.; Oliviero, C. Impact of Intestinal Microbiota on Growth Performance of Suckling and Weaned Piglets. Microbiol. Spectr. 2023, 11, e0374422. [Google Scholar] [CrossRef]
- Holman, D.B.; Kommadath, A.; Tingley, J.P.; Abbott, D.W. Novel Insights into the Pig Gut Microbiome Using Metagenome-Assembled Genomes. Microbiol. Spectr. 2022, 10, e0238022. [Google Scholar] [CrossRef]
- Le Sciellour, M.; Renaudeau, D.; Zemb, O. Longitudinal Analysis of the Microbiota Composition and Enterotypes of Pigs from Post-Weaning to Finishing. Microorganisms 2019, 7, 622. [Google Scholar] [CrossRef]
- Azouggagh, L.; Ibáñez-Escriche, N.; Martínez-Álvaro, M.; Varona, L.; Casellas, J.; Negro, S.; Casto-Rebollo, C. Characterization of microbiota signatures in Iberian pig strains using machine learning algorithms. Anim. Microbiome 2025, 7, 13. [Google Scholar] [CrossRef]
- Larzul, C.; Estellé, J.; Borey, M.; Blanc, F.; Lemonnier, G.; Billon, Y.; Thiam, M.G.; Quinquis, B.; Galleron, N.; Jardet, D.; et al. Driving gut microbiota enterotypes through host genetics. Microbiome 2024, 12, 116. [Google Scholar] [CrossRef]
- Saladrigas-García, M.; D’Angelo, M.; Ko, H.L.; Nolis, P.; Ramayo-Caldas, Y.; Folch, J.M.; Llonch, P.; Solà-Oriol, D.; Pérez, J.F.; Martín-Orúe, S.M. Understanding host-microbiota interactions in the commercial piglet around weaning. Sci. Rep. 2021, 11, 23488. [Google Scholar] [CrossRef] [PubMed]
- Holman, D.B.; Gzyl, K.E.; Mou, K.T.; Allen, H.K. Weaning Age and Its Effect on the Development of the Swine Gut Microbiome and Resistome. mSystems 2021, 6, e0068221. [Google Scholar] [CrossRef] [PubMed]
- Maltecca, C.; Dunn, R.; He, Y.; McNulty, N.P.; Schillebeeckx, C.; Schwab, C.; Shull, C.; Fix, J.; Tiezzi, F. Microbial composition differs between production systems and is associated with growth performance and carcass quality in pigs. Anim. Microbiome 2021, 3, 57. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Song, R.; Zhou, J.; Jia, Y.; Lu, J. Fermented Bamboo Fiber Improves Productive Performance by Regulating Gut Microbiota and Inhibiting Chronic Inflammation of Sows and Piglets during Late Gestation and Lactation. Microbiol. Spectr. 2023, 11, e0408422. [Google Scholar] [CrossRef]
- Yu, C.; Li, H.; Hua, L.; Che, L.; Feng, B.; Fang, Z.; Xu, S.; Zhuo, Y.; Li, J.; Wu, D.; et al. Deciphering the microbiome, lipopolysaccharides, and metabolome interplay: Unveiling putrescine’s mechanism for enhancing sperm quality in heat-stressed boars. Theriogenology 2025, 236, 60–73. [Google Scholar] [CrossRef]
- Su, J.; Li, J.; Azad, M.A.K.; Wang, W.; Luo, Z.; Wang, J.; Yin, J.; Yin, Y.; Tan, B.; Chen, J. Dynamic distribution of gut microbiota-metabolites during post-weaning longissimus dorsi muscle development in Ningxiang pigs. Microbiol. Spectr. 2024, 12, e0081324. [Google Scholar] [CrossRef]
- Li, Z.; Luo, Z.; Hu, D. Assessing Fecal Microbial Diversity and Hormone Levels as Indicators of Gastrointestinal Health in Reintroduced Przewalski’s Horses (Equus ferus przewalskii). Animals 2024, 14, 2616. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Duan, Y.; Zheng, C.; Yu, J.; Li, F.; Guo, Q.; Yin, Y. Long-Term Protein Restriction Modulates Lipid Metabolism in White Adipose Tissues and Alters Colonic Microbiota of Shaziling Pigs. Animals 2022, 12, 2944. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Xin, Y.; Ma, Y.; Xu, X.; Zhao, S.; Cao, J. Screening of Microbes Associated With Swine Growth and Fat Deposition Traits Across the Intestinal Tract. Front. Microbiol. 2020, 11, 586776. [Google Scholar] [CrossRef] [PubMed]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).