
Hybrid Whole Genomes of Brucella melitensis from Tunisian Animal Isolates: Virulence Factors, Antimicrobial Susceptibility, and Phylogeny

 , , , ,
, , , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Origin of Brucella Strains
2.2. Isolation and Identification of Brucella Strains
2.3. Antibiotic Susceptibility Testing (AST)
2.4. DNA Extraction, Library Preparation, and Sequencing
2.5. Whole-Genome Sequencing and Bioinformatic Analyses
3. Results
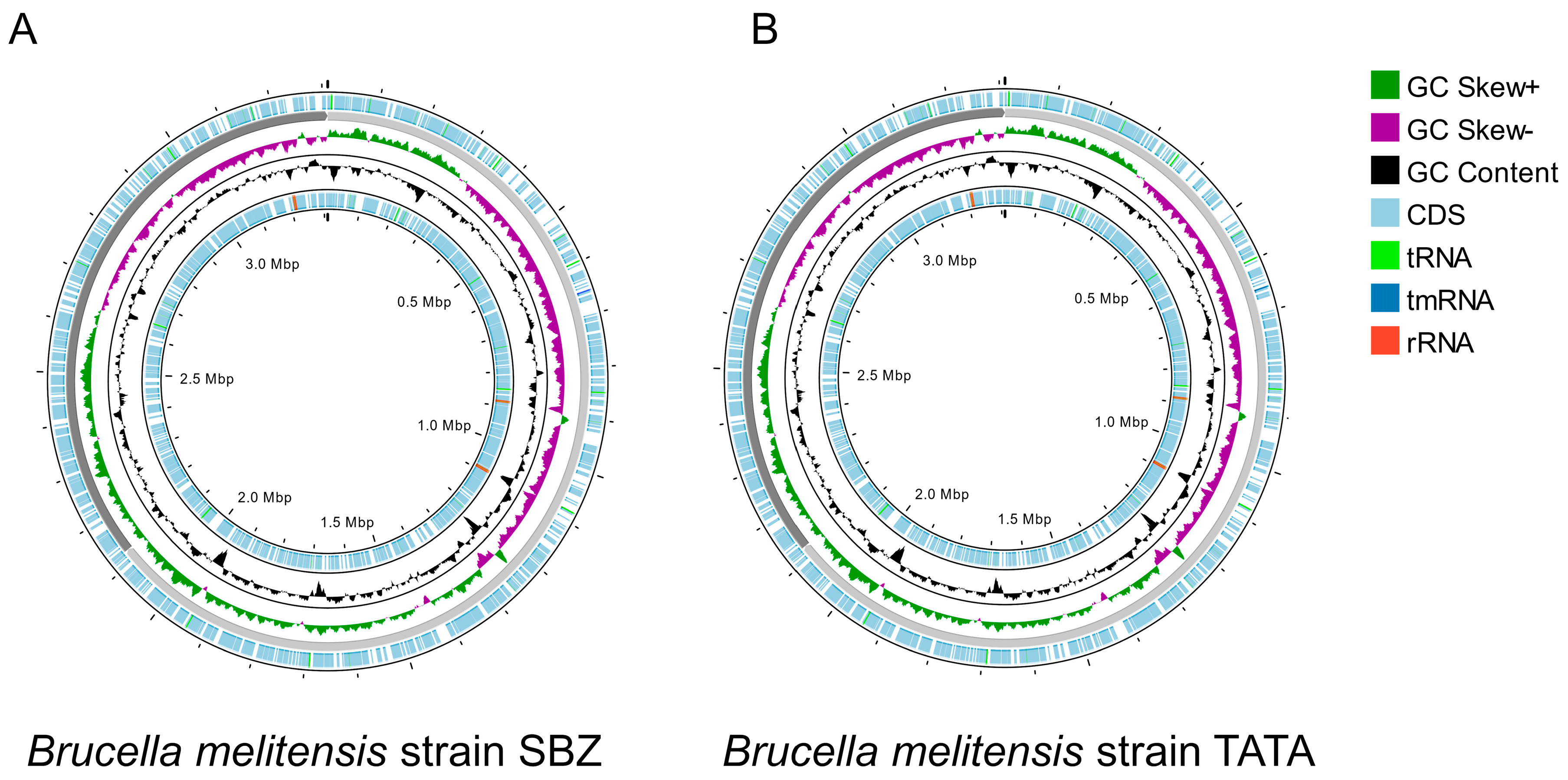
3.1. Brucella Growth, Identification, and Characterization
3.2. Antibiotic Susceptibility Testing (AST)
3.3. Identification of AMR and Virulence-Associated Genes
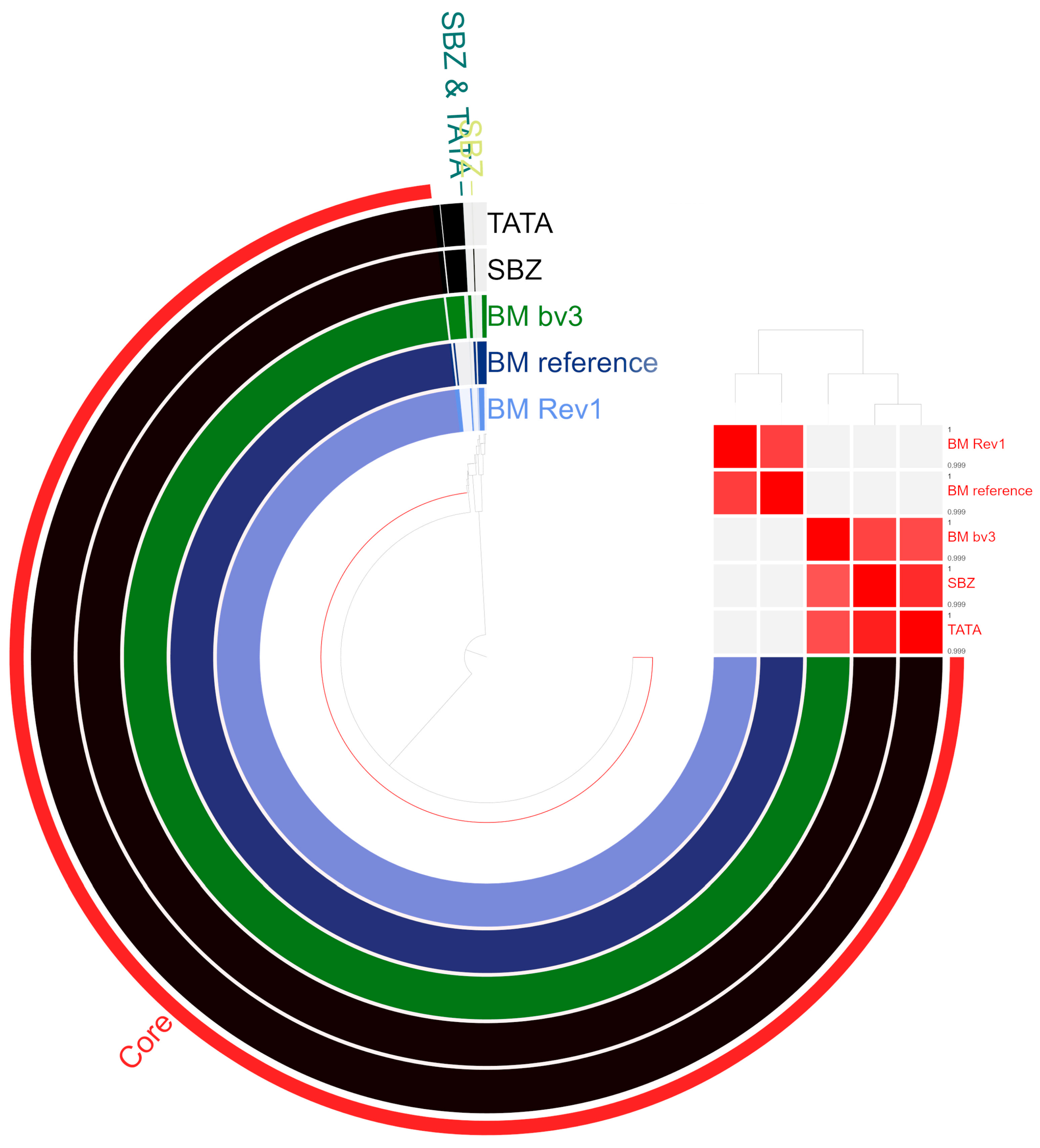
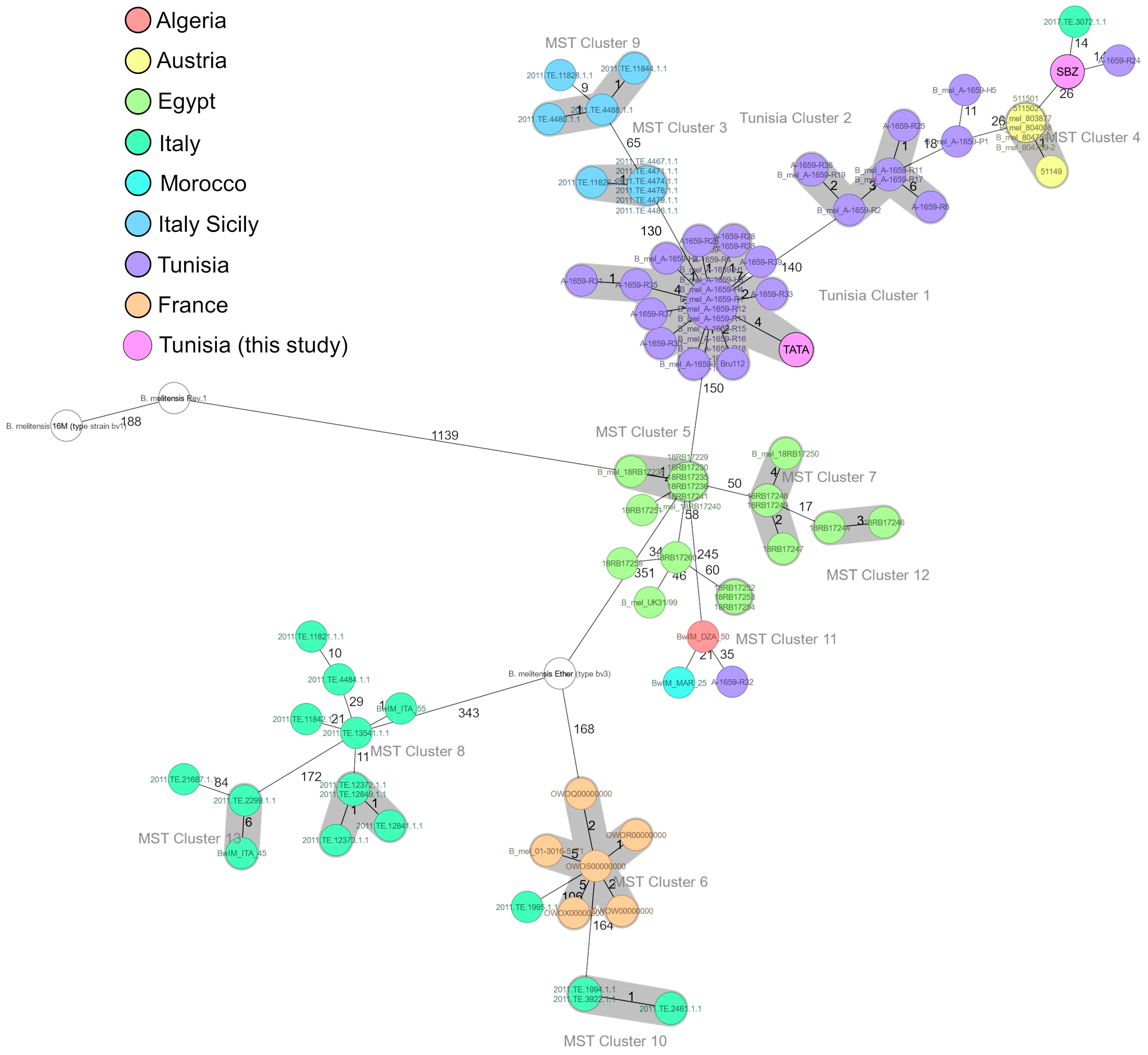
3.4. Core-Genome-Based MLST (cgMLST)
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Daugaliyeva, A.; Daugaliyeva, S.; Kydyr, N.; Peletto, S. Molecular typing methods to Characterize Brucella spp. from animals: A Review. Vet. World 2024, 17, 1778–1788. [Google Scholar] [CrossRef] [PubMed]
- Béjaoui, A.; Ben Abdallah, I.; Maaroufi, A. Brucella spp. Contamination in Artisanal Unpasteurized Dairy Products: An Emerging Foodborne Threat in Tunisia. Foods 2022, 11, 2269. [Google Scholar] [CrossRef]
- Lokamar, P.N.; Kutwah, M.A.; Atieli, H.; Gumo, S.; Ouma, C. Socio-economic impacts of brucellosis on livestock production and reproduction performance in Koibatek and Marigat regions, Baringo County, Kenya. BMC Vet. Res. 2020, 16, 61. [Google Scholar] [CrossRef]
- Shekhar, C. Impact of brucellosis on health and economy. Vet. Sci. Res. J. 2018, 9, 37–44. [Google Scholar] [CrossRef]
- Ibarra, M.; Campos, M.; Ibarra, C.; Gladys, U.; Huera, D.; Gutiérrez, M.; Chamorro, A.; Núñez, L. Financial Losses Associated with Bovine Brucellosis (Brucella abortus) in Carchi-Ecuador. Open J. Anim. Sci. 2023, 13, 205–216. [Google Scholar] [CrossRef]
- González-Espinoza, G.; Arce-Gorvel, V.; Mémet, S.; Gorvel, J.-P. Brucella: Reservoirs and Niches in Animals and Humans. Pathogens 2021, 10, 186. [Google Scholar] [CrossRef]
- Aragón-Aranda, B.; De Miguel, M.J.; Lázaro-Antón, L.; Salvador-Bescós, M.; Zúñiga-Ripa, A.; Moriyón, I.; Iriarte, M.; Muñoz, P.M.; Conde-Álvarez, R. Development of attenuated live vaccine candidates against swine brucellosis in a non-Zoonotic B. Suis Biovar 2 background. Vet. Res. 2020, 51, 92. [Google Scholar] [CrossRef]
- Sadaqat, M.H. pathogenesis, clinical manifestations, treatment and prevention of brucellosis in humans and animals. Afghan. J. Basic. Med. Sci. 2023, 1, 41–46. [Google Scholar] [CrossRef]
- Lindahl-Rajala, E.; Hoffman, T.; Fretin, D.; Godfroid, J.; Sattorov, N.; Boqvist, S.; Lundkvist, Å.; Magnusson, U. Detection and characterization of Brucella spp. in bovine milk in small-scale urban and peri-urban farming in Tajikistan. PLoS Negl. Trop. Dis. 2017, 11, e0005367. [Google Scholar] [CrossRef]
- Corbel, M.J.; Food and Agriculture Organization of the United Nations; World Health Organization. Brucellosis in Humans and Animals; World Health Organization: Geneva, Switzerland, 2006. Available online: https://iris.who.int/handle/10665/43597 (accessed on 20 December 2024).
- Rossetti, C.A.; Maurizio, E.; Rossi, U.A. Comparative Review of Brucellosis in Small Domestic Ruminants. Front. Vet. Sci. 2022, 9, 887671. [Google Scholar] [CrossRef]
- Azam, S.; Rao, S.B.; Jakka, P.; NarasimhaRao, V.; Bhargavi, B.; Gupta, V.K.; Radhakrishnan, G. Genetic Characterization and Comparative Genome Analysis of Brucella melitensis Isolates from India. Int. J. Genom. 2016, 2016, e3034756. [Google Scholar] [CrossRef]
- Hayoun, M.A.; Muco, E.; Shorman, M. Brucellosis. In StatPearls; StatPearls Publishing: St. Petersburg, FL, USA, 2024. Available online: http://www.ncbi.nlm.nih.gov/books/NBK441831/ (accessed on 12 January 2025).
- Barkallah, M.; Gharbi, Y.; Zormati, S.; Karkouch, N.; Mallek, Z.; Gautier, M.; Gdoura, R.; Fendri, I. A Mixed methods study of ruminant Brucellosis in central-eastern Tunisia. Trop. Anim. Health Prod. 2017, 49, 39–45. [Google Scholar] [CrossRef]
- Guesmi, K.; Kalthoum, S.; Mamlouk, A.; Baccar, M.N.; BelHajMohamed, B.; Hajlaoui, H.; Toumi, A.; Cherni, J.; Seghaier, C.; Messadi, L. Seroprevalence of zoonotic abortive diseases and their associated risk factors in Tunisian sheep. BMC Vet. Res. 2023, 19, 50. [Google Scholar] [CrossRef]
- Selmi, R.; Mamlouk, A.; Belkahia, H.; Ben Yahia, H.; Abdelaali, H.; Jemli, M.-H.; Ben Said, M.; Messadi, L. Serological and molecular survey of brucellosis and chlamydiosis in dromedary Camels from Tunisia. Comp. Immunol. Microbiol. Infect. Dis. 2024, 104, 102098. [Google Scholar] [CrossRef] [PubMed]
- Battikh, H.; Berriche, A.; Zayoud, R.; Ammari, L.; Abdelmalek, R.; Kilani, B.; Tiouiri Ben Aissa, H.; Zribi, M. Clinical and laboratory features of brucellosis in a university hospital in Tunisia. Infect. Dis. Now. 2021, 51, 547–551. [Google Scholar] [CrossRef]
- Khamassi Khbou, M.; Htira, S.; Harabech, K.; Benzarti, M. First case-control study of zoonotic brucellosis in Gafsa district, Southwest Tunisia. One Health 2018, 5, 21–26. [Google Scholar] [CrossRef]
- Guesmi, K.; Kalthoum, S.; Belhaj Mohamed, B.; Ben Aicha, I.; Hajlaoui, H.; Hrabech, K. Bilan de la brucellose animale et humaine en Tunisie: 2005–2018. Bull. Zoosanit. 2020, 20, 1–11. [Google Scholar]
- Eleiwa, A.; Nadal, J.; Vilaprinyo, E.; Marin-Sanguino, A.; Sorribas, A.; Basallo, O.; Lucido, A.; Richart, C.; Pena, R.N.; Ros-Freixedes, R.; et al. Hybrid assembly and comparative genomics unveil insights into the evolution and biology of the red-legged partridge. Sci. Rep. 2024, 14, 19531. [Google Scholar] [CrossRef]
- Ruan, Z.; Wu, J.; Chen, H.; Draz, M.S.; Xu, J.; He, F. Hybrid Genome Assembly and Annotation of a Pandrug-Resistant Klebsiella Pneumoniae Strain Using Nanopore and Illumina Sequencing. Infect. Drug Resist. 2020, 13, 199–206. [Google Scholar] [CrossRef]
- Pfeiffer, F.; Gröber, C.; Blank, M.; Händler, K.; Beyer, M.; Schultze, J.L.; Mayer, G. Systematic evaluation of error rates and causes in short samples in next-generation sequencing. Sci. Rep. 2018, 8, 10950. [Google Scholar] [CrossRef]
- Ma, X.; Shao, Y.; Tian, L.; Flasch, D.A.; Mulder, H.L.; Edmonson, M.N.; Liu, Y.; Chen, X.; Newman, S.; Nakitandwe, J.; et al. Analysis of error profiles in deep next-generation sequencing data. Genome Biol. 2019, 20, 50. [Google Scholar] [CrossRef]
- Magi, A.; Semeraro, R.; Mingrino, A.; Giusti, B.; D’Aurizio, R. Nanopore sequencing data analysis: State of the art, applications and challenges. Brief. Bioinform. 2017, 19, 1256–1272. [Google Scholar] [CrossRef] [PubMed]
- Weissensteiner, M.H.; Pang, A.W.C.; Bunikis, I.; Höijer, I.; Vinnere-Petterson, O.; Suh, A.; Wolf, J.B.W. Combination of short-read, long-read, and optical mapping assemblies reveals large-scale tandem repeat arrays with population genetic implications. Genome Res. 2017, 27, 697–708. [Google Scholar] [CrossRef]
- Probert, W.S.; Schrader, K.N.; Khuong, N.Y.; Bystrom, S.L.; Graves, M.H. Real-Time Multiplex PCR Assay for Detection of Brucella spp., B. abortus, and B. melitensis. J. Clin. Microbiol. 2004, 42, 1290–1293. [Google Scholar] [CrossRef] [PubMed]
- Tscherne, A.; Mantel, E.; Boskani, T.; Budniak, S.; Elschner, M.; Fasanella, A.; Feruglio, S.L.; Galante, D.; Giske, C.G.; Grunow, R.; et al. Adaptation of Brucella melitensis Antimicrobial Susceptibility Testing to the ISO 20776 Standard and Validation of the Method. Microorganisms 2022, 10, 1470. [Google Scholar] [CrossRef]
- Clinical and Laboratory Standards Institute. Methods for Antimicrobial Dilution and Disk Susceptibility Testing of Infrequently Isolated or Fastidious Bacteria, M45, 3rd ed.; CLSI Guideline M45-A2; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2016. [Google Scholar]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 25 May 2024).
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- De Coster, W.; Rademakers, R. NanoPack2: Population-scale evaluation of long-read sequencing data. Bioinformatics 2023, 39, btad311. [Google Scholar] [CrossRef]
- Wick, R.R. Filtlong: Quality filtering Tool for Long Reads. 2018. Available online: https://github.com/rrwick/Filtlong (accessed on 20 December 2024).
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 2017, 13, e1005595. [Google Scholar] [CrossRef]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef]
- Wick, R.R.; Schultz, M.B.; Zobel, J.; Holt, K.E. Bandage: Interactive visualization of de Novo genome assemblies. Bioinformatics 2015, 31, 3350–3352. [Google Scholar] [CrossRef] [PubMed]
- Wood, D.E.; Lu, J.; Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef]
- Ondov, B.D.; Bergman, N.H.; Phillippy, A.M. Interactive metagenomic visualization in a web browser. BMC Bioinform. 2011, 12, 385. [Google Scholar] [CrossRef]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST Server: Rapid annotations using subsystems technology. BMC Genom. 2008, 9, 75. [Google Scholar] [CrossRef] [PubMed]
- Grant, J.R.; Enns, E.; Marinier, E.; Mandal, A.; Herman, E.K.; Chen, C.; Graham, M.; Van Domselaar, G.; Stothard, P. Proksee: In-depth characterization and visualization of bacterial genomes. Nucleic Acids Res. 2023, 51, W484–W492. [Google Scholar] [CrossRef]
- Eren, A.M.; Kiefl, E.; Shaiber, A.; Veseli, I.; Miller, S.E.; Schechter, M.S.; Fink, I.; Pan, J.N.; Yousef, M.; Fogarty, E.C.; et al. Community-led, integrated, reproducible multi-omics with Anvi’o. Nat. Microbiol. 2020, 6, 3–6. [Google Scholar] [CrossRef]
- Jolley, K.A.; Bray, J.E.; Maiden, M.C.J. Open-access bacterial population genomics: BIGSdb software, the PubMLST.org website and their applications. Wellcome Open Res. 2018, 3, 124. [Google Scholar] [CrossRef]
- Feldgarden, M.; Brover, V.; Gonzalez-Escalona, N.; Frye, J.G.; Haendiges, J.; Haft, D.H.; Hoffmann, M.; Pettengill, J.B.; Prasad, A.B.; Tillman, G.E.; et al. AMRFinderPlus and the Reference Gene Catalog facilitate examination of the genomic links among antimicrobial resistance, stress response, and virulence. Sci. Rep. 2021, 11, 12728. [Google Scholar] [CrossRef]
- Jia, B.; Raphenya, A.R.; Alcock, B.; Waglechner, N.; Guo, P.; Tsang, K.K.; Lago, B.A.; Dave, B.M.; Pereira, S.; Sharma, A.N.; et al. CARD 2017: Expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res. 2017, 45, D566–D573. [Google Scholar] [CrossRef] [PubMed]
- Doster, E.; Lakin, S.M.; Dean, C.J.; Wolfe, C.; Young, J.G.; Boucher, C.; Belk, K.E.; Noyes, N.R.; Morley, P.S. MEGARes 2.0: A database for classification of antimicrobial drug, biocide and metal resistance determinants in metagenomic sequence data. Nucleic Acids Res. 2020, 48, D561–D569. [Google Scholar] [CrossRef]
- Bortolaia, V.; Kaas, R.S.; Ruppe, E.; Roberts, M.C.; Schwarz, S.; Cattoir, V.; Philippon, A.; Allesoe, R.L.; Rebelo, A.R.; Florensa, A.F.; et al. ResFinder 4.0 for predictions of phenotypes from genotypes. J. Antimicrob. Chemother. 2020, 75, 3491–3500. [Google Scholar] [CrossRef]
- Liu, B.; Zheng, D.; Zhou, S.; Chen, L.; Yang, J. VFDB 2022: A general classification scheme for bacterial virulence factors. Nucleic Acids Res. 2022, 50, D912–D917. [Google Scholar] [CrossRef] [PubMed]
- Schwengers, O.; Jelonek, L.; Dieckmann, M.A.; Beyvers, S.; Blom, J.; Goesmann, A. Bakta: Rapid and standardized annotation of bacterial genomes via alignment-free sequence identification. Microb. Genom. 2021, 7, 000685. [Google Scholar] [CrossRef]
- Rabinowitz, P.; Zilberman, B.; Motro, Y.; Roberts, M.C.; Greninger, A.; Nesher, L.; Ben-Shimol, S.; Yagel, Y.; Gdalevich, M.; Sagi, O.; et al. Whole Genome Sequence Analysis of Brucella melitensis Phylogeny and Virulence Factors. Microbiol. Res. 2021, 12, 698–710. [Google Scholar] [CrossRef]
- Janowicz, A.; De Massis, F.; Ancora, M.; Cammà, C.; Patavino, C.; Battisti, A.; Prior, K.; Harmsen, D.; Scholz, H.; Zilli, K.; et al. Core Genome Multilocus Sequence Typing and Single Nucleotide Polymorphism Analysis in the Epidemiology of Brucella melitensis Infections. J. Clin. Microbiol. 2018, 56, e00517-18. [Google Scholar] [CrossRef]
- Etemadi, A.; Moniri, R.; Neubauer, H.; Dasteh Goli, Y.; Alamian, S. Laboratory Diagnostic Procedures for Human Brucellosis: An Overview of Existing Approaches. Jundishapur J. Microbiol. 2019, 12, e91200. [Google Scholar] [CrossRef]
- Dadar, M.; Alamian, S.; Brangsch, H.; Elbadawy, M.; Elkharsawi, A.R.; Neubauer, H.; Wareth, G. Determination of Virulence-Associated Genes and Antimicrobial Resistance Profiles in Brucella Isolates Recovered from Humans and Animals in Iran Using NGS Technology. Pathogens 2023, 12, 82. [Google Scholar] [CrossRef]
- Wareth, G.; El-Diasty, M.; Abdel-Hamid, N.H.; Holzer, K.; Hamdy, M.E.R.; Moustafa, S.; Shahein, M.A.; Melzer, F.; Beyer, W.; Pletz, M.W.; et al. Molecular characterization and antimicrobial susceptibility testing of clinical and non-clinical Brucella melitensis and Brucella abortus isolates from Egypt. One Health 2021, 13, 100255. [Google Scholar] [CrossRef]
- Liang, P.-F.; Zhao, Y.; Zhao, J.-H.; Pan, D.-F.; Guo, Z.-Q. Human distribution and spatial-temporal clustering analysis of human brucellosis in China from 2012 to 2016. Infect. Dis. Poverty 2020, 9, 142. [Google Scholar] [CrossRef]
- Rauthan, K.; Goel, D.; Kumar, S. Annotation of a hypothetical protein (WP_002969292.1) from Brucella abortus. Bioinformation 2019, 15, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Long, G.S.; Hider, J.; Duggan, A.T.; Klunk, J.; Eaton, K.; Karpinski, E.; Giuffra, V.; Ventura, L.; Prowse, T.L.; Fornaciari, A.; et al. A 14th century CE Brucella melitensis genome and the recent expansion of the Western Mediterranean Clade. PLOS Pathog. 2023, 19, e1011538. [Google Scholar] [CrossRef] [PubMed]
- Johansen, T.B.; Scheffer, L.; Jensen, V.K.; Bohlin, J.; Feruglio, S.L. Whole-genome sequencing and antimicrobial resistance in Brucella melitensis from a Norwegian perspective. Sci. Rep. 2018, 8, 8538. [Google Scholar] [CrossRef] [PubMed]
- Dal, T.; Durmaz, R.; Ceylan, A.; Bacalan, F.; Karagoz, A.; Celebi, B.; Yasar, E.; Kilic, S.; Acikgoz, C. Molecular Investigation of the Transmission Dynamics of Brucellosis Observed Among Children in the Province of South—East Anatolia, Turkey. Jundishapur J. Microbiol. 2018, 11, e58857. [Google Scholar] [CrossRef]
- Van, T.T.H.; Yidana, Z.; Smooker, P.M.; Coloe, P.J. Antibiotic use in food animals worldwide, with a focus on Africa: Pluses and minuses. J. Glob. Antimicrob. Resist. 2020, 20, 170–177. [Google Scholar] [CrossRef]
- Abdel-Hamid, N.H.; Zaffan, M.R.; Hamdy, M.E.R.; Sabry, M.A.; Abdelhalem, M.H.; Elshiekh, A.Y.M.; Elmonir, W.; Hashad, M.E. Antimicrobial Susceptibility Testing and Molecular Genotyping of Brucella melitensis Isolates at the Human Animal Interface in Upper Egypt and Egyptian Boundaries. Egypt. J. Vet. Sci. 2024, 1–11. [Google Scholar] [CrossRef]
- Ötkün, S.; Gürbilek, S.E. Whole-genome sequencing-based analysis of Brucella species isolated from ruminants in various regions of Türkiye. BMC Infect. Dis. 2024, 24, 1220. [Google Scholar] [CrossRef]
- Ayoub, H.; Kumar, M.S.; Mehta, R.; Thomas, P.; Dubey, M.; Dhanze, H.; Ajantha, G.S.; Bhilegaonkar, K.N.; Salih, H.M.; Cull, C.A.; et al. Exploring genetic determinants of antimicrobial resistance in Brucella melitensis strains of human and animal origin from India. Front. Microbiol. 2024, 15, 1474957. [Google Scholar] [CrossRef]
- Arapović, J.; Kompes, G.; Dedić, K.; Teskeredžić, S.; Ostojić, M.; Travar, M.; Tihić, N.; Delić, J.; Skočibušić, S.; Zekiri-Sivro, M.; et al. Antimicrobial resistance profiles of Human Brucella melitensis isolates in three different microdilution broths: The first multicentre study in Bosnia and Herzegovina. J. Glob. Antimicrob. Resist. 2022, 29, 99–104. [Google Scholar] [CrossRef]
- European Committee on Antimicrobial Susceptibility Testing (EUCAST). Breakpoint Tables for Interpretation of MICs and Zone Diameters. Version 15.0. 2025. Available online: https://www.eucast.org/fileadmin/src/media/PDFs/EUCAST_files/Breakpoint_tables/v_15.0_Breakpoint_Tables.pdf (accessed on 23 May 2025).
- Khan, A.U.; Shell, W.S.; Melzer, F.; Sayour, A.E.; Ramadan, E.S.; Elschner, M.C.; Moawad, A.A.; Roesler, U.; Neubauer, H.; El-Adawy, H. Identification, Genotyping and Antimicrobial Susceptibility Testing of Brucella Spp. Isolated from Livestock in Egypt. Microorganisms 2019, 7, 603. [Google Scholar] [CrossRef] [PubMed]
- Morales-Estrada, A.; Hernández-Castro, R.; López-Merino, A.; Singh-Bedi, J.; Contreras-Rodríguez, A. Isolation, identification, and antimicrobial susceptibility of Brucella spp. cultured from cows and goats manure in Mexico. Aust. J. Vet. Sci. 2016, 48, 231. [Google Scholar] [CrossRef]
- Ravanel, N.; Gestin, B.; Maurin, M. In vitro selection of fluoroquinolone resistance in Brucella melitensis. Int. J. Antimicrob. Agents 2009, 34, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Valdezate, S.; Navarro, A.; Medina-Pascual, M.J.; Carrasco, G.; Saez-Nieto, J.A. Molecular screening for rifampicin and fluoroquinolone Resistance in a clinical population of Brucella melitensis. J. Antimicrob. Chemother. 2010, 65, 51–53. [Google Scholar] [CrossRef] [PubMed]
- Paulsen, I.T.; Seshadri, R.; Nelson, K.E.; Eisen, J.A.; Heidelberg, J.F.; Read, T.D.; Dodson, R.J.; Umayam, L.; Brinkac, L.M.; Beanan, M.J.; et al. The Brucella suis genome reveals fundamental similarities between animal and plant pathogens and symbionts. Proc. Natl. Acad. Sci. USA 2002, 99, 13148–13153. [Google Scholar] [CrossRef]
- Ferjani, A.; Buijze, H.; Kopprio, G.; Köhler, S.; Rehaiem, A.; Battikh, H.; Ammari, L.; Ferjani, S.; Kanzari, L.; Zribi, M.; et al. A Genomic Characterization of Clinical Brucella melitensis Isolates from Tunisia: Integration into the Global Population Structure. Microorganisms 2025, 13, 243. [Google Scholar] [CrossRef]
- Orsini, M.; Ianni, A.; Zinzula, L. Brucella ceti and Brucella pinnipedialis genome characterization unveils genetic features that highlight their zoonotic potential. Microbiol. Open 2022, 11, e1329. [Google Scholar] [CrossRef]
- Girault, G.; Freddi, L.; Jay, M.; Perrot, L.; Dremeau, A.; Drapeau, A.; Delannoy, S.; Fach, P.; Ferreira Vicente, A.; Mick, V.; et al. Combination of in silico and molecular techniques for discrimination and virulence characterization of marine Brucella ceti and Brucella pinnipedialis. Front. Microbiol. 2024, 15, 1437408. [Google Scholar] [CrossRef]
- Sibhat, B.; Adamu, H.; Asmare, K.; Lindahl, J.F.; Magnusson, U.; Sisay Tessema, T. Detection and Molecular Diversity of Brucella melitensis in Pastoral Livestock in North-Eastern Ethiopia. Pathogens 2024, 13, 1063. [Google Scholar] [CrossRef]
- Głowacka, P.; Żakowska, D.; Naylor, K.; Niemcewicz, M.; Bielawska-Drózd, A. Brucella—Virulence Factors, Pathogenesis and Treatment. Pol. J. Microbiol. 2018, 67, 151–161. [Google Scholar] [CrossRef]
- Bialer, M.G.; Sycz, G.; Muñoz González, F.; Ferrero, M.C.; Baldi, P.C.; Zorreguieta, A. Adhesins of Brucella: Their Roles in the Interaction with the Host. Pathogens 2020, 9, 942. [Google Scholar] [CrossRef]
- Hamdy, M.E.R.; Zaki, H.M. Detection of virulence-associated genes in Brucella melitensis biovar 3, the prevalent field strain in different animal species in egypt. Open Vet. J. 2018, 8, 112–117. [Google Scholar] [CrossRef]
- Janowicz, A.; De Massis, F.; Zilli, K.; Ancora, M.; Tittarelli, M.; Sacchini, F.; Di Giannatale, E.; Sahl, J.W.; Foster, J.T.; Garofolo, G. Evolutionary history and current distribution of the West Mediterranean lineage of Brucella melitensis in Italy. Microb. Genom. 2020, 6, e000446. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.A. Brucella lipopolysaccharide and pathogenicity: The core of the matter. Virulence 2018, 9, 379–382. [Google Scholar] [CrossRef] [PubMed]
- Mirnejad, R.; Jazi, F.M.; Mostafaei, S.; Sedighi, M. Molecular investigation of virulence factors of Brucella melitensis and Brucella abortus strains isolated from clinical and non-clinical samples. Microb. Pathog. 2017, 109, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Ke, Y.; Wang, Y.; Li, W.; Chen, Z. Type IV secretion system of Brucella spp. and its effectors. Front. Cell. Infect. Microbiol. 2015, 5, 72. [Google Scholar] [CrossRef]
- Martínez-Núñez, C.; Altamirano-Silva, P.; Alvarado-Guillén, F.; Moreno, E.; Guzmán-Verri, C.; Chaves-Olarte, E. The Two-Component System BvrR/BvrS Regulates the Expression of the Type IV Secretion System VirB in Brucella abortus. J. Bacteriol. 2010, 192, 5603–5608. [Google Scholar] [CrossRef]
- Arellano-Reynoso, B.; Lapaque, N.; Salcedo, S.; Briones, G.; Ciocchini, A.E.; Ugalde, R.; Moreno, E.; Moriyón, I.; Gorvel, J.-P. Cyclic β-1, 2-glucan is a Brucella virulence factor required for intracellular survival. Nat. Immunol. 2005, 6, 618–625. [Google Scholar] [CrossRef]
- Hashemifar, I.; Yadegar, A.; Jazi, F.M.; Amirmozafari, N. Molecular prevalence of putative virulence-associated genes in Brucella melitensis and Brucella abortus isolates from human and livestock specimens in Iran. Microb. Pathog. 2017, 105, 334–339. [Google Scholar] [CrossRef]
- Lee, J.J.; Kim, J.H.; Kim, D.G.; Kim, D.H.; Simborio, H.L.; Min, W.G.; Rhee, M.H.; Lim, J.H.; Chang, H.H.; Kim, S. Characterization of Betaine Aldehyde Dehydrogenase (BetB) as an Essential Virulence Factor of Brucella abortus. Vet. Microbiol. 2014, 168, 131–140. [Google Scholar] [CrossRef]
- Oueslati, I.; Berriche, A.; Ammari, L.; Abdelmalek, R.; Kanoun, F.; Kilani, B.; Tiouiri Benaissa, H. Epidemiological and clinical characteristics of neurobrucellosis case patients in Tunisia. MédecineMal. Infect. 2016, 46, 123–130. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Stats of Strains | SBZ | TATA | B. melitensis bv. 3 Ether | B. melitensis bv. 1 16 M | B. melitensis bv. 1 Rev.1 |
|---|---|---|---|---|---|
| Genome size (bp) | 3,310,738 | 3,310,809 | 3,310,727 | 3,294,931 | 3,299,187 |
| Chromosome I size (bp) | 2,122,905 | 2,122,935 | 2,122,766 | 2,117,144 | 2,121,370 |
| Chromosome II size (bp) | 1,187,833 | 1,187,874 | 1,187,961 | 1,177,787 | 1,177,817 |
| Num genes (Prodigal) | 3153 | 3152 | 3159 | 3152 | 3148 |
| L50 value | 1 | 1 | 1 | 1 | 1 |
| L75 value | 2 | 2 | 2 | 2 | 2 |
| L90 value | 2 | 2 | 2 | 2 | 2 |
| N50 value (bp) | 2,122,905 | 2,122,935 | 2,122,766 | 2,117,144 | 2,121,370 |
| N75 value (bp) | 1,187,833 | 1,187,874 | 1,187,961 | 1,177,787 | 1,177,817 |
| N90 value (bp) | 1,187,833 | 1,187,874 | 1,187,961 | 1,177,787 | 1,177,817 |
| GC content (bp) | 57.2 | 57.2 | 57.2 | 57.2 | 57.2 |
| Number of subsystems | 407 | 407 | 304 | 303 | 303 |
| Number of coding sequences | 3350 | 3340 | 3326 | 3326 | 3322 |
| Antibiotic | MIC Range (µg/mL) | MIC Values of SBZ Strain (µg/mL) | MIC Values of TATA Strain (µg/mL) | MIC Interpretive Criteria (µg/mL) | Phenotype of SBZ Strain | Phenotype of TATA Strain | ||
|---|---|---|---|---|---|---|---|---|
| S≤ | I | R≥ | ||||||
| Gentamicin | 0.004–8 | 0.5 | 0.5 | 4 | - | - | S | S |
| Streptomycin | 0.008–16 | 2 | 4 | 16 b | - | - | S | S |
| Doxycycline | 0.015–8 | 0.06 | 0.06 | 1 | . | - | S | S |
| Rifampicin a | 0.012–4 | 1 | 1 | 1 | 2 | 4 | S | S |
| Trimethoprim–Sulfamethoxazole | 0.002/0.04 to 4/76 | 0.5 | 0.5 | 2/38 | - | - | S | S |
| Ceftriaxone | 0.015–2 | 0.5 | 0.5 | ND * | ND * | ND * | - | - |
| Ciprofloxacin | 0.002–4 | 0.5 | 0.5 | ND * | ND * | ND * | - | - |
| Levofloxacin | 0.004–4 | 0.5 | 0.5 | ND * | ND * | ND * | - | - |
| Tetracycline | 0.25–8 | <0.25 | <0.25 | ND * | ND * | ND * | - | - |
| Identification of AMR Genes (% Identity) | |||||||
|---|---|---|---|---|---|---|---|
| Abricate (MEGARes) | AMRfinderPlus (BLASTX/(PARTIALX) | CARD (RGI) | |||||
| Type | AMR Genes | SBZ Strain | TATA Strain | SBZ Strain | TATA Strain | SBZ Strain | TATA Strain |
| Resistance–Nodulation–Cell Division (RND) efflux pumps (Drug_and_biocide resistance) | bepC | 99.64 | 99.64 | 99.78 | 99.78 | - | - |
| bepD | 99.92 | 99.92 | 99.75 | 99.75 | - | - | |
| bepE | 99.84 | 99.84 | 99.62 | 99.62 | - | - | |
| bepG | 99.29 | 99.29 | 99.38 | 99.38 | - | - | |
| bepF | 98.79 | 98.87 | 98.54 | 98.54 | - | - | |
| RND efflux pump (Fluoroquinolone and tetracycline resistance) | adeF | - | - | - | - | 46.73 | 46.73 |
| Antibiotic target alteration with defensin resistance (Cationic antibiotic resistance) | mprF | 99.58 | 99.58 | - | - | 99.54 | 99.54 |
| Antibiotic inactivation with Fosfomycin thiol transferase (Phosphonic acid antibiotic resistance) | fosxcc | - | - | - | - | 54.81 | 54.81 |
| Small multidrug resistance (SMR) antibiotic efflux pump/disinfecting agent and antiseptic resistance | qacG | - | - | - | - | 42.31 | 42.31 |
| Virulence Factors | Related Genes |
|---|---|
| Adhesines | bigA, bigB, bmaB/omA, bmaC |
| Lipopolysaccharide (LPS) | gmd, per, pgm, pmm, manAoAg, manCoAg, wzm, wzt, wbkB, wbkC, wbkA, wbpZ, wbpL, lpsA, acpXL, wboA, wbdA, lpsB, lpcC, manCcore, manBcore, fabZ, lpxA, lpxC, lpxD, lpxB, lpxE, lpxK, KdsA, kdsB, waaA/kdtA, htrB, manA, perA |
| Type IV secretion system (VirB) | virB1, virB2, virB3, virB4, virB5, virB6, virB7, virB8, virB9, virB10, virB11, virB12 |
| Type IV secretion system effectors | vceA, vceC, ricA, BPE005, BPE043, BPE275, BPE123, sepA, bspA, bspB, BspC, bspE, bspF, bspL |
| TIR domain-containing protein immune evasion | BtpA/Btp1/TcpB and BtpB |
| Two-component system BvrR/BvrS (TCS BvrRS) | bvrR and bvrS |
| CβG (cyclic β-1,2 glucan) | Cgs |
| Peptidoglycan | mviN |
| Biosynthesis of glycine betaine | betB |
| Outer membrane protein | omp19 |
| Urease | Ure |
| Proline racemases | prpA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ben Abdallah, I.; Kopprio, G.; Béjaoui, A.; Köhler, S.; Guesmi, K.; Kalthoum, S.; Gatz, J.; Arfaoui, A.; Lachtar, M.; Hajlaoui, H.; et al. Hybrid Whole Genomes of Brucella melitensis from Tunisian Animal Isolates: Virulence Factors, Antimicrobial Susceptibility, and Phylogeny. Microorganisms 2025, 13, 1651. https://doi.org/10.3390/microorganisms13071651
Ben Abdallah I, Kopprio G, Béjaoui A, Köhler S, Guesmi K, Kalthoum S, Gatz J, Arfaoui A, Lachtar M, Hajlaoui H, et al. Hybrid Whole Genomes of Brucella melitensis from Tunisian Animal Isolates: Virulence Factors, Antimicrobial Susceptibility, and Phylogeny. Microorganisms. 2025; 13(7):1651. https://doi.org/10.3390/microorganisms13071651
Chicago/Turabian StyleBen Abdallah, Ibtihel, Germán Kopprio, Awatef Béjaoui, Susanne Köhler, Kaouther Guesmi, Sana Kalthoum, Jacob Gatz, Amel Arfaoui, Monia Lachtar, Haikel Hajlaoui, and et al. 2025. "Hybrid Whole Genomes of Brucella melitensis from Tunisian Animal Isolates: Virulence Factors, Antimicrobial Susceptibility, and Phylogeny" Microorganisms 13, no. 7: 1651. https://doi.org/10.3390/microorganisms13071651
APA StyleBen Abdallah, I., Kopprio, G., Béjaoui, A., Köhler, S., Guesmi, K., Kalthoum, S., Gatz, J., Arfaoui, A., Lachtar, M., Hajlaoui, H., Baccar, M. N., Scholz, H., & Maaroufi, A. (2025). Hybrid Whole Genomes of Brucella melitensis from Tunisian Animal Isolates: Virulence Factors, Antimicrobial Susceptibility, and Phylogeny. Microorganisms, 13(7), 1651. https://doi.org/10.3390/microorganisms13071651