
Dietary Protein-Induced Changes in Archaeal Compositional Dynamics, Methanogenic Pathways, and Antimicrobial Resistance Profiles in Lactating Sheep


 , , and
, , and
Abstract

1. Introduction
2. Materials and Methods
2.1. Ethics Approval, Experimental Site, Research Design, Husbandry Practices, and Fecal Sample Collection
2.2. DNA Extraction
2.3. Polymerase Chain Reaction, Amplification, and 16S rDNA Sequencing
2.4. Metagenomics Sequencing
2.5. Bioinformatics Analysis
3. Results
3.1. Sequencing Reads
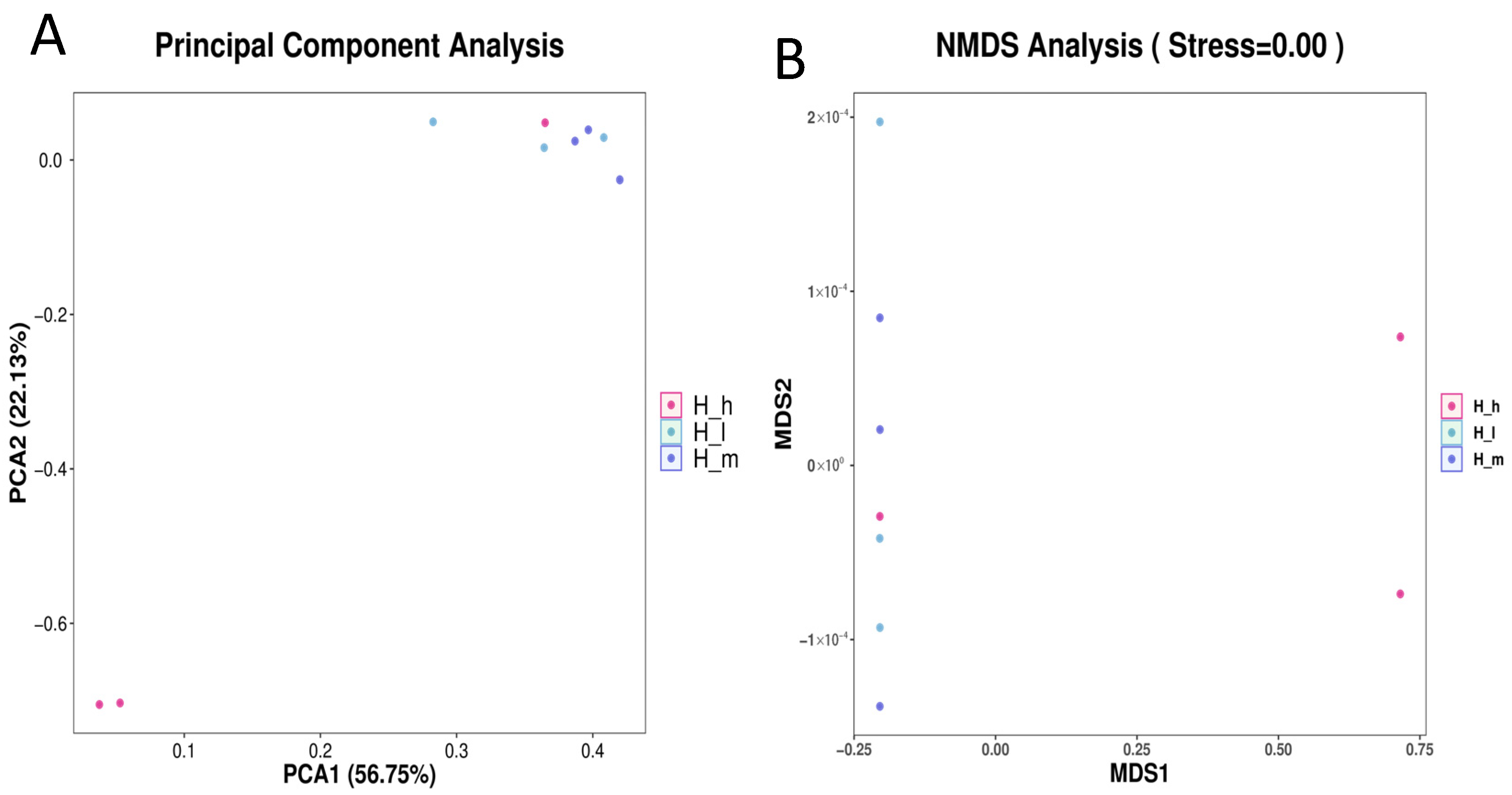
3.2. Fecal Archaeal Richness
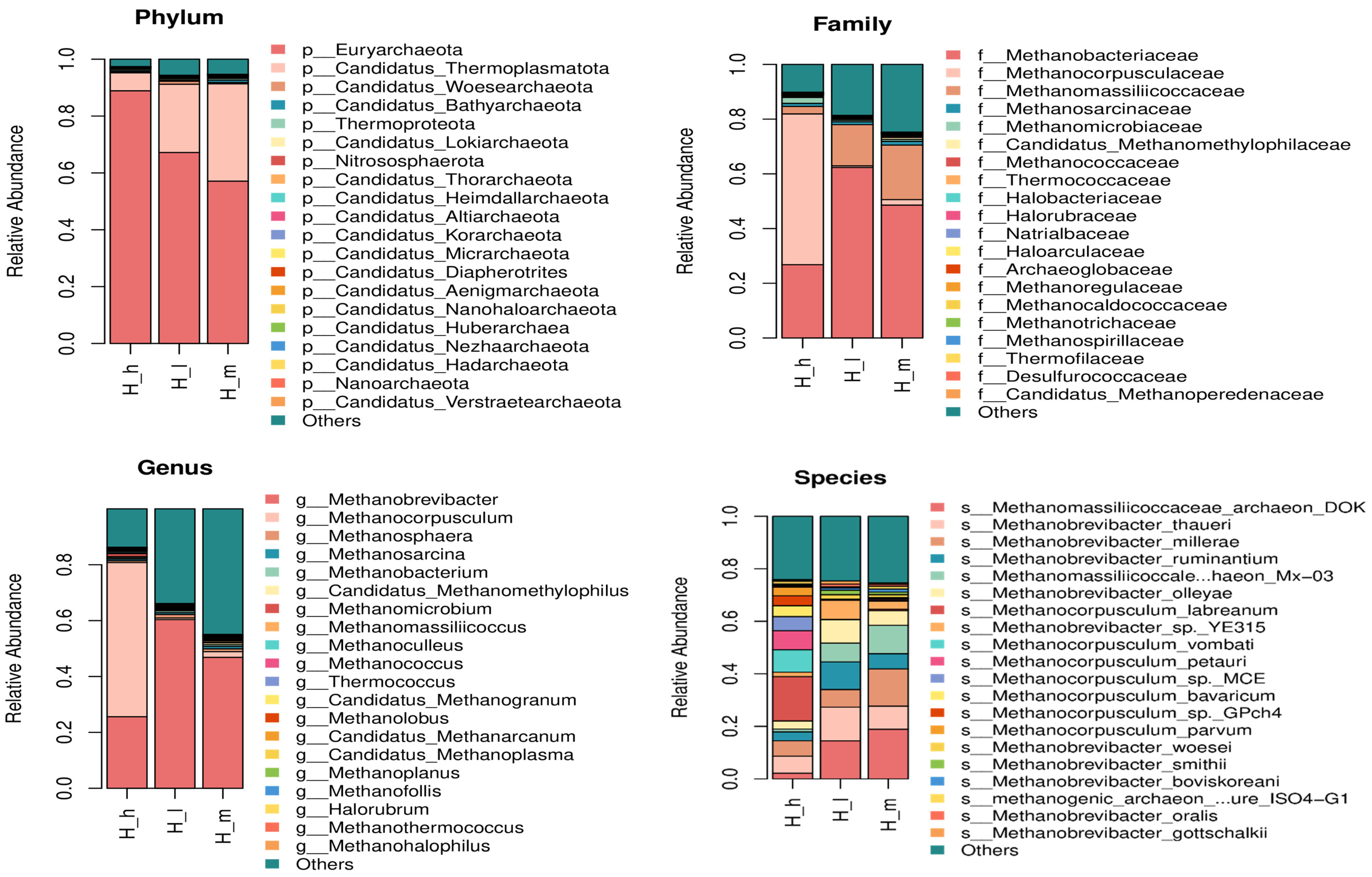
3.3. Archaeal Taxonomic Profiling
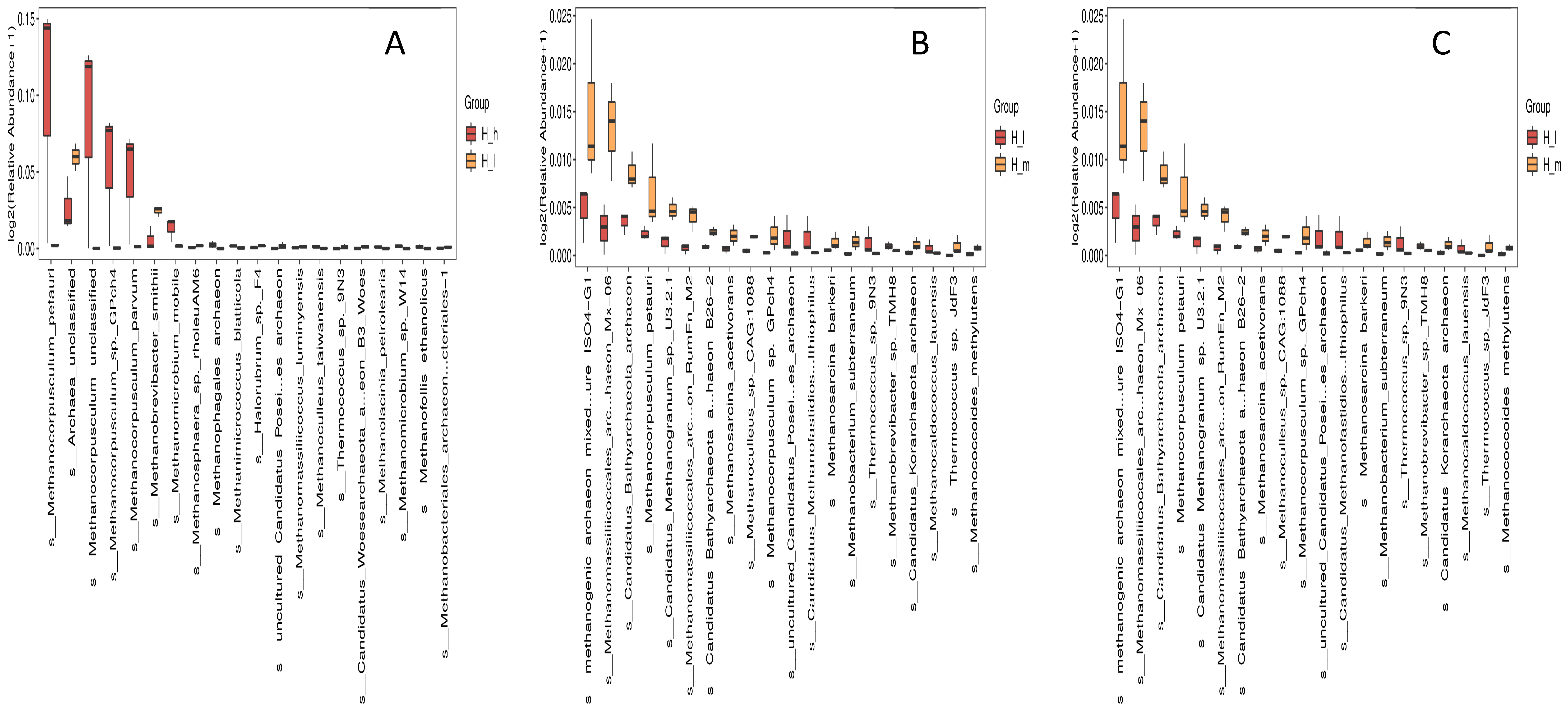
3.4. Comparative Taxonomic Profiling of Fecal Archaeal Species
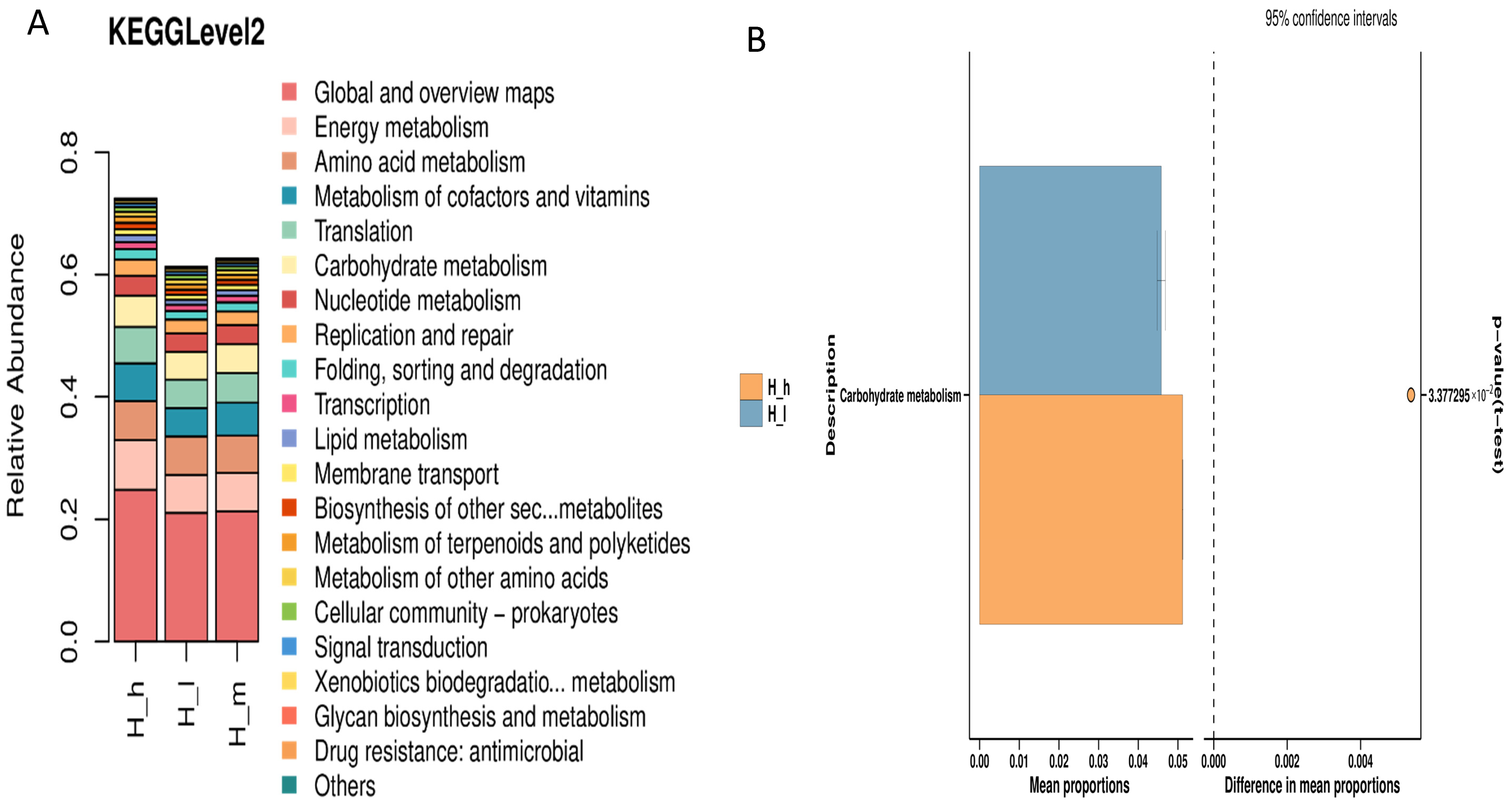
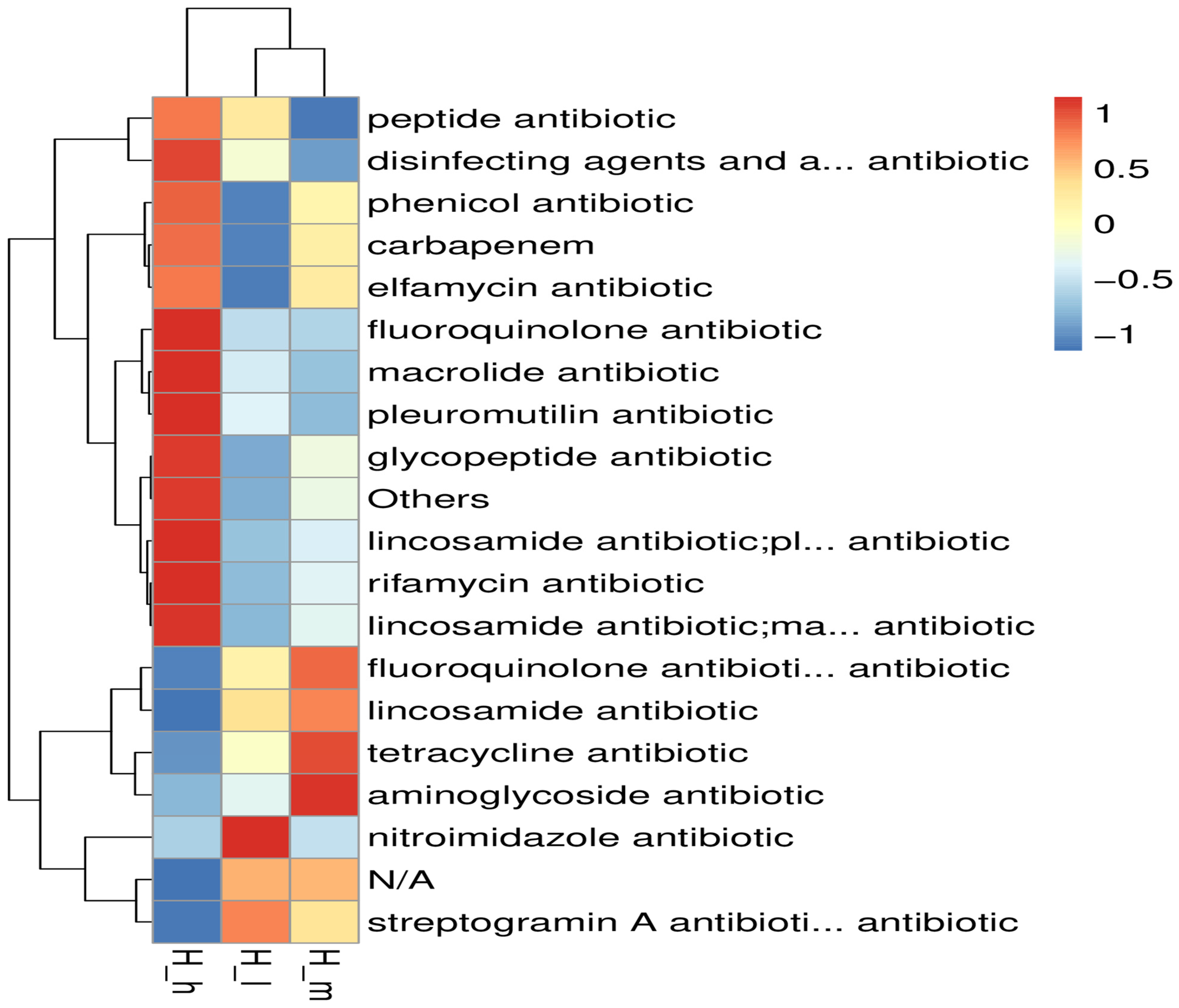
3.5. Functional Prediction of Archaeal Communities
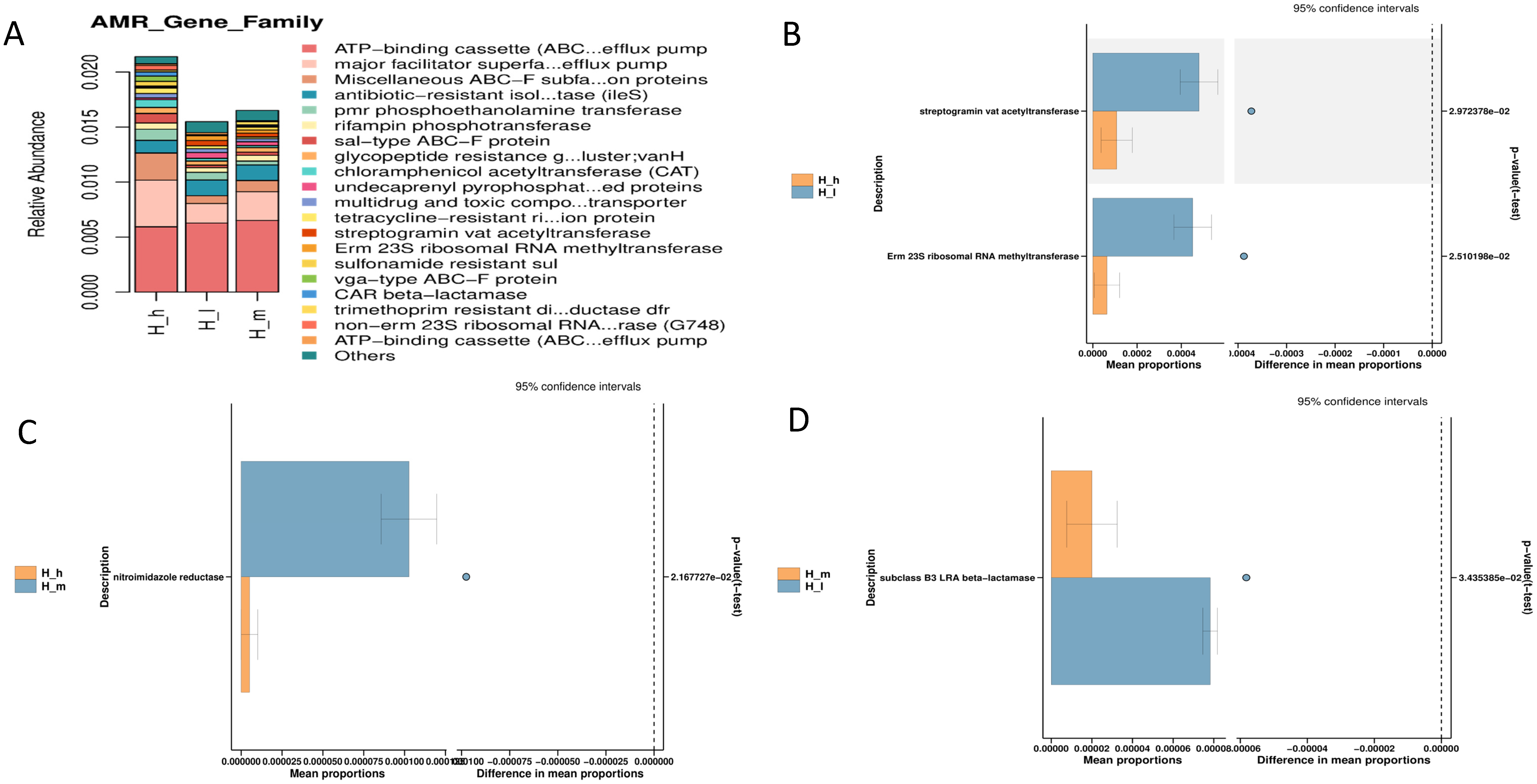
3.6. Resistome Analysis
3.7. Virulence Factor Profiles
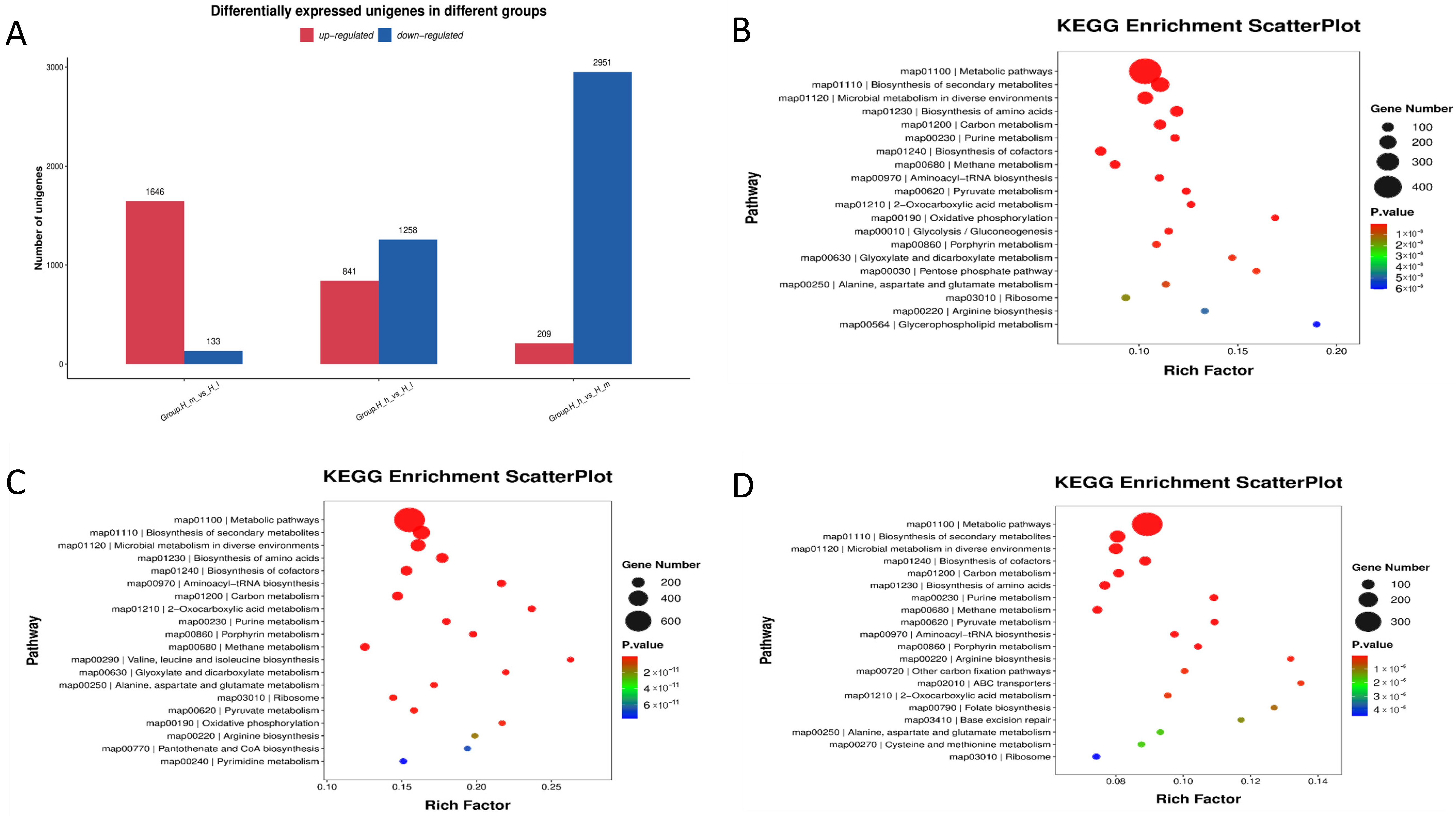
3.8. Differential Expression of Gut Microbial Functional Pathways
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AMR | Antimicrobial resistance |
| CAZyme | Carbohydrate-active enzymes |
| CARD | Comparative antibiotic-resistant database |
| PCR | Polymerase chain reaction |
| VFDB | Virulence factor database |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
References
- Morgavi, D.P.; Kelly, W.; Janssen, P.; Attwood, G. Rumen microbial (meta) genomics and its application to ruminant production. Animal 2013, 7, 184–201. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Wang, H.; Qiu, Y.; Ren, L.; Jiang, B. Microbial characteristics in anaerobic digestion process of food waste for methane production—A review. Bioresour. Technol. 2018, 248, 29–36. [Google Scholar] [CrossRef]
- Diether, N.E.; Willing, B.P. Microbial fermentation of dietary protein: An important factor in diet–microbe–host interaction. Microorganisms 2019, 7, 19. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Gong, Y.; Huang, S.; Ji, S.; Wang, W.; Wang, Y.; Yang, H.; Cao, Z.; Li, S. Effects of age, diet CP, NDF, EE, and starch on the rumen bacteria community and function in dairy cattle. Microorganisms 2021, 9, 1788. [Google Scholar] [CrossRef] [PubMed]
- Henderson, G.; Cox, F.; Ganesh, S.; Jonker, A.; Young, W.; Janssen, P.H. Rumen microbial community composition varies with diet and host, but a core microbiome is found across a wide geographical range. Sci. Rep. 2015, 5, 14567. [Google Scholar] [CrossRef]
- Tan, P.; Liu, H.; Zhao, J.; Gu, X.; Wei, X.; Zhang, X.; Ma, N.; Johnston, L.J.; Bai, Y.; Zhang, W. Amino acids metabolism by rumen microorganisms: Nutrition and ecology strategies to reduce nitrogen emissions from the inside to the outside. Sci. Total Environ. 2021, 800, 149596. [Google Scholar] [CrossRef]
- Broderick, G. Optimizing ruminant conversion of feed protein to human food protein. Animal 2018, 12, 1722–1734. [Google Scholar] [CrossRef]
- Tamminga, S. A review on environmental impacts of nutritional strategies in ruminants. J. Anim. Sci. 1996, 74, 3112–3124. [Google Scholar] [CrossRef]
- McLoughlin, S.; Spillane, C.; Campion, F.P.; Claffey, N.; Sosa, C.C.; McNicholas, Y.; Smith, P.E.; Diskin, M.G.; Waters, S.M. Breed and ruminal fraction effects on bacterial and archaeal community composition in sheep. Sci. Rep. 2023, 13, 3336. [Google Scholar] [CrossRef]
- Leahy, S.; Kelly, W.; Ronimus, R.; Wedlock, N.; Altermann, E.; Attwood, G. Genome sequencing of rumen bacteria and archaea and its application to methane mitigation strategies. Animal 2013, 7, 235–243. [Google Scholar] [CrossRef]
- Li, F.; Guan, L.L. Metatranscriptomic profiling reveals linkages between the active rumen microbiome and feed efficiency in beef cattle. Appl. Environ. Microbiol. 2017, 83, e00061-17. [Google Scholar] [CrossRef]
- Li, F.; Li, C.; Chen, Y.; Liu, J.; Zhang, C.; Irving, B.; Fitzsimmons, C.; Plastow, G.; Guan, L.L. Host genetics influence the rumen microbiota and heritable rumen microbial features associate with feed efficiency in cattle. Microbiome 2019, 7, 92. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Hitch, T.C.; Chen, Y.; Creevey, C.J.; Guan, L.L. Comparative metagenomic and metatranscriptomic analyses reveal the breed effect on the rumen microbiome and its associations with feed efficiency in beef cattle. Microbiome 2019, 7, 6. [Google Scholar] [CrossRef]
- McLoughlin, S.; Spillane, C.; Claffey, N.; Smith, P.E.; O’Rourke, T.; Diskin, M.G.; Waters, S.M. Rumen microbiome composition is altered in sheep divergent in feed efficiency. Front. Microbiol. 2020, 11, 1981. [Google Scholar] [CrossRef]
- Shen, J.; Zheng, L.; Chen, X.; Han, X.; Cao, Y.; Yao, J. Metagenomic analyses of microbial and carbohydrate-active enzymes in the rumen of dairy goats fed different rumen degradable starch. Front. Microbiol. 2020, 11, 1003. [Google Scholar] [CrossRef] [PubMed]
- Abrahamsen, F.; Min, B.; Gurung, R.; Gurung, N.; Abebe, W. Altering the gut microbiome of meat goats: Feeding varying levels of hempseed meal on animal performance, rumen microbiome abundance, and methanogen community changes of meat goats. BAOJ Microbiol. 2022, 6, 1006. [Google Scholar]
- Wang, L.; Liu, K.; Wang, Z.; Bai, X.; Peng, Q.; Jin, L. Bacterial community diversity associated with different utilization efficiencies of nitrogen in the gastrointestinal tract of goats. Front. Microbiol. 2019, 10, 239. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Meng, Q.; Li, S.; Jiang, L.; Wu, H. Effect of urea-supplemented diets on the ruminal bacterial and archaeal community composition of finishing bulls. Appl. Microbiol. Biotechnol. 2017, 101, 6205–6216. [Google Scholar] [CrossRef]
- Mavrommatis, A.; Skliros, D.; Sotirakoglou, K.; Flemetakis, E.; Tsiplakou, E. The effect of forage-to-concentrate ratio on schizochytrium spp.-supplemented goats: Modifying rumen microbiota. Animals 2021, 11, 2746. [Google Scholar] [CrossRef]
- Zhang, J.; Shi, H.; Wang, Y.; Cao, Z.; Yang, H.; Li, S. Effect of limit-fed diets with different forage to concentrate ratios on fecal bacterial and archaeal community composition in Holstein heifers. Front. Microbiol. 2018, 9, 976. [Google Scholar] [CrossRef]
- Kolte, A.P.; Dhali, A.; Malik, P.; Samanta, A.; Bhatta, R. Rumen metagenomics and its implications in animal nutrition: A review. Indian J. Anim. Nutr. 2017, 34, 124–134. [Google Scholar] [CrossRef]
- Subedi, U.; Kader, K.; Jayawardhane, K.N.; Poudel, H.; Chen, G.; Acharya, S.; Camargo, L.S.; Bittencourt, D.M.d.C.; Singer, S.D. The potential of novel gene editing-based approaches in forages and rumen archaea for reducing livestock methane emissions. Agriculture 2022, 12, 1780. [Google Scholar] [CrossRef]
- Mizrahi, I.; Wallace, R.J.; Moraïs, S. The rumen microbiome: Balancing food security and environmental impacts. Nat. Rev. Microbiol. 2021, 19, 553–566. [Google Scholar] [CrossRef]
- Zhao, H.; Bai, S.; Tan, J.; Liu, M.; Zhao, Y.; Jiang, L. Can meta-omics revolutionize our understanding of rumen methane emissions? Anim. Nutr. 2024, 1, e14. [Google Scholar] [CrossRef]
- International, A. Official Methods of Analysis of AOAC International; AOAC International: Rockville, MD, USA, 2000; Volume 17. [Google Scholar]
- Zhao, X.; Ali, S.; Hassan, M.F.; Bashir, M.A.; Ni, X.; Lv, C.; Yang, H.; Danzeng, B.; Quan, G. Effects of graded levels of dietary protein supplementation on milk yield, body weight gain, blood biochemical parameters, and gut microbiota in lactating ewes. Front. Vet. Sci. 2023, 10, 1223450. [Google Scholar] [CrossRef]
- Russell, J.B.; Hespell, R.B. Microbial rumen fermentation. J. Dairy Sci. 1981, 64, 1153–1169. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Zhang, X.; Wang, M.; Zhou, C.; Jiao, J.; Tan, Z. Enhancing metabolic efficiency through optimizing metabolizable protein profile in a time progressive manner with weaned goats as a model: Involvement of gut microbiota. Microbiol. Spectr. 2022, 10, e02545-21. [Google Scholar] [CrossRef] [PubMed]
- Cui, K.; Qi, M.; Wang, S.; Diao, Q.; Zhang, N. Dietary energy and protein levels influenced the growth performance, ruminal morphology and fermentation and microbial diversity of lambs. Sci. Rep. 2019, 9, 16612. [Google Scholar] [CrossRef]
- Wang, X.; Xu, T.; Zhang, X.; Zhao, N.; Hu, L.; Liu, H.; Zhang, Q.; Geng, Y.; Kang, S.; Xu, S. The response of ruminal microbiota and metabolites to different dietary protein levels in Tibetan sheep on the Qinghai-Tibetan plateau. Front. Vet. Sci. 2022, 9, 922817. [Google Scholar] [CrossRef]
- Zhang, Z.; Wei, W.; Yang, S.; Huang, Z.; Li, C.; Yu, X.; Qi, R.; Liu, W.; Loor, J.J.; Wang, M. Regulation of dietary protein solubility improves ruminal nitrogen metabolism in vitro: Role of bacteria–protozoa interactions. Nutrients 2022, 14, 2972. [Google Scholar] [CrossRef]
- Hook, S.E.; Wright, A.-D.G.; McBride, B.W. Methanogens: Methane producers of the rumen and mitigation strategies. Archaea 2010, 2010, 945785. [Google Scholar] [CrossRef] [PubMed]
- Saminathan, M.; Sieo, C.C.; Gan, H.M.; Abdullah, N.; Wong, C.M.V.L.; Ho, Y.W. Effects of condensed tannin fractions of different molecular weights on population and diversity of bovine rumen methanogenic archaea in vitro, as determined by high-throughput sequencing. Anim. Feed. Sci. Technol. 2016, 216, 146–160. [Google Scholar] [CrossRef]
- Kostopoulou, E.; Chioti, A.G.; Tsioni, V.; Sfetsas, T. Microbial dynamics in anaerobic digestion: A review of operational and environmental factors affecting microbiome composition and function. J. Clean. Prod. 2023, 22, 2235–2252. [Google Scholar]
- Tang, Y.; Liu, X.; Zhu, S.; Jia, M.; Liu, J.-X.; Sun, H.-Z. New insights into the enteric methane production based on the archaeal genome atlas of ruminant gastrointestinal tract. J. Adv. Res. 2024. online ahead of print. [Google Scholar] [CrossRef]
- Poulsen, M.; Schwab, C.; Borg Jensen, B.; Engberg, R.M.; Spang, A.; Canibe, N.; Højberg, O.; Milinovich, G.; Fragner, L.; Schleper, C. Methylotrophic methanogenic Thermoplasmata implicated in reduced methane emissions from bovine rumen. Nat. Commun. 2013, 4, 1428. [Google Scholar] [CrossRef] [PubMed]
- Mi, J.; Jing, X.; Ma, C.; Shi, F.; Cao, Z.; Yang, X.; Yang, Y.; Kakade, A.; Wang, W.; Long, R. A metagenomic catalogue of the ruminant gut archaeome. Nat. Commun. 2024, 15, 9609. [Google Scholar] [CrossRef]
- Palmonari, A.; Federiconi, A.; Formigoni, A. Animal board invited review: The effect of diet on rumen microbial composition in dairy cows. Animal 2024, 18, 101319. [Google Scholar] [CrossRef]
- Dai, X.; Faciola, A.P. Evaluating strategies to reduce ruminal protozoa and their impacts on nutrient utilization and animal performance in ruminants–a meta-analysis. Front. Microbiol. 2019, 10, 2648. [Google Scholar] [CrossRef] [PubMed]
- Tan, R.; Jin, M.; Shao, Y.; Yin, J.; Li, H.; Chen, T.; Shi, D.; Zhou, S.; Li, J.; Yang, D. High-sugar, high-fat, and high-protein diets promote antibiotic resistance gene spreading in the mouse intestinal microbiota. Gut Microbes 2022, 14, 2022442. [Google Scholar] [CrossRef]
- Mefferd, C.C.; Bhute, S.S.; Phan, J.R.; Villarama, J.V.; Do, D.M.; Alarcia, S.; Abel-Santos, E.; Hedlund, B.P. A high-fat/high-protein, Atkins-type diet exacerbates Clostridioides (Clostridium) difficile infection in mice, whereas a high-carbohydrate diet protects. mSystems 2020, 5, e00765-19. [Google Scholar] [CrossRef]
- Zhang, C.; Yu, M.; Yang, Y.; Mu, C.; Su, Y.; Zhu, W. Differential effect of early antibiotic intervention on bacterial fermentation patterns and mucosal gene expression in the colon of pigs under diets with different protein levels. Appl. Microbiol. Biotechnol. 2017, 101, 2493–2505. [Google Scholar] [CrossRef] [PubMed]
- Porcheron, G.; Dozois, C.M. Interplay between iron homeostasis and virulence: Fur and RyhB as major regulators of bacterial pathogenicity. Vet. Microbiol. 2015, 179, 2–14. [Google Scholar] [CrossRef] [PubMed]
- de Macario, E.C.; Macario, A.J. Methanogenic archaea in health and disease: A novel paradigm of microbial pathogenesis. Int. J. Med. Microbiol. 2009, 299, 99–108. [Google Scholar]
- Wallace, R.J.; Rooke, J.A.; McKain, N.; Duthie, C.-A.; Hyslop, J.J.; Ross, D.W.; Waterhouse, A.; Watson, M.; Roehe, R. The rumen microbial metagenome associated with high methane production in cattle. BMC Genom. 2015, 16, 839. [Google Scholar] [CrossRef]
- Lu, Z.; Xu, Z.; Shen, Z.; Tian, Y.; Shen, H. Dietary energy level promotes rumen microbial protein synthesis by improving the energy productivity of the ruminal microbiome. Front. Microbiol. 2019, 10, 847. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variables | Treatments 1 | ||
|---|---|---|---|
| H_l | H_m | H_h | |
| Corn silage (%) | 40.00 | 35.00 | 34.00 |
| Wheat straw (%) | 13.00 | 10.00 | 10.00 |
| Bean powder (%) | 2.00 | 10.00 | 11.00 |
| Corn grain (%) | 28.20 | 26.00 | 19.05 |
| Soybean meal (%) | 5.40 | 8.65 | 18.60 |
| Corn starch (%) | 8.65 | 7.60 | 4.70 |
| Bean powder (%) | 2.00 | 10.00 | 11.00 |
| Premix (%) 2 | 1.00 | 1.00 | 1.00 |
| Calcium carbonate (%) | 0.55 | 0.60 | 0.65 |
| Calcium hydrogen phosphate (%) | 0.55 | 0.50 | 0.35 |
| Salt (%) | 0.30 | 0.30 | 0.30 |
| Baking soda (%) | 0.35 | 0.35 | 0.35 |
| Forage to concentrate ratio | 55:45 | 55:45 | 55:45 |
| Chemical composition | |||
| Dry matter (%) | 89.06 | 88.92 | 88.75 |
| ME (MJ/kg) | 9.45 | 9.47 | 9.47 |
| Crude protein (%) | 8.58 | 10.34 | 13.93 |
| NDF (%) | 32.17 | 32.01 | 32.52 |
| ADF (%) | 17.24 | 17.71 | 18.44 |
| EE (%) | 2.39 | 2.32 | 2.43 |
| Ash (%) | 9.06 | 9.27 | 9.62 |
| Calcium (%) | 0.69 | 0.71 | 0.71 |
| Phosphorus (%) | 0.38 | 0.39 | 0.39 |
| Group | Observed Species | Shannon | Chao1 | Simpson | Goods Coverage |
|---|---|---|---|---|---|
| H_l | 298 ± 25.53 b | 4.30 ± 0.59 | 317.18 ± 20.22 b | 0.89 ± 0.04 | 0.99 ± 0.00 |
| H_m | 394.67 ± 8.5 a | 4.79 ± 0.26 | 417.82 ± 4.39 a | 0.91 ± 0.02 | 0.99 ± 0.00 |
| H_h | 389.33 ± 39.07 a | 4.28 ± 0.67 | 425.91 ± 23.72 a | 0.89 ± 0.03 | 0.99 ± 0.00 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mushtaq, M.; Ni, X.; Khan, M.; Zhao, X.; Yang, H.; Danzeng, B.; Ali, S.; Zafar, M.H.; Quan, G. Dietary Protein-Induced Changes in Archaeal Compositional Dynamics, Methanogenic Pathways, and Antimicrobial Resistance Profiles in Lactating Sheep. Microorganisms 2025, 13, 1560. https://doi.org/10.3390/microorganisms13071560
Mushtaq M, Ni X, Khan M, Zhao X, Yang H, Danzeng B, Ali S, Zafar MH, Quan G. Dietary Protein-Induced Changes in Archaeal Compositional Dynamics, Methanogenic Pathways, and Antimicrobial Resistance Profiles in Lactating Sheep. Microorganisms. 2025; 13(7):1560. https://doi.org/10.3390/microorganisms13071560
Chicago/Turabian StyleMushtaq, Maida, Xiaojun Ni, Muhammad Khan, Xiaoqi Zhao, Hongyuan Yang, Baiji Danzeng, Sikandar Ali, Muhammad Hammad Zafar, and Guobo Quan. 2025. "Dietary Protein-Induced Changes in Archaeal Compositional Dynamics, Methanogenic Pathways, and Antimicrobial Resistance Profiles in Lactating Sheep" Microorganisms 13, no. 7: 1560. https://doi.org/10.3390/microorganisms13071560
APA StyleMushtaq, M., Ni, X., Khan, M., Zhao, X., Yang, H., Danzeng, B., Ali, S., Zafar, M. H., & Quan, G. (2025). Dietary Protein-Induced Changes in Archaeal Compositional Dynamics, Methanogenic Pathways, and Antimicrobial Resistance Profiles in Lactating Sheep. Microorganisms, 13(7), 1560. https://doi.org/10.3390/microorganisms13071560