
Whole-Genome Sequencing of a Potentially Novel Aeromonas Species Isolated from Diseased Siberian Sturgeon (Acipenser baerii) Using Oxford Nanopore Sequencing

 , , , , and
, , , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Sampling
2.2. 16S rRNA Gene Sequence Analysis
2.3. Genome Sequencing and Phylogenomic Analysis
2.4. Assessment of Antimicrobial Resistance Gene Detection and Pathogenicity Prediction
3. Results
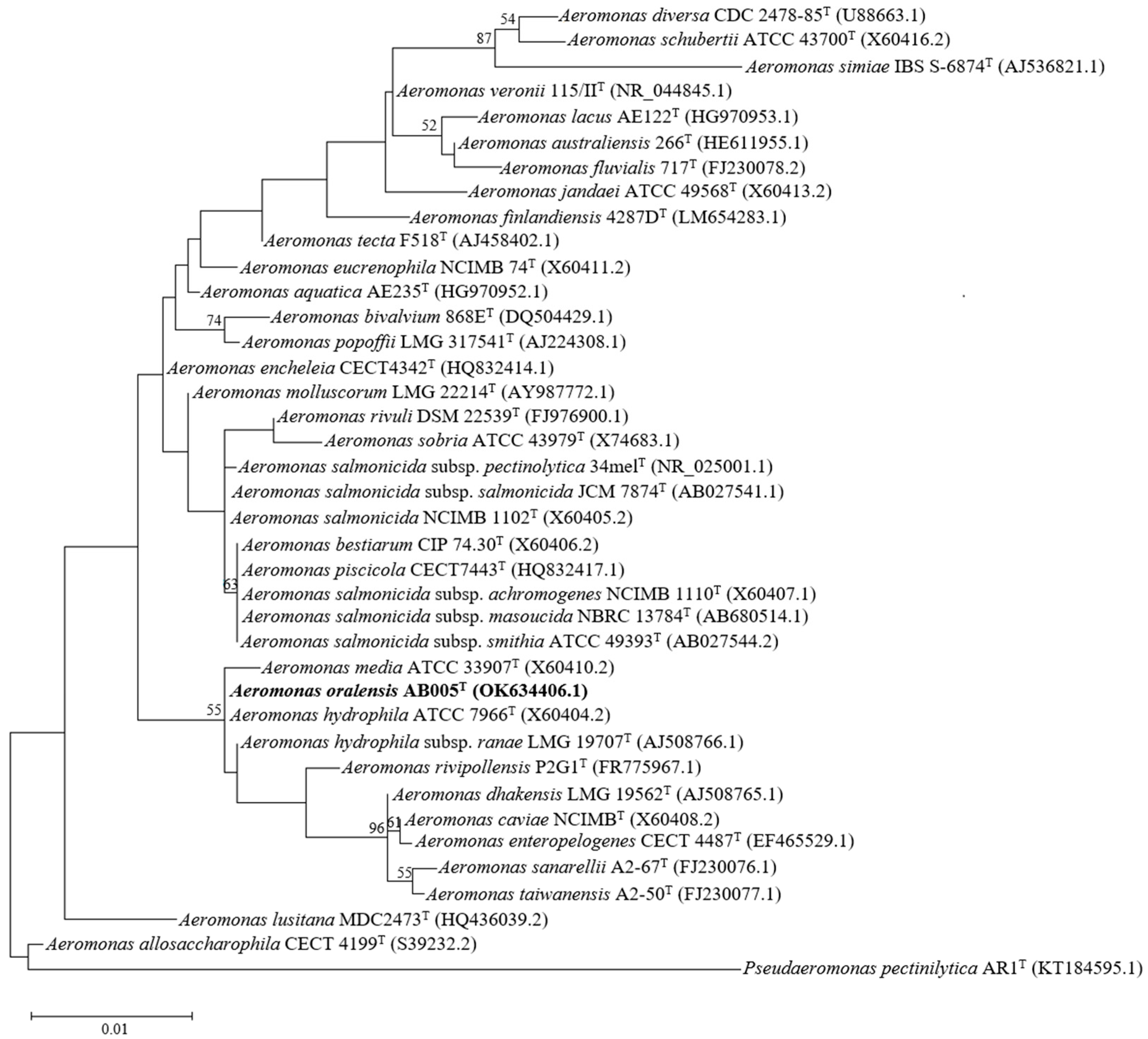
3.1. Phylogenetic Identification
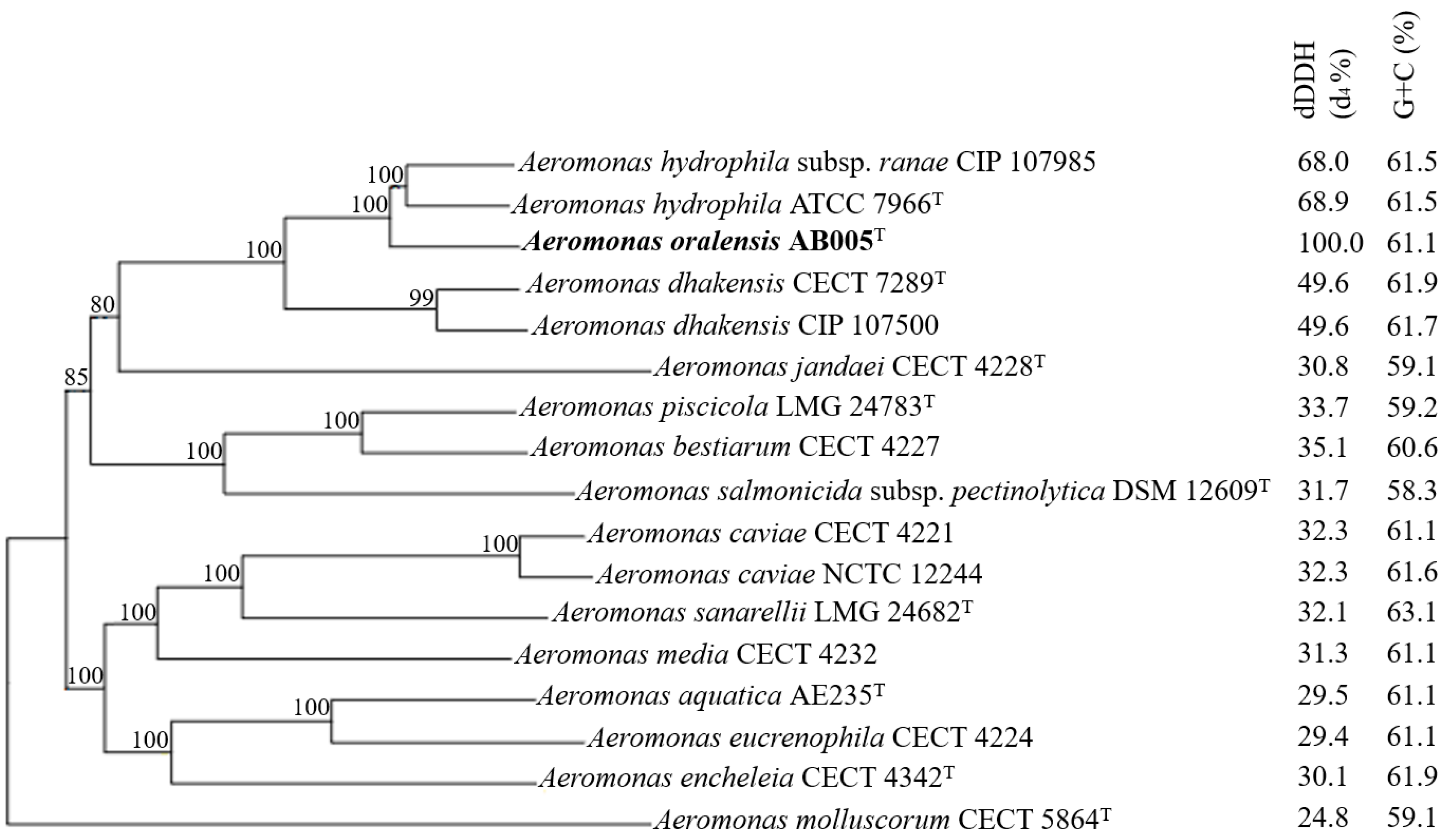
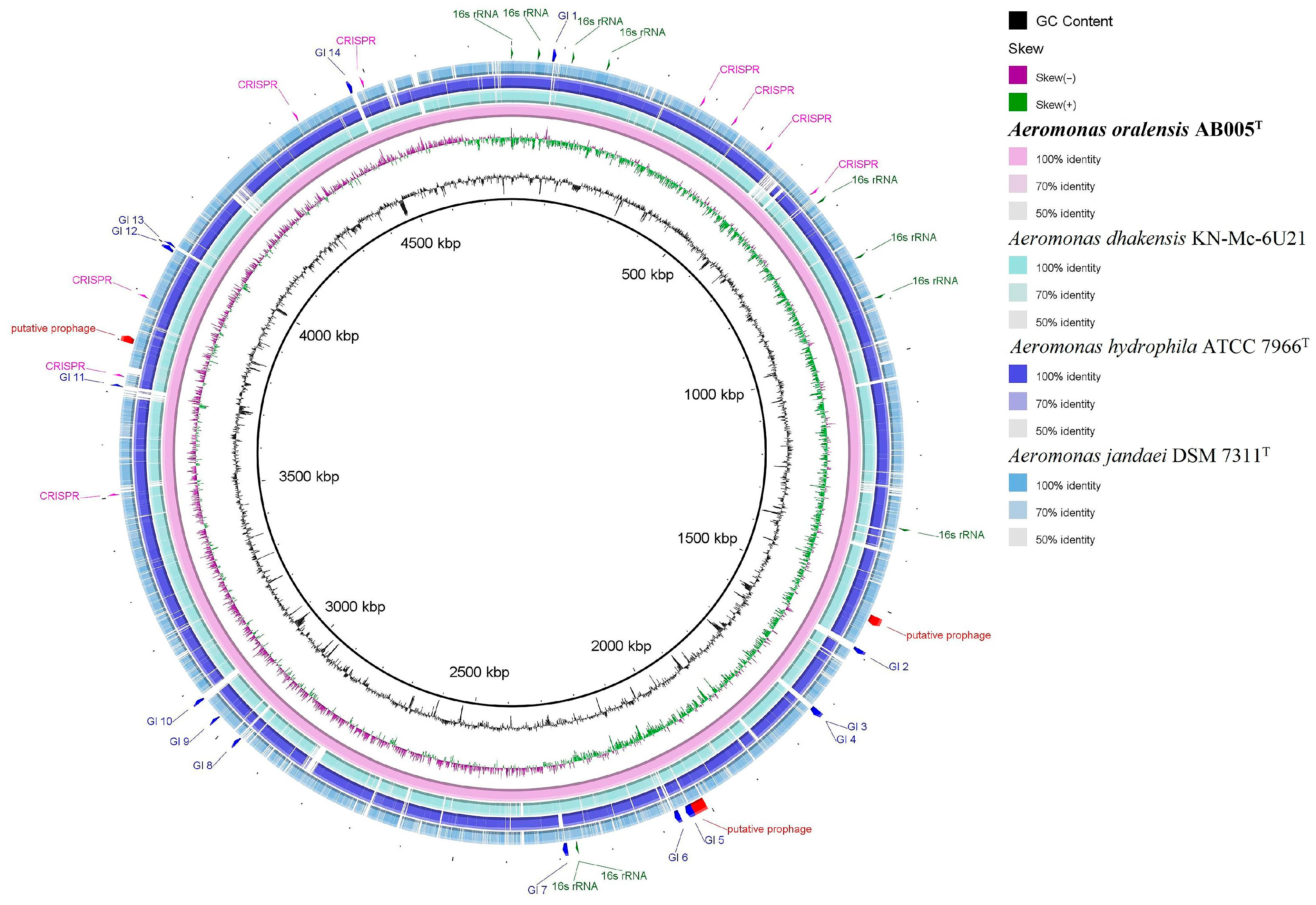
3.2. Genome Characteristics and Phylogenomic Analysis
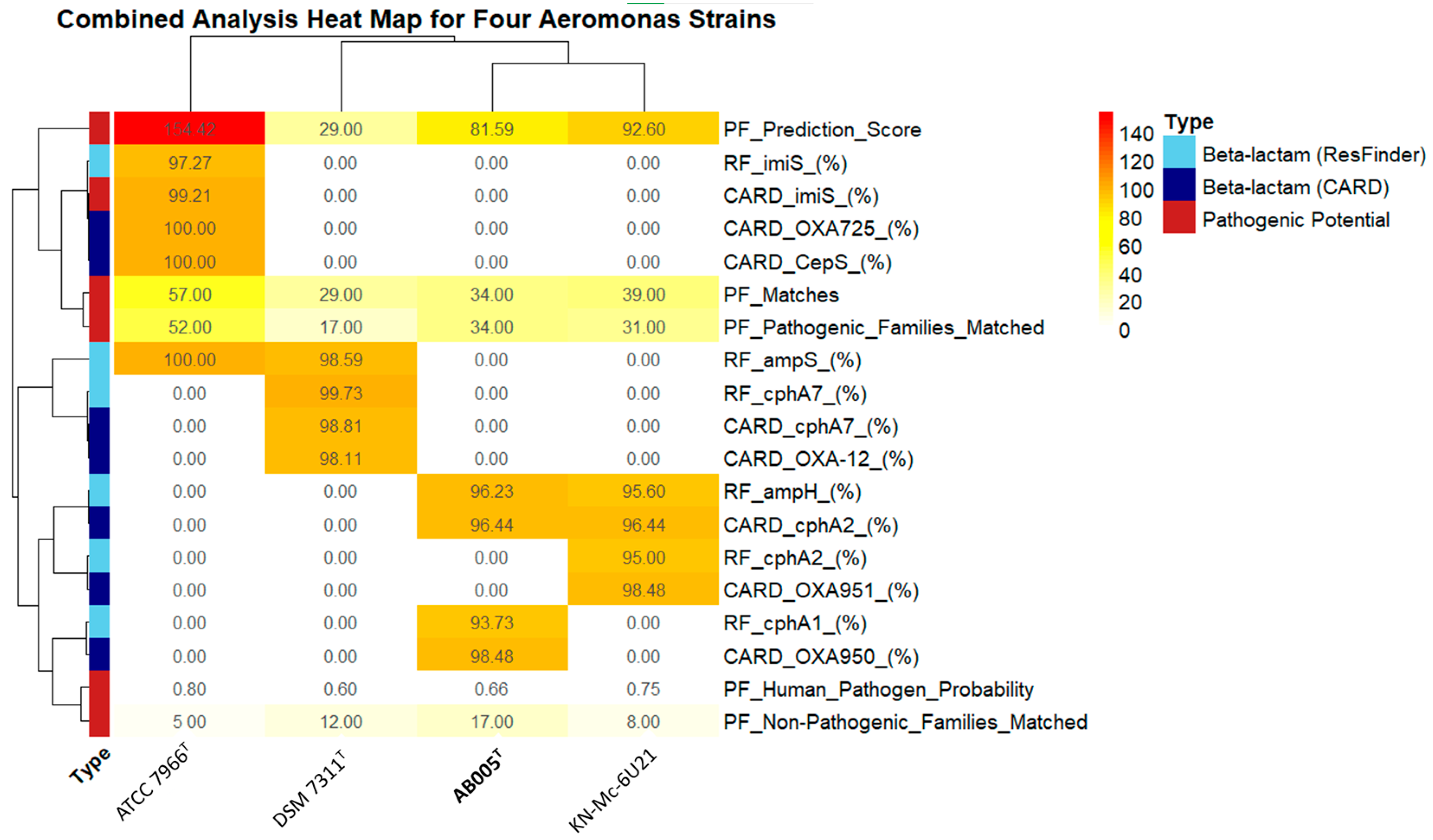
3.3. Antimicrobial Resistance Genes and Pathogenicity
4. Discussion
5. Conclusions
6. Description of Aeromonas oralensis sp. nov.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Parte, A.C.; Sarda Carbasse, J.; Meier-Kolthoff, J.P.; Reimer, L.C.; Göker, M. List of Prokaryotic Names with Standing in Nomenclature (LPSN) Moves to the DSMZ. Int. J. Syst. Evol. Microbiol. 2020, 70, 5607–5612. [Google Scholar] [CrossRef]
- Figueras, M.J.; Latif-Eugenin, F.; Ballester, F.; Pujol, I.; Tena, D.; Berg, K.; Hossain, M.J.; Beaz-Hidalgo, R.; Liles, M.R. Aeromonas intestinalis and Aeromonas enterica Isolated from Human Faeces, ‘Aeromonas crassostreae’ from Oyster and ‘Aeromonas aquatilis’ Isolated from Lake Water Represent Novel Species. New Microbes New Infect. 2017, 15, 74–76. [Google Scholar] [CrossRef] [PubMed]
- Joerg, G. Aeromonas; Caister Academic Press: Poole, UK, 2015; pp. 187–210. [Google Scholar]
- Abu-Elala, N.; Abdelsalam, M.; Marouf, S.; Setta, A. Comparative Analysis of Virulence Genes, Antibiotic Resistance and gyrB-Based Phylogeny of Motile Aeromonas Species Isolates from Nile Tilapia and Domestic Fowl. Lett. Appl. Microbiol. 2015, 61, 429–436. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Yu, L.; Nan, Z.; Zhang, P.; Kan, B.; Yan, D.; Su, J. Taxonomy, virulence genes and antimicrobial resistance of Aeromonas isolated from extra-intestinal and intestinal infections. BMC Infect. Dis. 2019, 19, 158. [Google Scholar] [CrossRef] [PubMed]
- Silva, L.C.A.D.; Leal-Balbino, T.C.; Melo, B.S.T.; Mendes-Marques, C.L.; Rezende, A.M.; Almeida, A.M.P.; Leal, N.C. Genetic diversity and virulence potential of clinical and environmental Aeromonas spp. isolates from a diarrhea outbreak. BMC Microbiol. 2017, 17, 179. [Google Scholar] [CrossRef]
- Igbinosa, I.H.; Igumbor, E.U.; Aghdasi, F.; Tom, M.; Okoh, A.I. Emerging Aeromonas Species Infections and Their Significance in Public Health. Sci. World J. 2012, 2012, 625023. [Google Scholar] [CrossRef]
- Bhowmick, U.D.; Bhattacharjee, S. Bacteriological, Clinical and Virulence Aspects of Aeromonas-Associated Diseases in Humans. Pol. J. Microbiol. 2018, 67, 137–149. [Google Scholar] [CrossRef]
- Wang, D.; Sun, F.; Li, Z.; Hu, Y.; Xu, R. Acute septic arthritis of shoulder joint caused by Aeromonas veronii biotype sobria. Orthopade 2018, 47, 1027–1031. [Google Scholar] [CrossRef]
- Chen, H.; Wang, M.; Lin, X.; Shi, C.; Liu, Z. Bacterial Microbiota Profile in Gills of Modified Atmosphere-Packaged Oysters Stored at 4 °C. Food Microbiol. 2017, 61, 58–65. [Google Scholar] [CrossRef]
- Fernández-Bravo, A.; Figueras, M.J. An Update on the Genus Aeromonas: Taxonomy, Epidemiology, and Pathogenicity. Microorganisms 2020, 8, 129. [Google Scholar] [CrossRef]
- Abbott, S.L.; Cheung, W.K.; Janda, J.M. The Genus Aeromonas: Biochemical Characteristics, Atypical Reactions, and Phenotypic Identification Schemes. J. Clin. Microbiol. 2003, 41, 2348–2357. [Google Scholar] [CrossRef] [PubMed]
- Parker, J.L.; Shaw, J.G. Aeromonas spp. clinical microbiology and disease. J. Infect. 2011, 62, 109–118. [Google Scholar] [CrossRef]
- Martínez-Murcia, A.J.; Saavedra, M.J.; Mota, V.R.; Maier, T.; Stackebrandt, E.; Cousin, S. Aeromonas aquariorum sp. nov., Isolated from Aquaria of Ornamental Fish. Int. J. Syst. Evol. Microbiol. 2008, 58, 1169–1175. [Google Scholar] [CrossRef]
- Figueras, M.J. Clinical Relevance of Aeromonas. Rev. Med. Microbiol. 2005, 16, 145–153. [Google Scholar] [CrossRef]
- Ormen, O.; Granum, P.E.; Lassen, J.; Figueras, M.J. Lack of Agreement between Biochemical and Genetic Identification of Aeromonas spp. APMIS 2005, 113, 203–207. [Google Scholar] [CrossRef] [PubMed]
- Abd El-Ghany, W.A. A review on Aeromoniasis in poultry: A bacterial disease of zoonotic nature. J. Infect. Dev. Ctries 2023, 17, 1–9. [Google Scholar] [CrossRef]
- Saavedra, M.J.; Figueras, M.J.; Martínez-Murcia, A.J. Updated Phylogeny of the Genus Aeromonas. Int. J. Syst. Evol. Microbiol. 2006, 56, 2481–2487. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Murcia, A.J.; Benlloch, S.; Collins, M.D. Phylogenetic Interrelationships of Members of the Genera Aeromonas and Plesiomonas as Determined by 16S Ribosomal DNA Sequencing: Lack of Congruence with Results of DNA-DNA Hybridizations. Int. J. Syst. Bacteriol. 1992, 42, 412–421. [Google Scholar] [CrossRef]
- Yáñez, M.A.; Catalán, V.; Apráiz, D.; Figueras, M.J.; Martínez-Murcia, A.J. Phylogenetic analysis of members of the genus Aeromonas based on gyrB gene sequences. Int. J. Syst. Evol. Microbiol. 2003, 53 Pt 3, 875–883. [Google Scholar] [CrossRef]
- Soler, L.; Yáñez, M.A.; Chacon, M.R.; Aguilera-Arreola, M.G.; Catalán, V.; Figueras, M.J.; Martínez-Murcia, A.J. Phylogenetic analysis of the genus Aeromonas based on two housekeeping genes. Int. J. Syst. Evol. Microbiol. 2004, 54 Pt 5, 1511–1519. [Google Scholar] [CrossRef]
- Nhung, P.H.; Hata, H.; Ohkusu, K.; Noda, M.; Shah, M.M.; Goto, K.; Ezaki, T. Use of the Novel Phylogenetic Marker dnaJ and DNA-DNA Hybridization to Clarify Interrelationships within the Genus Aeromonas. Int. J. Syst. Evol. Microbiol. 2007, 57, 1232–1237. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Storesund, J.E.; Lunestad, B.T.; Hoel, S.; Lerfall, J.; Jakobsen, A.N. Whole genome sequence analysis of Aeromonas spp. isolated from ready-to-eat seafood: Antimicrobial resistance and virulence factors. Front. Microbiol. 2023, 30, 1175304. [Google Scholar] [CrossRef] [PubMed]
- Bakiyev, S.; Smekenov, I.; Zharkova, I.; Kobegenova, S.; Sergaliyev, N.; Battistuzzi, F.U. Isolation, Identification, and Characterization of Pathogenic Aeromonas hydrophila from Critically Endangered Acipenser baerii. Aquac. Rep. 2022, 26, 101311. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef] [PubMed]
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef]
- Thompson, J.D.; Gibson, T.J.; Plewniak, F.; Jeanmougin, F.; Higgins, D.G. The CLUSTAL_X Windows Interface: Flexible Strategies for Multiple Sequence Alignment Aided by Quality Analysis Tools. Nucleic Acids Res. 1997, 25, 4876–4882. [Google Scholar] [CrossRef]
- Tamura, K.; Nei, M. Estimation of the Number of Nucleotide Substitutions in the Control Region of Mitochondrial DNA in Humans and Chimpanzees. Mol. Biol. Evol. 1993, 10, 512–526. [Google Scholar]
- Felsenstein, J. Confidence Limits on Phylogenies: An Approach Using the Bootstrap. Evolution 1985, 39, 783–791. [Google Scholar] [CrossRef]
- Wick, R.R.; Judd, L.M.; Holt, K.E. Performance of Neural Network Basecalling Tools for Oxford Nanopore Sequencing. Genome Biol. 2019, 20, 129. [Google Scholar] [CrossRef]
- Kolmogorov, M.; Yuan, J.; Lin, Y.; Pevzner, P.A. Assembly of Long, Error-Prone Reads Using Repeat Graphs. Nat. Biotechnol. 2019, 37, 540–546. [Google Scholar] [CrossRef]
- Overbeek, R.; Olson, R.; Pusch, G.D.; Olsen, G.J.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Parrello, B.; Shukla, M.; et al. The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucleic. Acids. Res. 2014, 42, D206–D214. [Google Scholar] [CrossRef] [PubMed]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the Quality of Microbial Genomes Recovered from Isolates, Single Cells, and Metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Carbasse, J.S.; Peinado-Olarte, R.L.; Göker, M. TYGS and LPSN: A Database Tandem for Fast and Reliable Genome-Based Classification and Nomenclature of Prokaryotes. Nucleic Acids Res. 2022, 50, D801–D807. [Google Scholar] [CrossRef]
- Yoon, S.-H.; Ha, S.-M.; Lim, J.; Kwon, S.; Chun, J. A Large-Scale Evaluation of Algorithms to Calculate Average Nucleotide Identity. Antonie Leeuwenhoek 2017, 110, 1281–1286. [Google Scholar] [CrossRef] [PubMed]
- Alikhan, N.F.; Petty, N.K.; Ben Zakour, N.L.; Beatson, S.A. BLAST Ring Image Generator (BRIG): Simple Prokaryote Genome Comparisons. BMC Genomics. 2011, 12, 402. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Lu, F.; Luo, Y.; Bie, L.; Xu, L.; Wang, Y. OrthoVenn3: An Integrated Platform for Exploring and Visualizing Orthologous Data across Genomes. Nucleic Acids Res. 2023, 51, W397–W403. [Google Scholar] [CrossRef]
- Rouli, L.; Merhej, V.; Fournier, P.-E.; Raoult, D. The Bacterial Pangenome as a New Tool for Analysing Pathogenic Bacteria. New Microbes New Infect. 2015, 7, 72–85. [Google Scholar] [CrossRef]
- Bortolaia, V.; Kaas, R.S.; Ruppe, E.; Roberts, M.C.; Schwarz, S.; Cattoir, V.; Philippon, A.; Allesoe, R.L.; Rebelo, A.R.; Ferrer Florensa, A.; et al. ResFinder 4.0 for Predictions of Phenotypes from Genotypes. J. Antimicrob. Chemother. 2020, 75, 3491–3500. [Google Scholar] [CrossRef]
- Clausen, P.T.L.C.; Aarestrup, F.M.; Lund, O. Rapid and Precise Alignment of Raw Reads against Redundant Databases with KMA. BMC Bioinformatics 2018, 19, 307. [Google Scholar] [CrossRef]
- Alcock, B.P.; Huynh, W.; Chalil, R.; Smith, K.W.; Raphenya, A.R.; Wlodarski, M.; Edalatmand, A.; Petkau, A.; A Syed, S.; Tsang, K.K.; et al. CARD 2023: Expanded Curation, Support for Machine Learning, and Resistome Prediction at the Comprehensive Antibiotic Resistance Database. Nucleic Acids Res. 2023, 51, D690–D699. [Google Scholar] [CrossRef]
- Barbour, J.D.; Cosentino, S.; Voldby Larsen, M.; Møller Aarestrup, F.; Lund, O. PathogenFinder—Distinguishing Friend from Foe Using Bacterial Whole Genome Sequence Data. PLoS ONE 2013, 8, e77302. [Google Scholar]
- Benson, D.A.; Cavanaugh, M.; Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2017, 45, D37–D42. [Google Scholar] [CrossRef] [PubMed]
- Lefort, V.; Desper, R.; Gascuel, O. FastME 2.0: A Comprehensive, Accurate, and Fast Distance-Based Phylogeny Inference Program. Mol. Biol. Evol. 2015, 32, 2798–2800. [Google Scholar] [CrossRef]
- Farris, J.S. Estimating Phylogenetic Trees from Distance Matrices. Am. Nat. 1972, 106, 645–668. [Google Scholar] [CrossRef]
- Kreft, Ł.; Botzki, A.; Coppens, F.; Vandepoele, K.; Van Bel, M. PhyD3: A Phylogenetic Tree Viewer with Extended PhyloXML Support for Functional Genomics Data Visualization. Bioinformatics 2017, 33, 2946–2947. [Google Scholar] [CrossRef]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A Better, Faster Version of the PHAST Phage Search Tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef]
- Couvin, D.; Bernheim, A.; Toffano-Nioche, C.; Touchon, M.; Michalik, J.; Néron, B.; Rocha, E.P.C.; Vergnaud, G.; Gautheret, D.; Pourcel, C. CRISPRCasFinder, an Update of CRISPRFinder, Includes a Portable Version, Enhanced Performance, and Integrates Search for Cas Proteins. Nucleic Acids Res. 2018, 46, W246–W251. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Scientific Name | Aeromonas oralensis AB005T | Aeromonas hydrophila ATCC 7966T | Aeromonas dhakensis KN-Mc-6U21 | Aeromonas jandaei DSM 7311T |
|---|---|---|---|---|
| Genome size (Mb) | 4.7 | 4.7 | 4.9 | 4.6 |
| G + C content (%) | 61.2 | 61.5 | 61.5 | 59 |
| N50 (Mb) | 4.7 | 4.7 | 4.9 | 4.6 |
| L50 | 1 | 1 | 1 | 1 |
| Number of contigs | 1 | 1 | 1 | 1 |
| Number of coding sequences | 4607 | 4161 | 4316 | 3967 |
| Number of tRNAs | 128 | 126 | 126 | 124 |
| 5S rRNA | 11 | 11 | 11 | 11 |
| 16S rRNA | 10 | 10 | 10 | 10 |
| 23S rRNA | 10 | 10 | 10 | 10 |
| NCBI Accession no. | CP187186.2 | CP000462.1 | CP023141.1 | CP149571.1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mashzhan, A.; Smekenov, I.; Bakiyev, S.; Utegenova, K.; Samatkyzy, D.; Daniyarov, A.; Kairov, U.; Sarbassov, D.; Bissenbaev, A. Whole-Genome Sequencing of a Potentially Novel Aeromonas Species Isolated from Diseased Siberian Sturgeon (Acipenser baerii) Using Oxford Nanopore Sequencing. Microorganisms 2025, 13, 1680. https://doi.org/10.3390/microorganisms13071680
Mashzhan A, Smekenov I, Bakiyev S, Utegenova K, Samatkyzy D, Daniyarov A, Kairov U, Sarbassov D, Bissenbaev A. Whole-Genome Sequencing of a Potentially Novel Aeromonas Species Isolated from Diseased Siberian Sturgeon (Acipenser baerii) Using Oxford Nanopore Sequencing. Microorganisms. 2025; 13(7):1680. https://doi.org/10.3390/microorganisms13071680
Chicago/Turabian StyleMashzhan, Akzhigit, Izat Smekenov, Serik Bakiyev, Kalamkas Utegenova, Diana Samatkyzy, Asset Daniyarov, Ulykbek Kairov, Dos Sarbassov, and Amangeldy Bissenbaev. 2025. "Whole-Genome Sequencing of a Potentially Novel Aeromonas Species Isolated from Diseased Siberian Sturgeon (Acipenser baerii) Using Oxford Nanopore Sequencing" Microorganisms 13, no. 7: 1680. https://doi.org/10.3390/microorganisms13071680
APA StyleMashzhan, A., Smekenov, I., Bakiyev, S., Utegenova, K., Samatkyzy, D., Daniyarov, A., Kairov, U., Sarbassov, D., & Bissenbaev, A. (2025). Whole-Genome Sequencing of a Potentially Novel Aeromonas Species Isolated from Diseased Siberian Sturgeon (Acipenser baerii) Using Oxford Nanopore Sequencing. Microorganisms, 13(7), 1680. https://doi.org/10.3390/microorganisms13071680