
Conserved and Specific Root-Associated Microbiome Reveals Close Correlation Between Fungal Community and Growth Traits of Multiple Chinese Fir Genotypes

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Material and Methods
2.1. Sample Collection and Treatment
2.2. DNA Extraction and ITS Amplicon Sequencing
2.3. Amplicon Bioinformatics Analysis
3. Results
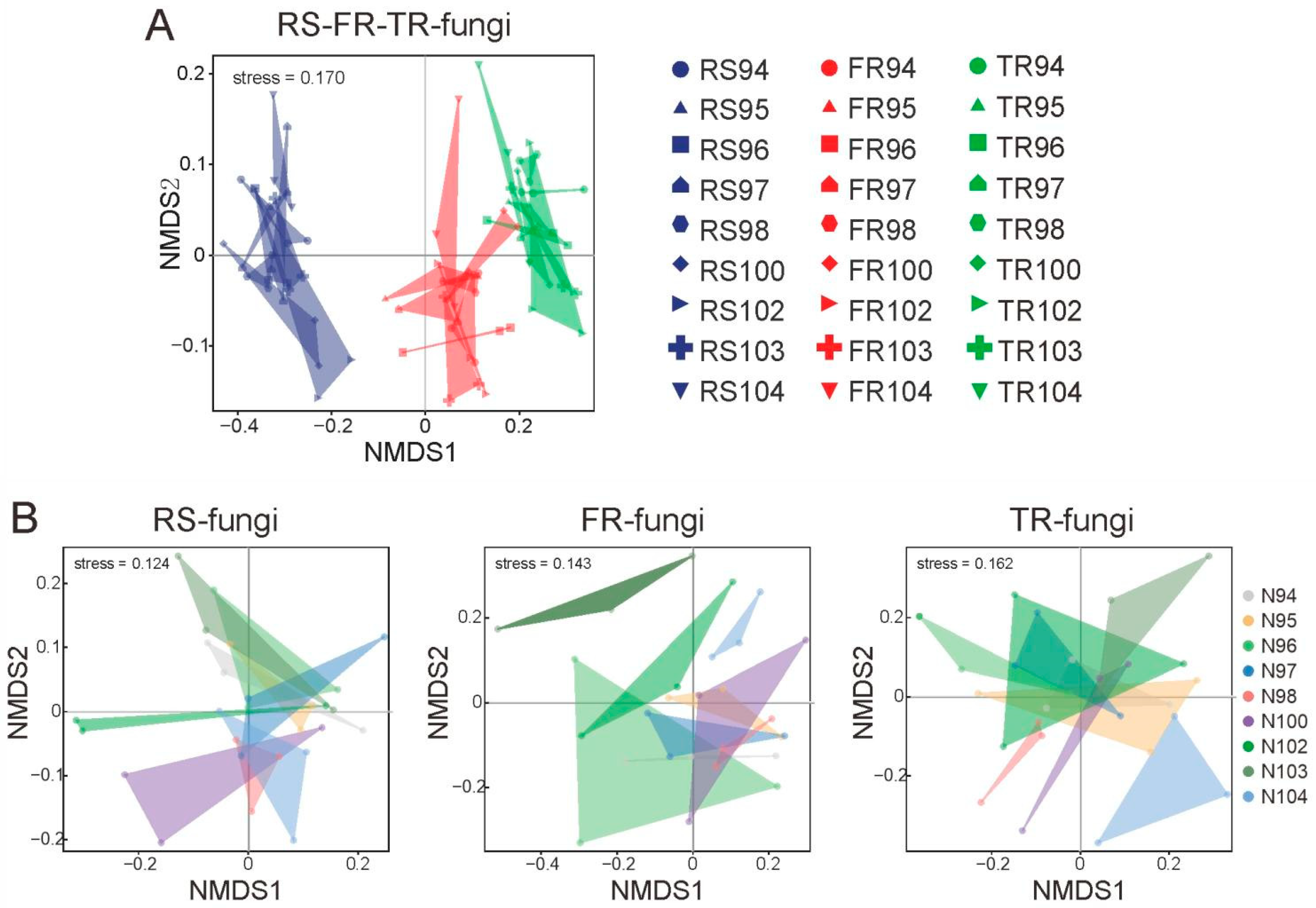
3.1. Divergence of the Fungal Community from Rhizosphere Soil, Fine Roots, and Thick Roots
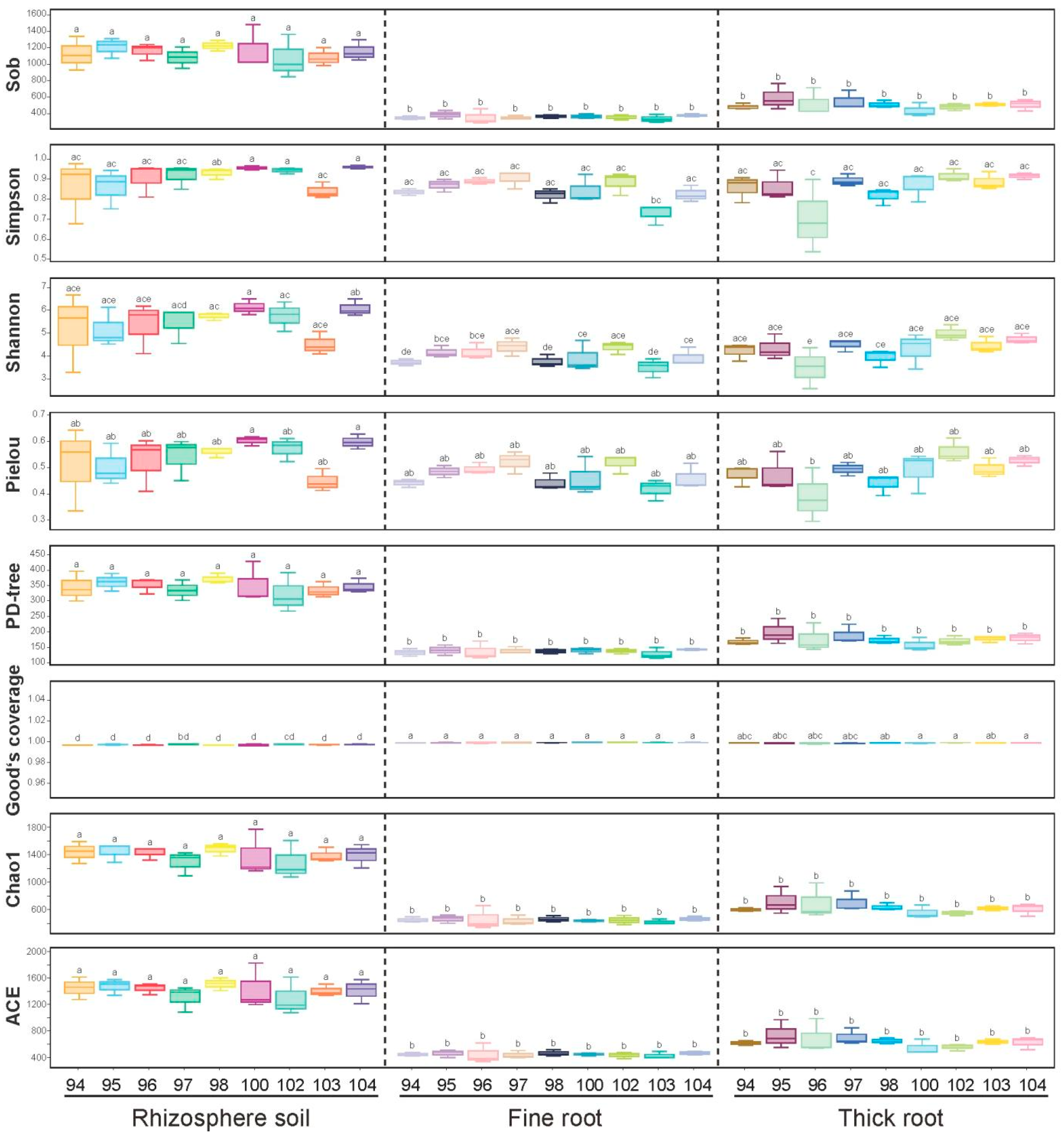
3.2. The α-Diversity of the Fungal Community from Different Tissues
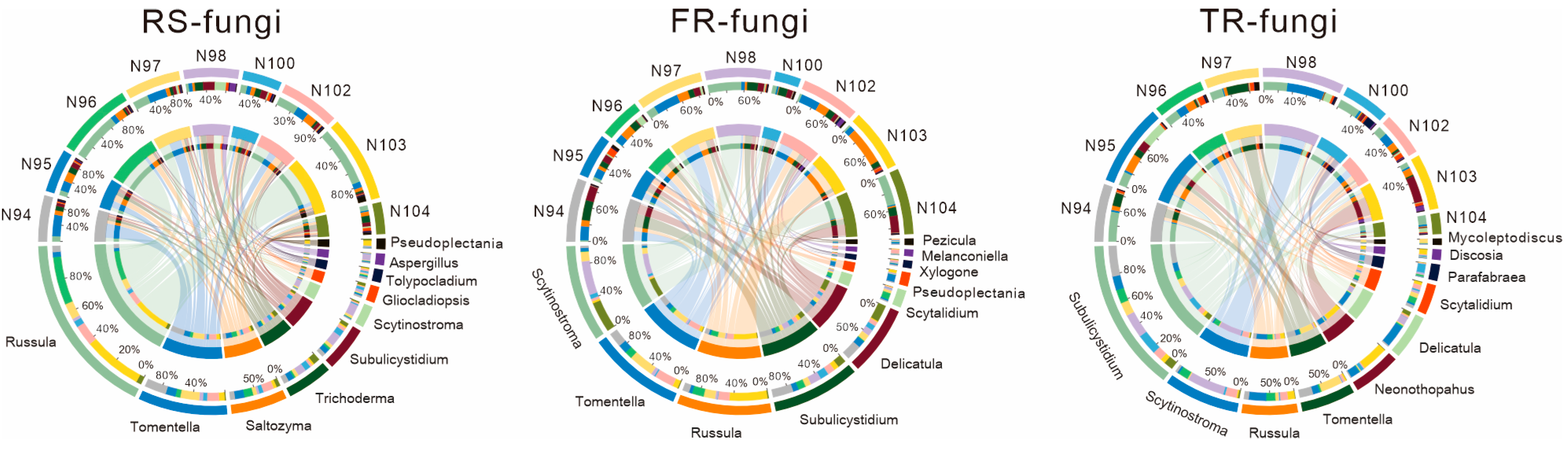
3.3. Genus-Level Fungal Community Structure Analysis
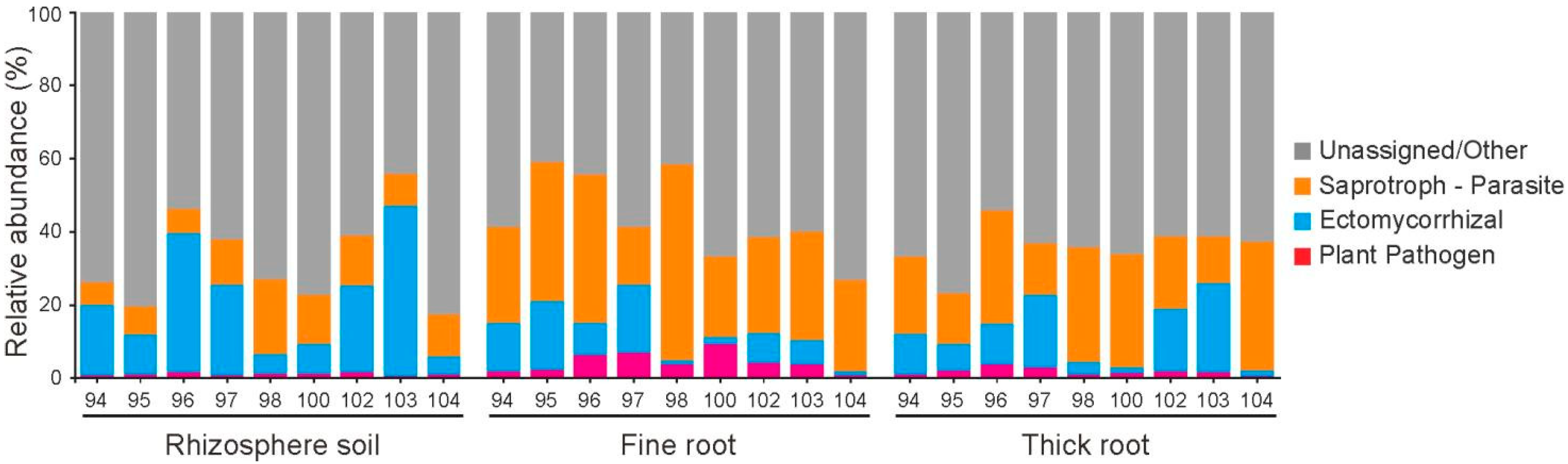
3.4. Functional Prediction Reveals Tissue-Specific and Genotype-Specific Functions
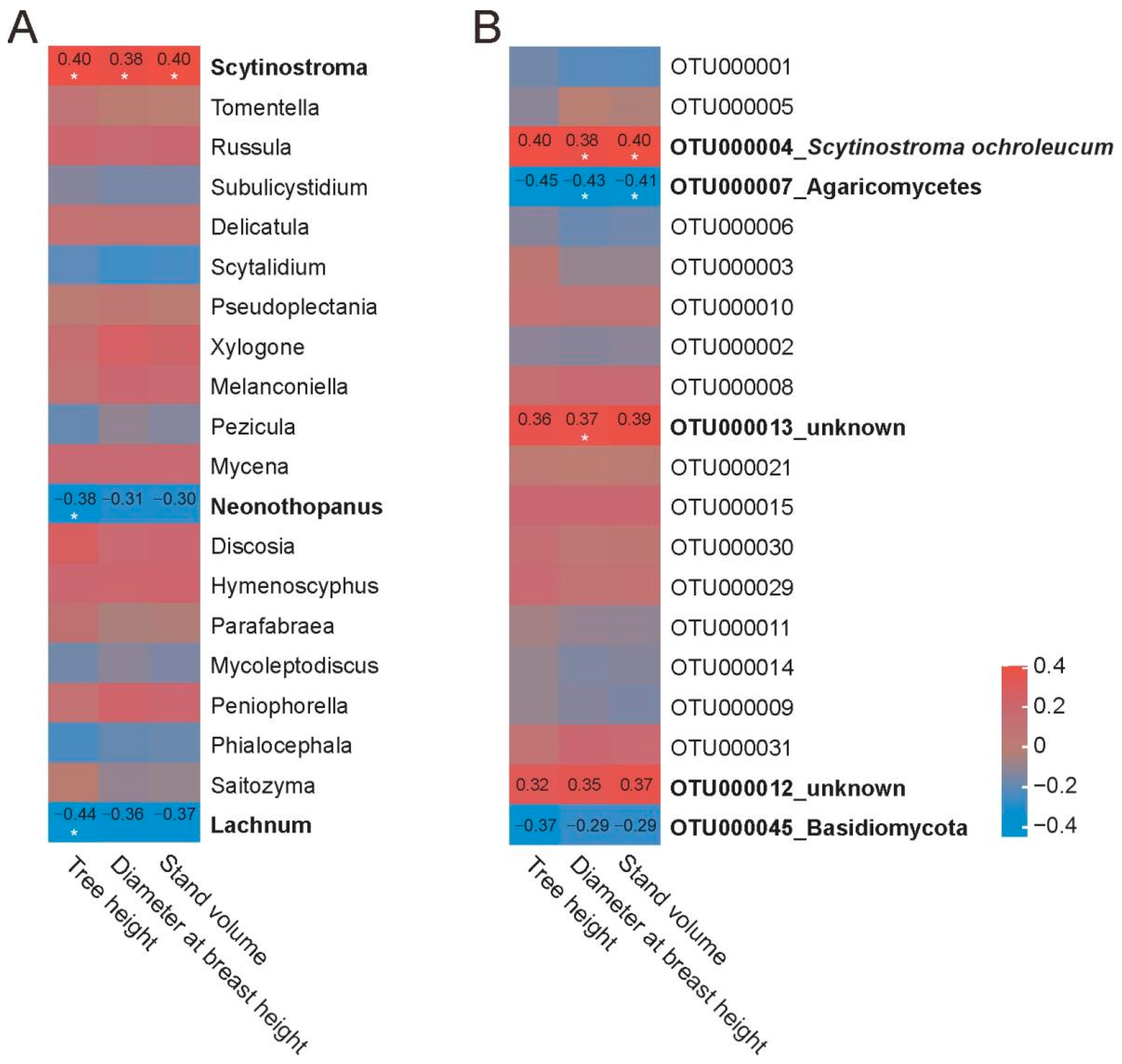
3.5. Correlation of Fungal Taxonomic Abundance with Chinese Fir Growth
4. Discussion
4.1. The Decreasing Trend of Fungal Community from the Rhizosphere to the Root Interior
4.2. Rhizosphere Soil Is a Critical Fungal Reservoir for Root Colonization
4.3. Tissue and Host-Genotype Specificity in Structure and Function of Fungal Community
4.4. Key Fungal Genera Influencing Chinese Fir Growth
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| NMDS | Non-metric multidimensional scaling |
| FRs | fine roots |
| RS | rhizosphere soil |
| TRs | thick roots |
| OTUs | Operational Taxonomic Units |
| AMFs | arbuscular mycorrhizal fungi |
| PRRs | pattern recognition receptors |
| ISR | inducing systemic resistance |
| ECMs | ectomycorrhizal fungi |
References
- Llado, S.; Lopez-Mondejar, R.; Baldrian, P. Forest soil bacteria: Diversity, involvement in ecosystem processes, and response to global change. Microbiol. Mol. Biol. Rev. 2017, 81, e00063-16. [Google Scholar] [CrossRef] [PubMed]
- Bai, B.; Liu, W.; Qiu, X.; Zhang, J.; Zhang, J.; Bai, Y. The root microbiome: Community assembly and its contributions to plant fitness. J. Integr. Plant Biol. 2022, 64, 230–243. [Google Scholar] [CrossRef] [PubMed]
- Harbort, C.J.; Hashimoto, M.; Inoue, H.; Niu, Y.; Guan, R.; Rombola, A.D.; Kopriva, S.; Voges, M.; Sattely, E.S.; Garrido-Oter, R.; et al. Root-secreted coumarins and the microbiota interact to improve iron nutrition in Arabidopsis. Cell Host Microbe 2020, 28, 825–837.e6. [Google Scholar] [CrossRef] [PubMed]
- Wahab, A.; Muhammad, M.; Munir, A.; Abdi, G.; Zaman, W.; Ayaz, A.; Khizar, C.; Reddy, S.P.P. Role of arbuscular mycorrhizal fungi in regulating growth, enhancing productivity, and potentially influencing ecosystems under abiotic and biotic stresses. Plants 2023, 12, 3102. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.; He, X.; Baer, M.; Beirinckx, S.; Tian, T.; Moya, Y.A.T.; Zhang, X.; Deichmann, M.; Frey, F.P.; Bresgen, V.; et al. Plant flavones enrich rhizosphere Oxalobacteraceae to improve maize performance under nitrogen deprivation. Nat. Plants 2021, 7, 481–499. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.; Zou, Y.-N.; Kuča, K.; Hashem, A.; Abd_Allah, E.F.; Wu, Q.-S. Elucidating the mechanisms underlying enhanced drought tolerance in plants mediated by arbuscular mycorrhizal fungi. Front. Microbiol. 2021, 12, 809473. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; La, S.; Zhang, X.; Gao, L.; Tian, Y. Salt-induced recruitment of specific root-associated bacterial consortium capable of enhancing plant adaptability to salt stress. ISME J. 2021, 15, 2865–2882. [Google Scholar] [CrossRef] [PubMed]
- Finkel, O.M.; Salas-Gonzalez, I.; Castrillo, G.; Conway, J.M.; Law, T.F.; Teixeira, P.; Wilson, E.D.; Fitzpatrick, C.R.; Jones, C.D.; Dangl, J.L. A single bacterial genus maintains root growth in a complex microbiome. Nature 2020, 587, 103–108. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, P.; Colaianni, N.R.; Law, T.F.; Conway, J.M.; Gilbert, S.; Li, H.; Salas-Gonzalez, I.; Panda, D.; Del Risco, N.M.; Finkel, O.M.; et al. Specific modulation of the root immune system by a community of commensal bacteria. Proc. Natl. Acad. Sci. USA 2021, 118, e2100678118. [Google Scholar] [CrossRef] [PubMed]
- Romeralo, C.; Martín-García, J.; Martínez-Álvarez, P.; Muñoz-Adalia, E.J.; Gonçalves, D.R.; Torres, E.; Witzell, J.; Diez, J.J. Pine species determine fungal microbiome composition in a common garden experiment. Fungal Ecol. 2022, 56, 101137. [Google Scholar] [CrossRef]
- Zhao, X.; Guo, M.; Zhang, T.; Bai, S.; Meng, Y.; Tian, Y.; Yang, J.; Ma, F. Spatiotemporal dynamics of root exudates drive microbial adaptation mechanisms under day-night alterations in constructed wetlands. Chem. Eng. J. 2023, 477, 147311. [Google Scholar] [CrossRef]
- Wang, Q.; Wang, Z.; Du, W.; Liu, Y.; Hong, L.; Wu, P.; Ma, X.; Wang, K. Stable diversity but distinct metabolic activity of microbiome of roots from adult and young Chinese fir trees. Forests 2024, 15, 2140. [Google Scholar] [CrossRef]
- Wang, K.; Wang, Q.; Hong, L.; Liu, Y.; Yang, J.; Asiegbu, F.O.; Wu, P.; Huang, L.; Ma, X. Distribution and characterization of endophytic and rhizosphere bacteriome of below-ground tissues in Chinese fir plantation. Tree Physiol. 2024, 44, tpae137. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Wei, Y.; Chen, J.; Dong, Y.; Hou, L.; Jiao, R. Screening, identification and growth-promotion products of multifunctional bacteria in a Chinese fir plantation. Forests 2021, 12, 120. [Google Scholar] [CrossRef]
- Zhou, F.; Zhang, H.; Yang, H.; Wang, S.; Zou, B.; Zhou, L.; Yang, Z.-J.; Zheng, Y. Tree growth is linked to the diversity of belowground fungal functional guilds across nine Chinese fir plantations in subtropics. Appl. Soil Ecol. 2025, 211, 106149. [Google Scholar] [CrossRef]
- Tian, Y.; Xu, J.; Lian, X.; Wei, B.; Ma, X.; Wu, P. Effect of Glomus intraradices on root morphology, biomass production and phosphorous use efficiency of Chinese fir seedlings under low phosphorus stress. Front. Plant Sci. 2022, 13, 1095772. [Google Scholar] [CrossRef] [PubMed]
- Hong, L.; Wang, Q.; Zhang, J.; Chen, X.; Liu, Y.; Asiegbu, F.O.; Wu, P.; Ma, X.; Wang, K. Advances in the beneficial endophytic fungi for the growth and health of woody plants. For. Res. 2024, 4, e028. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Magoč, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef] [PubMed]
- Bokulich, N.A.; Subramanian, S.; Faith, J.J.; Gevers, D.; Gordon, J.I.; Knight, R.; Mills, D.A.; Caporaso, J.G. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat. Methods 2013, 10, 57–59. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef] [PubMed]
- Ondov, B.; Bergman, N.; Phillippy, A. Interactive metagenomic visualization in a Web browser. BMC Bioinform. 2011, 12, 385. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H. ggplot2. Wiley Interdiscip. Rev. Comput. Stat. 2011, 3, 180–185. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed]
- Oksanen, J.A.I.; Blanchet, F.G.; Kindt, R.; Legendre, P.; O’Hara, R.; Simpson, G.; Minchin, P.E.H.; O’Hara, R. Vegan: Community Ecology Package, R package version 1.8-5; R Foundation for Statistical Computing: Vienna, Austria, 2007. [Google Scholar]
- Nguyen, N.H.; Song, Z.; Bates, S.T.; Branco, S.; Tedersoo, L.; Menke, J.; Schilling, J.S.; Kennedy, P.G. FUNGuild: An open annotation tool for parsing fungal community datasets by ecological guild. Fungal Ecol. 2016, 20, 241–248. [Google Scholar] [CrossRef]
- William, R. Psych: Procedures for Psychological, Psychometric, and Personality Research; Northwestern University: Evanston, IL, USA, 2024. [Google Scholar] [CrossRef]
- Li, T.; Ren, J.; He, W.; Wang, Y.; Wen, X.; Wang, X.; Ye, M.; Chen, G.; Zhao, K.; Hou, G.; et al. Anatomical structure interpretation of the effect of soil environment on fine root function. Front. Plant Sci. 2022, 13, 993127. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Liang, Q.; Yao, Q.; Bai, Y.; Zhu, H. The role of the rhizobiome recruited by root exudates in plant disease resistance: Current status and future directions. Environ. Microbiome 2024, 19, 91. [Google Scholar] [CrossRef] [PubMed]
- Afridi, M.S.; Kumar, A.; Javed, M.A.; Dubey, A.; de Medeiros, F.H.V.; Santoyo, G. Harnessing root exudates for plant microbiome engineering and stress resistance in plants. Microbiol. Res. 2024, 279, 127564. [Google Scholar] [CrossRef] [PubMed]
- Schlüter, S.; Blaser, S.; Weber, M.; Schmidt, V.; Vetterlein, D. Quantification of root growth patterns from the soil perspective via root distance models. Front. Plant Sci. 2018, 9, 1084. [Google Scholar] [CrossRef] [PubMed]
- Tian, P.; Razavi, B.S.; Zhang, X.; Wang, Q.; Blagodatskaya, E. Microbial growth and enzyme kinetics in rhizosphere hotspots are modulated by soil organics and nutrient availability. Soil Biol. Biochem. 2020, 141, 107662. [Google Scholar] [CrossRef]
- Jiang, G.; Zhang, Y.; Chen, M.; Ramoneda, J.; Han, L.; Shi, Y.; Peyraud, R.; Wang, Y.; Shi, X.; Chen, X.; et al. Effects of plant tissue permeability on invasion and population bottlenecks of a phytopathogen. Nat. Commun. 2024, 15, 62. [Google Scholar] [CrossRef] [PubMed]
- Waqar, S.; Bhat, A.A.; Khan, A.A. Endophytic fungi: Unravelling plant-endophyte interaction and the multifaceted role of fungal endophytes in stress amelioration. Plant Physiol. Biochem. 2024, 206, 108174. [Google Scholar] [CrossRef] [PubMed]
- de Lamo, F.J.; Šimkovicová, M.; Fresno, D.H.; de Groot, T.; Tintor, N.; Rep, M.; Takken, F.L.W. Pattern-triggered immunity restricts host colonization by endophytic fusaria, but does not affect endophyte-mediated resistance. Mol. Plant Pathol. 2021, 22, 204–215. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Zeng, J.; Wan, X.; Wang, Y.; Lan, S.; Zou, S.; Qian, X. Variation in community structure of the root-associated fungi of Cinnamomum camphora forest. J. Fungi 2022, 8, 1210. [Google Scholar] [CrossRef] [PubMed]
- Davidson, H.; Shrestha, R.; Cornulier, T.; Douglas, A.; Travis, T.; Johnson, D.; Price, A.H. Spatial effects and GWA mapping of root colonization assessed in the interaction between the rice diversity Panel 1 and an arbuscular mycorrhizal fungus. Front. Plant Sci. 2019, 10, 633. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Jin, Z.; Wang, Z.; Liao, Y.; Yu, L.; Li, X. Host Plant selection imprints structure and assembly of fungal community along the soil-root continuum. mSystems 2022, 7, e00361-22. [Google Scholar] [CrossRef] [PubMed]
- Ling, N.; Wang, T.; Kuzyakov, Y. Rhizosphere bacteriome structure and functions. Nat. Commun. 2022, 13, 836. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Xu, Z.; Chen, L.; Xun, W.; Shu, X.; Chen, Y.; Sun, X.; Wang, Z.; Ren, Y.; Shen, Q.; et al. Root colonization by beneficial rhizobacteria. FEMS Microbiol. Rev. 2024, 48, fuad066. [Google Scholar] [CrossRef] [PubMed]
- Duret, M.; Wallner, A.; Buée, M.; Aziz, A. Rhizosphere microbiome assembly, drivers and functions in perennial ligneous plant health. Microbiol. Res. 2024, 287, 127860. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Lin, Y.-C.J.; Chen, Y.-L.; Zhou, C.; Li, S.; De Ridder, N.; Oliveira, D.M.; Zhang, L.; Zhang, B.; Wang, J.P.; et al. Woody plant cell walls: Fundamentals and utilization. Mol. Plant 2024, 17, 112–140. [Google Scholar] [CrossRef] [PubMed]
- Lebreton, A.; Zeng, Q.; Miyauchi, S.; Kohler, A.; Dai, Y.-C.; Martin, F.M. Evolution of the mode of nutrition in symbiotic and saprotrophic fungi in forest ecosystems. Annu. Rev. Ecol. Evol. Syst. 2021, 52, 385–404. [Google Scholar] [CrossRef]
- Martins, B.R.; Radl, V.; Treder, K.; Michałowska, D.; Pritsch, K.; Schloter, M. The rhizosphere microbiome of 51 potato cultivars with diverse plant growth characteristics. FEMS Microbiol. Ecol. 2024, 100, fiae088. [Google Scholar] [CrossRef] [PubMed]
- Conti Taguali, S.; Pöter, R.; Aloi, F.; Fernández-Trujillo, C.; Acedo, A.; La Spada, F.; Li Destri Nicosia, M.G.; Pane, A.; Schena, L.; Cacciola, S.O. Influence of environmental and agronomic variables on soil microbiome in citrus orchards: A comparative analysis of organic and conventional farming system. Microbiol. Res. 2025, 299, 128260. [Google Scholar] [CrossRef] [PubMed]
- Xun, W.; Liu, Y.; Ma, A.; Yan, H.; Miao, Y.; Shao, J.; Zhang, N.; Xu, Z.; Shen, Q.; Zhang, R. Dissection of rhizosphere microbiome and exploiting strategies for sustainable agriculture. New Phytol. 2024, 242, 2401–2410. [Google Scholar] [CrossRef] [PubMed]
- Adaskaveg, J.E.; Blanchette, R.A.; Gilbertson, R.L. Decay of date palm wood by white-rot and brown-rot fungi. Can. J. Bot. 1991, 69, 615–629. [Google Scholar] [CrossRef]
- Carvalho, M.L.; Maciel, V.F.; Bordonal, R.d.O.; Carvalho, J.L.N.; Ferreira, T.O.; Cerri, C.E.P.; Cherubin, M.R. Stabilization of organic matter in soils: Drivers, mechanisms, and analytical tools—A literature review. Rev. Bras. Ciência Solo 2023, 47, e0230130. [Google Scholar] [CrossRef]
- Chakrawal, A.; Lindahl, B.D.; Manzoni, S. Modelling optimal ligninolytic activity during plant litter decomposition. New Phytol. 2024, 243, 866–880. [Google Scholar] [CrossRef] [PubMed]
- Adamczyk, B.; Heinonsalo, J.; Simon, J. Mechanisms of carbon sequestration in highly organic ecosystems—Importance of chemical ecology. ChemistryOpen 2020, 9, 464–469. [Google Scholar] [CrossRef] [PubMed]
- Shakhova, E.S.; Karataeva, T.A.; Markina, N.M.; Mitiouchkina, T.; Palkina, K.A.; Perfilov, M.M.; Wood, M.G.; Hoang, T.T.; Hall, M.P.; Fakhranurova, L.I.; et al. An improved pathway for autonomous bioluminescence imaging in eukaryotes. Nat. Methods 2024, 21, 406–410. [Google Scholar] [CrossRef] [PubMed]
- Šoln, K.; Koce, J.D. Oxidative stress in roots: Detection of lipid peroxidation and total antioxidative capacity. Methods Mol. Biol. 2022, 2447, 221–231. [Google Scholar] [CrossRef] [PubMed]
- Vieira, M.B.B.; Oliveira, I.C.; Oliveira, M.d.D.A.d.; Costa Júnior, J.S.d.; Andrade, T.d.J.A.d.S.; Feitosa, C.M.; Rai, M.; Lima, N.M.; Silva, D.d.C. A review on bioluminescent fungus Neonothopanus gardneri. Res. Soc. Dev. 2022, 11, e16811528009. [Google Scholar] [CrossRef]
- Wang, Y.; Su, N.; Hou, G.; Li, J.; Ye, M. Hypoglycemic and hypolipidemic effects of a polysaccharide from Lachnum YM240 and its derivatives in mice, induced by a high fat diet and low dose STZ. Medchemcomm 2017, 8, 964–974. [Google Scholar] [CrossRef] [PubMed]
- Stadler, M.; Anke, H.; Arendholz, W.R.; Hansske, F.; Anders, U.; Sterner, O.; Bergquist, K.E. Lachnumon and lachnumol a, new metabolites with nematicidal and antimicrobial activities from the ascomycete Lachnum papyraceum (Karst.) Karst. I. Producing organism, fermentation, isolation and biological activities. J. Antibiot. 1993, 46, 961–967. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, C.; Liang, W.; Liu, S. Rhizosphere Microbiome: The emerging barrier in plant-pathogen interactions. Front. Microbiol. 2021, 12, 772420. [Google Scholar] [CrossRef] [PubMed]
- Spear, E.R.; Broders, K.D. Host-generalist fungal pathogens of seedlings may maintain forest diversity via host-specific impacts and differential susceptibility among tree species. New Phytol. 2021, 231, 460–474. [Google Scholar] [CrossRef] [PubMed]
- Whitham, T.G.; Gehring, C.A.; Lamit, L.J.; Wojtowicz, T.; Evans, L.M.; Keith, A.R.; Smith, D.S. Community specificity: Life and afterlife effects of genes. Trends Plant Sci. 2012, 17, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Lamit, L.J.; Holeski, L.M.; Flores-Rentería, L.; Whitham, T.G.; Gehring, C.A. Tree genotype influences ectomycorrhizal fungal community structure: Ecological and evolutionary implications. Fungal Ecol. 2016, 24, 124–134. [Google Scholar] [CrossRef]
- Fiorilli, V.; Martinez-Medina, A.; Pozo, M.J.; Lanfranco, L. Plant immunity modulation in arbuscular mycorrhizal symbiosis and its impact on pathogens and pests. Annu. Rev. Phytopathol. 2024, 62, 127–156. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, X.; Wang, Z.; Du, W.; Zhang, J.; Liu, Y.; Hong, L.; Wang, Q.; Zhou, C.; Wu, P.; Ma, X.; et al. Conserved and Specific Root-Associated Microbiome Reveals Close Correlation Between Fungal Community and Growth Traits of Multiple Chinese Fir Genotypes. Microorganisms 2025, 13, 1741. https://doi.org/10.3390/microorganisms13081741
Chen X, Wang Z, Du W, Zhang J, Liu Y, Hong L, Wang Q, Zhou C, Wu P, Ma X, et al. Conserved and Specific Root-Associated Microbiome Reveals Close Correlation Between Fungal Community and Growth Traits of Multiple Chinese Fir Genotypes. Microorganisms. 2025; 13(8):1741. https://doi.org/10.3390/microorganisms13081741
Chicago/Turabian StyleChen, Xuan, Zhanling Wang, Wenjun Du, Junhao Zhang, Yuxin Liu, Liang Hong, Qingao Wang, Chuifan Zhou, Pengfei Wu, Xiangqing Ma, and et al. 2025. "Conserved and Specific Root-Associated Microbiome Reveals Close Correlation Between Fungal Community and Growth Traits of Multiple Chinese Fir Genotypes" Microorganisms 13, no. 8: 1741. https://doi.org/10.3390/microorganisms13081741
APA StyleChen, X., Wang, Z., Du, W., Zhang, J., Liu, Y., Hong, L., Wang, Q., Zhou, C., Wu, P., Ma, X., & Wang, K. (2025). Conserved and Specific Root-Associated Microbiome Reveals Close Correlation Between Fungal Community and Growth Traits of Multiple Chinese Fir Genotypes. Microorganisms, 13(8), 1741. https://doi.org/10.3390/microorganisms13081741