Abstract
Crossbreeding often results in heterosis. In this study, we generated hybrids from largemouth bass from geographically isolated populations. Growth, digestive enzyme activity, and muscle nutrient composition were compared between the hybrid groups (NC and CN) and the purebred groups (NN and CC), and the gut microbiota was investigated. The heterosis rates of body length, body height, and body thickness in hybrid largemouth bass were statistically significant. Digestive enzyme activity was higher in the hybrid groups than in the purebred groups. Compared with the CN and CC groups, the NC group had significantly higher levels of essential amino acids and total amino acids in the muscle. The polyunsaturated fatty acid content was lower in the hybrid groups than in the purebred groups. The gut microbiota in the hybrid groups predominantly exhibited a non-additive inheritance pattern, characterized by a reduced abundance of Proteobacteria and an increased abundance of Fusobacteria. Microbial taxa showing differences in abundance between the hybrid and purebred groups harbored genes enriched in multiple amino acid and fatty acid metabolism pathways. Cetobacterium, Pseudomonas, and Stenotrophomonas were more abundant in the hybrids, and were positively correlated with multiple amino acids and fatty acids. These results highlight the critical role of gut microbiota in heterosis.
1. Introduction
Heterosis refers to the phenomenon where the F1 hybrid generation derived from parents with distinct genetic backgrounds exhibits enhanced growth rates or superior stress resistance compared with its parental lines [1]. Hybridization has emerged as a routine and effective strategy in the breeding of various livestock [2,3], crops [4,5,6], and aquatic animals [7,8,9], significantly enhancing breeding efficiency and production performance. An understanding of the mechanism of heterosis is critical for determining its potential benefits in agricultural technology and biotechnology. Extensive research has focused on the molecular mechanism of heterosis from a genetic perspective [10,11,12]. Nevertheless, it is likely that many other factors also contribute to heterosis.
The complex array of microbes in the gut, collectively termed the gut microbiota, maintains a long-term, intimate, and intricate relationship with the host [13]. The gut microbiota represents a highly specialized and structurally complex microbial ecosystem, which is shaped by interactions among microorganisms, the host, dietary intake, and environmental factors. Host phylogeny and dietary patterns are recognized as two predominant factors influencing the composition and structure of the gut microbial community in animals [14,15]. The gut microbiota plays a crucial role in the development of the host’s nervous system, behavioral regulation, immune function, food digestion, and metabolic processes [16]. It also influences host reproduction by affecting gametic integrity and embryo viability, potentially playing a critical role in the formation of new species [17,18]. Previous research has shown that the gut microbiota can impact the host genome, thereby altering the host’s physiological and metabolic activities. This enables hosts to expand their dietary niches and access novel nutritional opportunities, and suggests that gut microbiota may contribute to the process of speciation [17]. For example, Acetitomaculum was found to be significantly increased in the crossbred progeny of sika deer (Cervus nippon) and elk (Cervus elaphus), enhancing their nutrient absorption and utilization [19]. Huang et al. [20] proposed that alterations in the intestinal microbial structure of crossbred progeny represent a key factor contributing to heterosis. Consequently, the gut microbiota is one of the potential physiological mechanisms underlying heterosis.
The gut microbiota plays a crucial role in facilitating the digestion and absorption of nutrients, thereby influencing the nutrient composition of muscle. Muscle nutrient composition is a critical parameter for evaluating the nutritional value of aquatic products [21]. The lipid content in muscle significantly influences its elasticity and texture [22]. The presence of certain amino acids and fatty acids not only determines whether the muscle can supply essential nutrients required by humans, but also plays a crucial role in determining muscle flavor [23,24], providing significant advantages in terms of quality and marketability. Breeding to improve the nutritional profile of muscle holds significant potential. Several studies have shown that the crossbred progeny exhibit marked differences from their parental lines in terms of lipid, amino acid, and fatty acid contents in muscle [25,26], suggesting that hybridization is an effective method for breeding progeny with improved muscle nutritional profiles. These findings imply that there are critical relationships among the gut microbiota, genome, and phenotype. The genome of hybrid progeny may allow them to develop distinct gut microbiota, which contributes to the heterosis of certain phenotypic traits and muscle nutrient composition.
Largemouth bass (Micropterus salmoides) is an economically important fish species. Its advantages include rapid growth, strong stress resistance, and flavorful and nutritious meat [27]. Nevertheless, reduced genetic diversity and differentiation in genetic structure have been widely observed in the breeding populations of largemouth bass [28]. Although several cross-breeding studies have aimed to improve the genetics of largemouth bass [29,30], they did not fully elucidate the mechanisms of heterosis in this fish. Although research into the crossbreeding of aquatic animals has been ongoing since the last century, the theoretical foundation underlying heterosis remains enigmatic [31,32,33]. In this study, largemouth bass from geographically isolated populations were utilized as parental lines. The phenotypic traits, muscle nutrient composition, and gut microbial structure of the crossbred progeny were systematically analyzed and compared. The aims of this study were to elucidate the mechanistic basis of heterosis in largemouth bass from the perspective of gut microbiota, and to investigate the influence of hybridization on the muscle nutrient profile. The results of this study provide a theoretical foundation for the genetic improvement of largemouth bass in both research and practical settings.
2. Materials and Methods
2.1. Experimental Populations
Two geographically isolated populations of largemouth bass were utilized as parental lines for cross-breeding: the US-introduced protospecies (Wild population in the freshwater basin of Florida) and the farmed population from Jiangsu, China. Two crossbred progeny populations were generated through hybridization. Specifically, one crossbred progeny group (NC) was produced with the US-introduced protospecies as the paternal parent and the farmed population from Jiangsu as the maternal parent. The other crossbred progeny group (CN) was generated with the farmed population from Jiangsu as the paternal parent and the US-introduced protospecies as the maternal parent. The progeny groups resulting from the interbreeding of two parental groups were utilized as the controls. The purebred group derived from the US-introduced protospecies was designated as NN, and the purebred group derived from the farmed population was designated as CC.
In accordance with the principle of pairing one female fish with two male fish, groups of three fish were placed into the breeding facility for spawning based on the hybridization design described above. Each experimental group was established using 20 distinct parental pairs to ensure population genetic diversity and enhance the reliability of the experimental results. After the fish fry were reared to a length of 10 cm, 180 individuals with an average weight of 4.82 ± 0.02 g were selected for the breeding experiment in each group. Twelve cages (2 m × 2 m × 2 m) were installed in the breeding pond, with 60 individuals placed in each cage. Each group consisted of three replicate cages. During the experiment, the dissolved oxygen concentration in the water remained above 6 mg/L, the total ammonia nitrogen was 0.50 ± 0.12 mg/L, and the pH was 7.67 ± 0.26. Fish were fed a complete diet of commercial feed (with a crude protein content of ≥48% and a crude fat content of ≥6%) twice daily (at 8:00 a.m. and 5:00 p.m.). The experiment lasted for 180 days.
2.2. Phenotypic Measurement and Sample Collection
At the end of the 180-day experimental period, the fish were fasted for 24 h, and then 30 fish per replicate (90 fish per group) were randomly selected for analysis. Growth traits, including body length, body height, and body thickness, were measured. Heterosis, expressed as a percentage (H%), was calculated for each phenotypic trait as described by Mai et al. [10], with the following formula:
where F1, PM, and PF represent the average phenotypic values of the hybrid population, paternal line, and maternal line, respectively. The significance of the H% value was tested using the one-way ANOVA followed by the Tukey test, with a significance level set at p < 0.05. After the morphological indices were recorded, 12 fish were randomly selected from each group. The fish were anesthetized using MS-222, and muscle tissue samples (four pieces per parallel) were collected from the dorsal muscle at 2 cm posterior to the head. The muscle samples were stored at −20 °C until nutrient composition analysis. In addition, 15 fish were randomly selected for analyses of digestive enzyme activity and the gut microbiome. The intestinal tissue was promptly dissected, and its surface was moistened with 8.5‰ w/v normal saline solution. Then, the contents of the posterior half of the intestine were collected, immediately frozen in liquid nitrogen, and stored at −80 °C until the gut microbial analysis. After removing the intestinal contents, the cleaned intestinal tissue was frozen in liquid nitrogen and stored at −80 °C until digestive enzyme activity assays.
2.3. Determination of Digestive Enzyme Activity
The intestinal tissue was homogenized with nine volumes of normal saline to prepare a 10% w/v tissue homogenate. The homogenate was centrifuged at 2500 rpm for 15 min at 4 °C using a high-speed centrifuge (Model 17/17R, Thermo Fisher, Waltham, MA, USA). The supernatant was collected, and the activities of amylase, lipase, and proteinase were measured using commercial assay kits (Jiancheng Institute, Nanjing, China) according to the manufacturer’s instructions.
2.4. Proximate Composition of Muscle Samples
Muscle nutrient composition was analyzed as described previously [22]. The muscle moisture content was determined by the drying method, specifically by calculating the difference in weight between before and after drying under 101.3 kPa air pressure at 105 °C. The muscle crude protein content (with a 6.25 N-to-protein conversion factor) was measured using the Kjeldahl method. The crude fat content was assessed by Soxhlet extraction. The muscle ash content was determined after incineration at 550 °C using a furnace (Carbolite Gero, Sheffield, UK).
2.5. Determination of Muscle Amino Acid and Fatty Acid Composition
The amino acid concentration in muscle was determined using an automatic amino acid analyzer (Model LA8080, Hitachi, Tokyo, Japan). Lyophilized muscle samples were hydrolyzed with 6 mol/L hydrochloric acid at 110 °C. After two cycles of drying and redissolution, each sample was dissolved in 1 mL of diluent and the pH was adjusted to 2.2 with sodium citrate buffer. The resulting solution was then filtered through a 0.22 μm membrane prior to analysis. Amino acid concentrations were quantified by calculating the peak areas using the external standard method.
The lyophilized muscle sample was hydrolyzed using a hydrochloric acid solution, then neutralized with an ethanol solution, and extracted with an ether-petroleum ether mixture. The extracts were then concentrated and dried using a rotary evaporator (Model N-1300S, EYELA, Tokyo Rikakikai Co., Ltd., Tokyo, Japan) to obtain total muscle lipids. Fatty acid methyl esters (FAMEs) were prepared by reacting the mixture with 2% w/v sodium hydroxide in formaldehyde, using triglyceride nondecarbonate as the internal standard. Qualitative analysis of FAMEs was performed using a gas chromatograph (Model 7890A, Agilent, Santa Clara, CA, USA) equipped with a capillary column (length: 100 m, inner diameter: 0.25 mm, film thickness: 0.2 μm). The gas chromatography procedure followed the method described by Wang et al. [22]. The fatty acid content was quantified using the peak area percentage method. The significance testing of muscle nutrition was analyzed using SPSS 26.0 (SPSS Inc., Chicago, IL, USA). Homogeneity of variance and normality were performed for the data. One-way analysis of variance (ANOVA) was conducted for the data meeting the criteria. Significant differences were assessed using Duncan’s multiple comparison test, with p < 0.05 considered statistically significant.
2.6. Nucleic Acid Extraction and High-Throughput Sequencing
Gut microbes were analyzed as described by Wang et al. [34]. Genomic DNA was extracted from intestinal microbial samples using the EZNA Stool DNA Kit (D4015, Omega Inc., Norcross, GA, USA). The DNA extracted from three fish from the same group were mixed to prepare the sequencing library (five replicates for each group). Following confirmation of DNA quality, the extracted DNA was amplified using universal primers for the v3-v4 region: 341F (5′-ACTCCTACGGGAGGCAGCA-3′) and 805R (5′-GGACTACHVGGGTWTCTAAT-3′). After amplification, the PCR products were purified using AMPure XP beads (Beckman Coulter Genomics, Danvers, MA, USA) and then quantified using a Qubit fluorometer (Invitrogen, Carlsbad, CA, USA). The sequencing libraries were prepared using the Pacific Biosciences SMRTbellTM Template Prep Kit (Kapa Biosciences, Woburn, MA, USA). Library quality was assessed using an Agilent 2100 Bioanalyzer (Agilent), and sequencing was performed on the Illumina NovaSeq platform (Illumina, San Diego, CA, USA).
2.7. Data Preprocessing
After sequencing, the raw reads were quality-filtered using fqtrim (v0.94) to obtain high-quality clean reads. The sequences were denoised following the QIIME2 DADA2 workflow to eliminate invalid mosaic sequences and extract amplicon sequence variants (ASVs). Subsequently, clustering was performed using Vsearch software v2.3.4., and the feature sequences of each ASV were classified using the classify-sklearn algorithm in QIIME2. The α-diversity of microbiota was calculated based on the ASV abundance table using QIIME2. The Kruskal–Wallis rank sum test and Dunn’s test were used to assess the significance of differences among groups. Additionally, QIIME2 was used to compute the β-diversity of microbiota, and parametric and non-parametric statistical tests were performed using R packages. Multi-level taxonomic classification of the gut microbiota was conducted by referencing the NT-16S database and utilizing SILVA138 (https://www.arb-silva.de/documentation/release-138/; accessed on 6 October 2024), with the significance evaluated accordingly.
2.8. Inheritance Patterns of Microbial Abundance
A Kruskal–Wallis H test was conducted on all gut microbiota detected at the gene level to evaluate differences in their relative abundance between the hybrid and purebred groups. The microflora showing significant differences in abundance between these groups were selected for further analysis of their inheritance patterns. Following the methods of Huang et al. [20] and Mai et al. [10], the microbial taxa were categorized into three patterns of inheritance: additive, non-additive (dominant), or nonadditive (over-dominant). Based on the differences in microbial abundance between the hybrid and purebred groups, the microbiota was categorized into 12 paternal-cross-maternal abundance patterns. Specifically, when there was a significant difference in microbial abundance between the parental groups, and the abundance in hybrid groups was between that in the two parents, this inheritance pattern was referred to as additive (Patterns I and XII). When the microbial abundance in the hybrid population did not significantly differ from that of one parent population but was significantly higher or lower than that in the other parent population, its inheritance pattern was classified as dominant (Patterns II, IV, IX, and XI). When the microbial abundance in the hybrid groups was significantly higher or lower than that of both parental populations, its inheritance pattern was defined as over-dominant (Patterns V, VI, VIII, III, VII, and X).
2.9. Correlation Analysis
Key bacterial genera that exhibited a significantly increased abundance in the hybrid group were identified. The Mantel test was employed to conduct correlation analyses between microbial taxa and the contents of muscle amino acids and fatty acids, as well as digestive enzyme activities. The correlation analysis between specific amino acids or fatty acids was performed using the Spearman rank correlation test. A significant correlation was defined as occurring when p < 0.05.
3. Results
3.1. Enhancement of Growth Performance
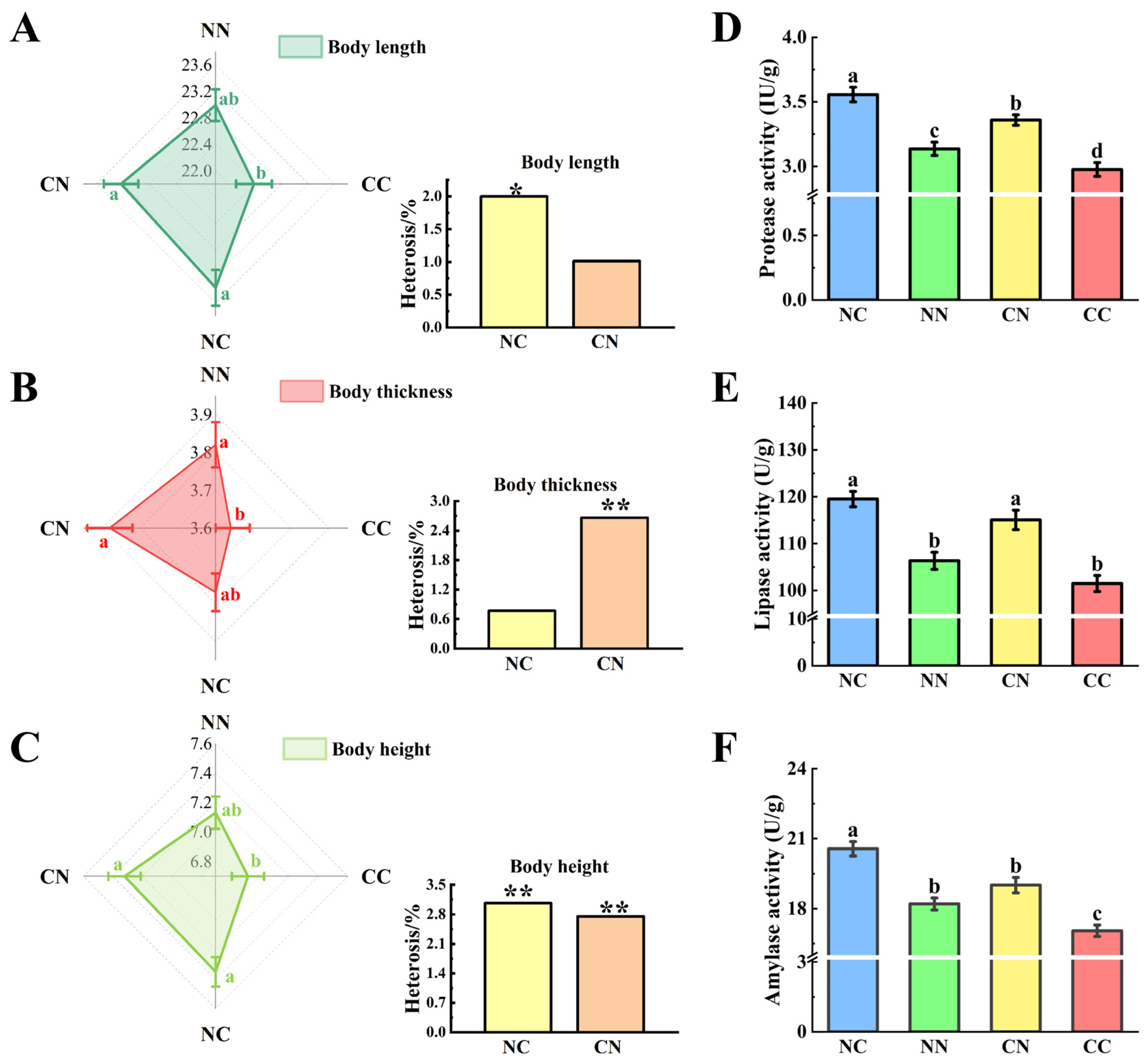
The phenotypic characteristics of the hybrid groups were significantly different from those of the purebred groups (Figure 1A–C). Specifically, the values of body length, body thickness, and body height were markedly higher in the hybrid groups (NC and CN) than in the CC group (p < 0.05). The values of these phenotypes were also higher in the NC and CN than in the NN group; however, the differences were not statistically significant (p > 0.05). The calculation of heterosis rates for each phenotype revealed significant heterosis of body length and body height in the NC group (p < 0.05); and body thickness and body height in the CN group (p < 0.05). Notably, body height exhibited the highest heterosis rate (3%).
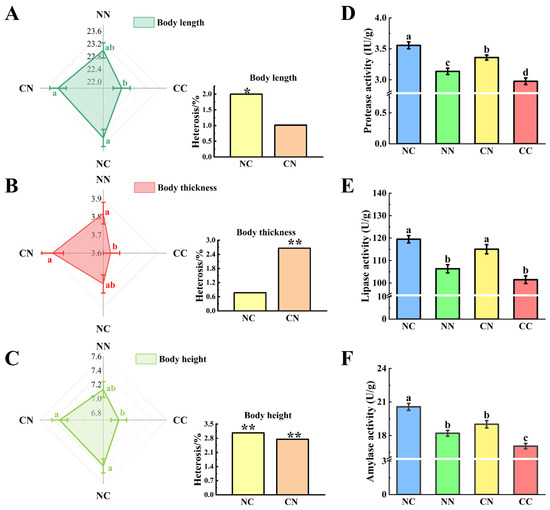
Figure 1.
Comparative analysis of growth and digestive enzyme activities between hybrid and purebred groups. Assessment of differences and heterosis (%) in terms of body length (A), body thickness (B) and body height (C). Different small letters indicate statistically significant differences among four groups. Values marked with * and ** indicate statistically significant significance at p < 0.05 and p < 0.01. Enzyme activity of (D) protease, (E) lipase and (F) amylase. NC and CN, hybrid groups; CC and NN, purebred groups.
We analyzed the digestive enzyme activities in gut, including protease activity (Figure 1D), lipase activity (Figure 1E), and amylase activity (Figure 1F). The enzymatic activities of the hybrid group were significantly different from those of the purebred group (p < 0.05). Notably, the activities of all three digestive enzymes were significantly higher in the NC group than in the two purebred groups (p < 0.05); and the activities of protease and lipase were significantly higher in the CN group than in the two purebred groups (p < 0.05).
3.2. Differences in Proximate Composition of Muscle
Proximate analyses of muscle samples revealed that (Table 1) the water and protein contents in the muscle were similar among the four groups, with no significant differences (p > 0.05). The fat content in the muscle was higher in the hybrid groups than in the purebred groups (p < 0.05). The ash content was significantly higher in the NC group than in the CN group (p < 0.05).

Table 1.
Muscle proximate composition of four populations of largemouth bass.
3.3. Differences in Amino Acid and Fatty Acid Composition of Muscle
The amino acid composition of the muscle of largemouth bass in the four populations is shown in Table 2. With the exception of Gly, all the other amino acids showed significant differences in their contents in muscle among the four groups (p < 0.05). The amino acid profiles of the NC group and NN group were similar, and all amino acids were present at higher levels in these groups than in the CC group (p < 0.05). The levels of Ser, Glu, Tyr, Arg, Pro, Cys, and Met did not differ significantly between the CN group and the NC or NN groups (p > 0.05). However, the levels of other amino acids were significantly lower in the CN group than in the NC and NN groups (p < 0.05). In addition, the levels of essential amino acids, delicious amino acids (DAA), and total amino acids were significantly higher in the NC and NN groups than in the CN and CC groups (p < 0.05). Notably, the CC group exhibited the lowest levels of amino acids, which were significantly lower than their corresponding levels in the CN group (p < 0.05). The ratio of essential amino acids to total amino acids was comparable across all groups, with no significant differences (p > 0.05).
Table 2.
Muscle amino acid structure of various largemouth bass populations.
The fatty acid composition of the muscle in largemouth bass for each group is shown in Table 3. A total of 15 fatty acids were identified in this study. The saturated fatty acid content was significantly higher (p < 0.05) in the NC group than in the other groups. The monounsaturated fatty acid content was highest in the CN group, significantly higher than that in the NC, NN, and CC groups (p < 0.05). Additionally, the polyunsaturated fatty acid content in the muscle was significantly lower in the hybrid groups than in the purebred groups (p < 0.05). The fatty acid composition profile of the NC group was similar to that of the NN group, but was similar between the CN group and the CC group. Compared with the other groups, the NC group demonstrated significantly lower levels of n-6 polyunsaturated fatty acids (p < 0.05), and the CN group exhibited significantly reduced levels of n-3 polyunsaturated fatty acids (p < 0.05).
Table 3.
Muscle fatty acid structure of various largemouth bass populations (%).
3.4. Differences in Gut Microbial Community Composition
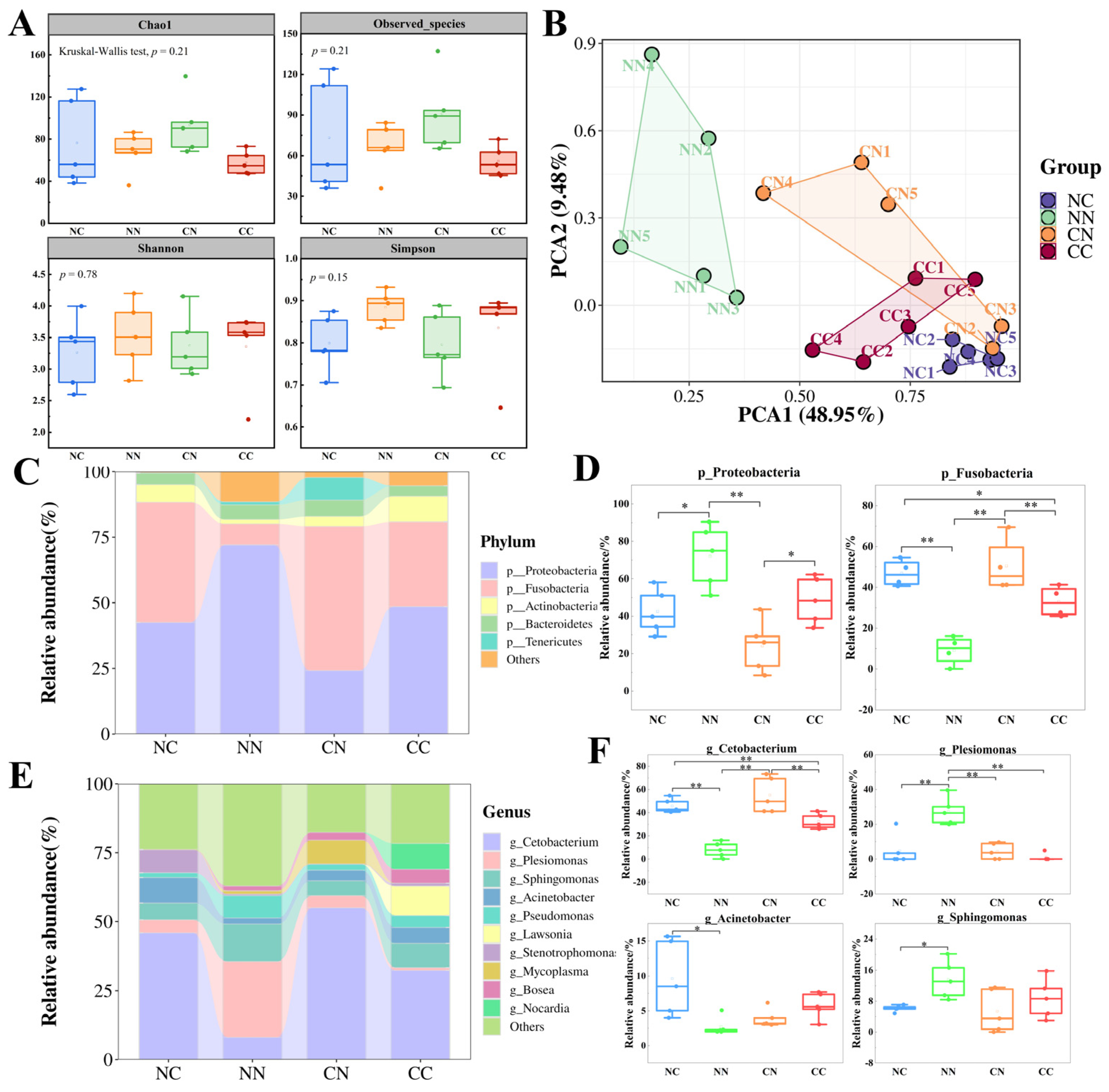
The α-diversity of the intestinal microbial communities was compared on the basis of the Observed_species index and Chao1 index, which are indicators of the species richness of gut microbiota (Figure 2A). The values of these indexes were similar in all the groups, with no significant differences (p > 0.05). Shannon’s index and Simpson’s index, which are indexes of biodiversity, were higher in the CC and NN groups than in the NC and CN groups, but the differences were not statistically significant (p > 0.05). We conducted a principal coordinates analysis based on weighted UniFrac distances to evaluate the β-diversity of intestinal microbiota. As shown in Figure 2B, the samples from the four groups formed distinct clusters. Specifically, the NN group formed a cluster that was separate from the other groups, whereas the clusters for the NC, CN, and CC groups were located close together, with partial overlap between the CC and CN groups. These findings suggest that the structure of the intestinal microbial community differed among the four groups. Consequently, we further investigated the composition of gut microbiota in the four groups.
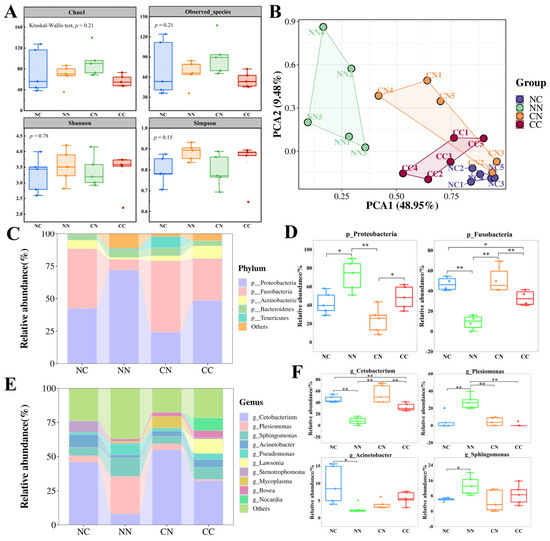
Figure 2.
Comparison of gut microbial community composition between hybrid groups and purebred groups. (A) Analysis of gut microbial α-diversity in hybrid and purebred largemouth bass. Measures of α-diversity included Chao1 index, observed species index, Shannon index and Simpson index. (B) Principal component analysis based on weighted_unifrac distance calculation. (C) Overall composition of microbial community at phylum level; top 5 taxa in terms of relative abundance are shown. (D) Relative abundance of dominant phylum-level bacteria. * and ** indicate statistically significant difference at p < 0.05 and p < 0.01. (E) Overall composition of microbial communities at genus level. (F) Relative abundance of dominant bacterial genera.
First, the structure of the intestinal microbiome in the hybrid groups and purebred groups was analyzed at the phylum level (Figure 2C, Table S1). The results showed that Proteobacteria and Fusobacteria were the most important microbial taxa in the largemouth bass intestine. The abundance of these two phyla differed significantly between the hybrid groups and the purebred groups (p < 0.05, Figure 2D). Specifically, Proteobacteria exhibited the highest relative abundance in the NN group (72%), which was significantly higher than that in the other groups (p < 0.05). The relative abundance of Proteobacteria was lowest in the CN group (24.16%). The relative abundance of Fusobacteria was significantly higher in the NC group (45.87%) and CN group (54.99%) than in the NN group (8.05%) and CC group (32.35%) (p < 0.05). Additionally, the NC and CC groups exhibited a higher abundance of Actinobacteria, while the CN group exhibited a higher abundance of Tenericutes.
Next, the composition of gut microbiota was analyzed at the genus level (Table S2). Cetobacterium was the predominant genus in the largemouth bass intestine (Figure 2E); its relative abundance was significantly higher (p < 0.05) in the NC and CN groups (45.87% and 54.98%, respectively) than in the NN and CC groups (8.04% and 32.35%, respectively). The highest relative abundance of Plesiomonas was in the NN group, at 27.44%, which was significantly higher (p < 0.05) than its abundance in the two hybrid groups and the CC group. Additionally, Stenotrophomonas accounted for 8.3% of gut bacteria in the NC population, whereas its abundance was nearly negligible in the other groups. Except for Cetobacterium and Plesiomonas, among the four genera with the highest abundance (Figure 2F), Acinetobacter exhibited the highest abundance in the NC group, significantly higher than in the NN group (p < 0.05). Sphingomonas showed the highest abundance in the NN group, significantly higher than in the NC group (p < 0.05). However, the abundance of Sphingomonas did not differ significantly between the CN group and the CC group (p > 0.05).
3.5. Inheritance Patterns of the Microbiome in Hybrid Groups
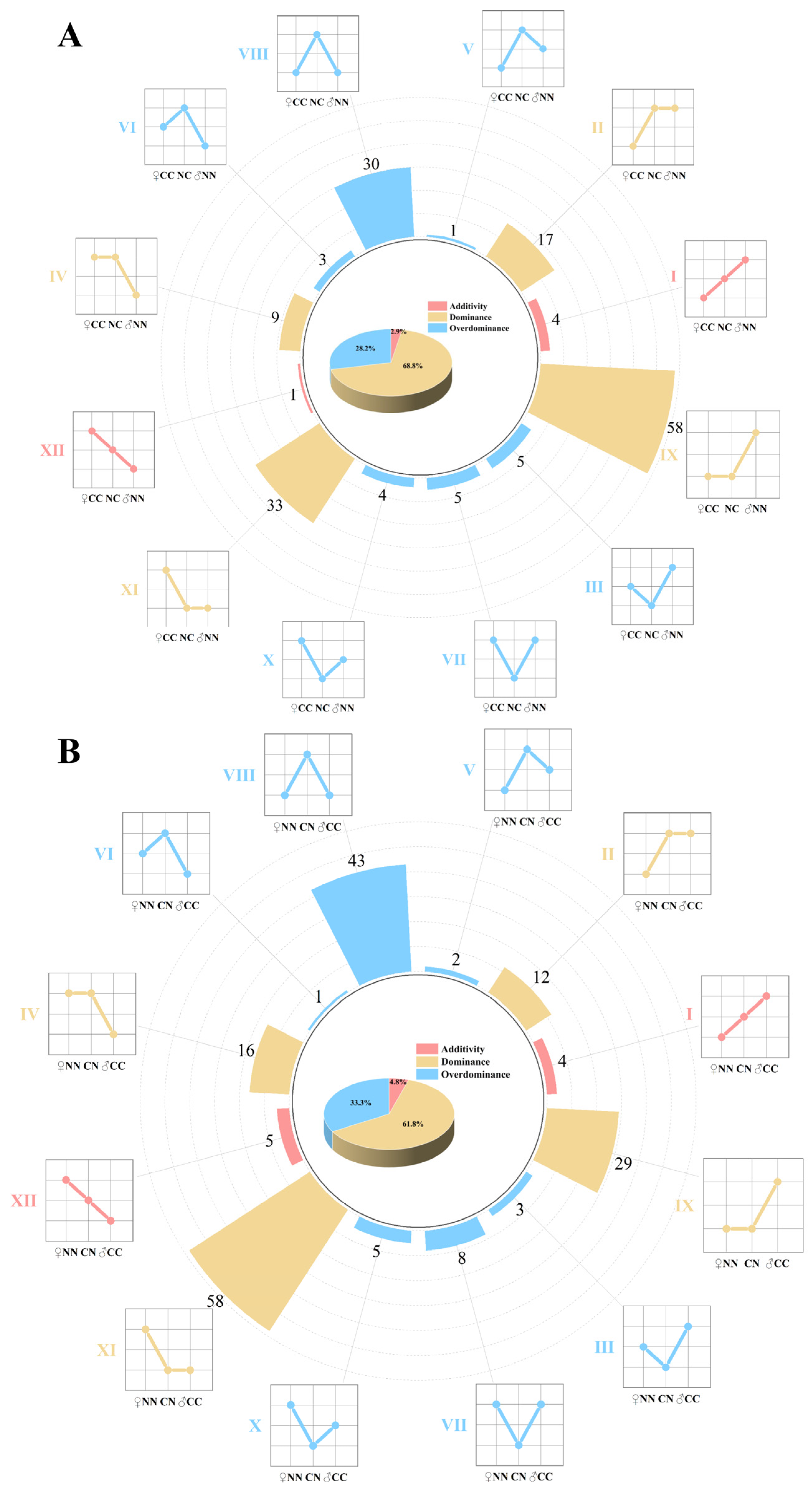
The relative abundance of intestinal microbes in four groups of largemouth bass was analyzed at the genus level. A total of 228 genera with significant differences in abundance among the groups were identified. Based on the variations in abundance among the different groups, 170 bacterial genera in the NC group and 186 bacterial genera in the CN group were categorized into 12 distinct abundance patterns. These 12 patterns were further classified into three inheritance types: additive (I and XII), dominant (II, IV, IX, and XI), and over-dominant (III, V, VI, VII, VIII, and X). In the NC group (Figure 3A), five, 117, and 48 microbial taxa were categorized into additive, dominant, and over-dominant modules, respectively, and these accounted for 2.9%, 68.8%, and 28.2% of the total bacterial community, respectively. In the CN group (Figure 3B), nine, 115, and 62 microbial taxa were classified into additive, dominant, and over-dominant modules, representing 4.8%, 61.8%, and 33.3% of the total count, respectively. Notably, the dominant modules were most prevalent in the hybrid groups.
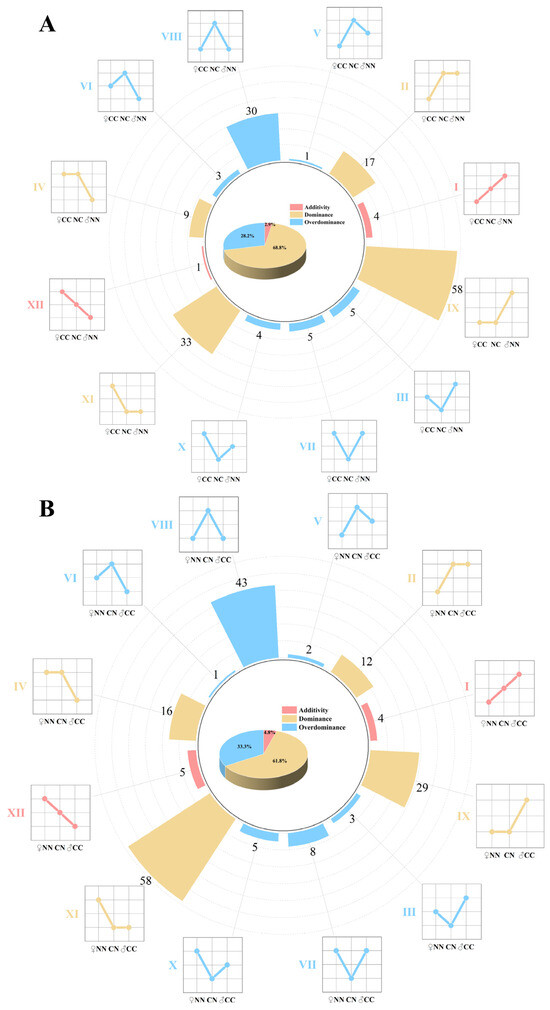
Figure 3.
Inheritance patterns of gut microbiome. (A) Inheritance patterns between NC group and parental lines. (B) Inheritance patterns between CN group and purebred group. Microbiome was divided into 12 inheritance patterns based on differences in abundance; additive (I, XII), dominant (II, IV, IX and XI), and over-dominant (III, V, VI, VII, VIII and X). Line charts illustrate differences in abundance between hybrid and purebred groups.
3.6. Functional Analysis of Microbiome
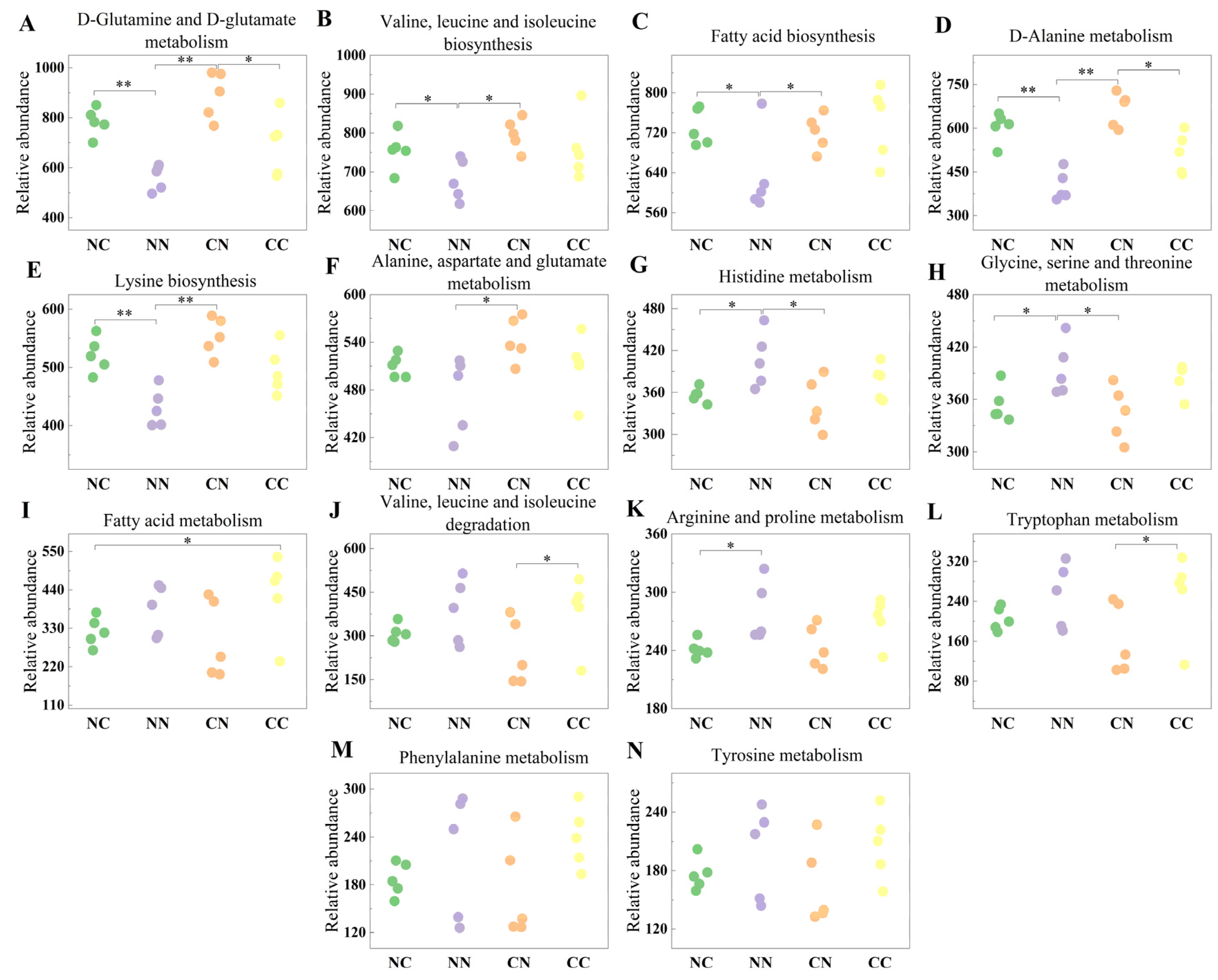
To further investigate the roles of gut microbiota, we employed PICRUSt2 software v2.6.0. to derive relative gene abundance information from the DNA sequence data. The obtained sequence information was compared against the KEGG database (Table S3). The results showed that the gut microbiota was associated with numerous pathways regulating amino acid and fatty acid metabolism (Figure 4). Specifically, compared with the purebred groups, the hybrid groups showed an increased abundance of microbial genes involved in D-Glutamine and D-glutamate metabolism; valine, leucine, and isoleucine biosynthesis; fatty acid biosynthesis; D-Alanine metabolism; and lysine biosynthesis (p < 0.05). The relative abundance of microbial genes involved in histidine metabolism and glycine, serine, and threonine metabolism was significantly higher in the NN group than in the hybrid groups (p < 0.05). In addition, the hybrid and purebred groups exhibited differences in the relative abundance of microbial genes involved in other pathways associated with amino acid metabolism, although these differences were not statistically significant (p > 0.05).
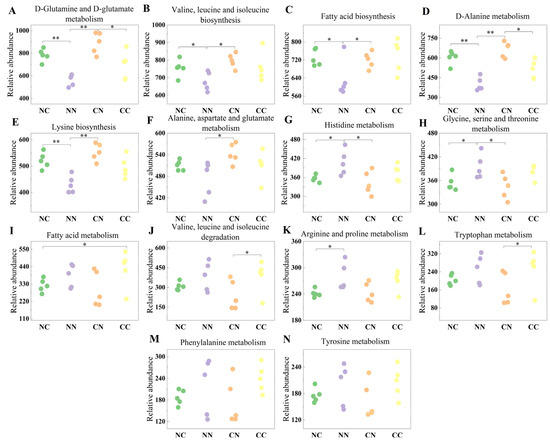
Figure 4.
Functional analysis of gut microbial genes showing significant differences in abundance in between hybrid and purebred groups. Relative abundance of microbial genes involved in (A) D-Glutamine and D-glutamate metabolism; (B) Valine, leucine and isoleucine biosynthesis; (C) Fatty acid biosynthesis; (D) D-Alanine metabolism; (E) Lysine biosynthesis; (F) Alanine, aspartate and glutamate metabolism; (G) Histidine metabolism; (H) Glycine, serine and threonine metabolism; (I) Fatty acid metabolism; (J) Valine, leucine and isoleucine degradation; (K) Arginine and proline metabolism; (L) Tryptophan metabolism; (M) Phenylalanine metabolism; (N) Tyrosine metabolism. Values marked with * and ** indicate statistical significance of p < 0.05 and p < 0.01.
3.7. Correlations Between Gut Microbiome and Muscle Nutrient Composition
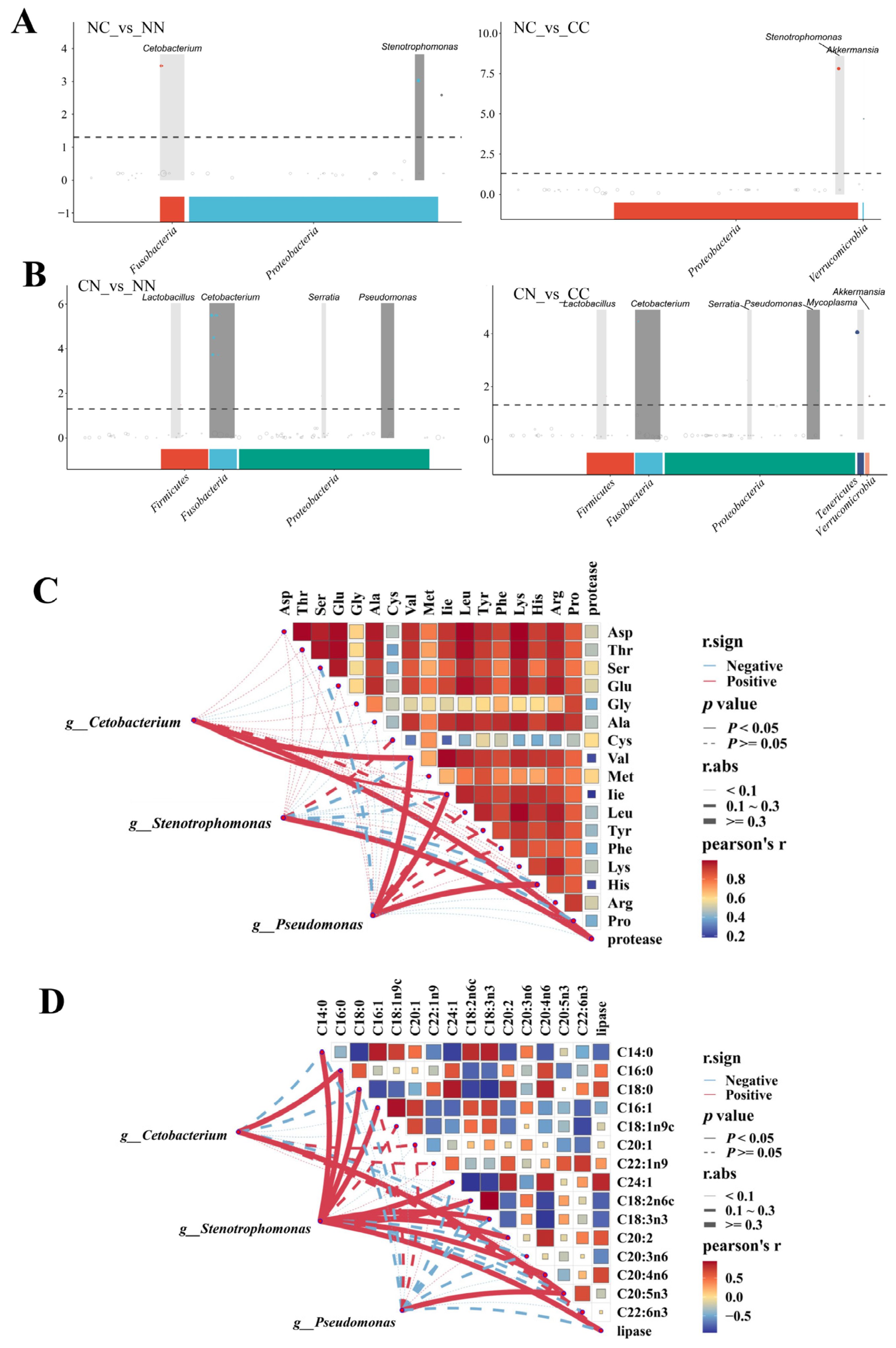
Based on the MetagenomeSeq analysis, we identified key microbiota taxa exhibiting significantly increased abundance in the hybrid groups. As shown in Figure 5A, the abundance of Cetobacterium was significantly higher in the NC group than in the purebred groups, and the abundance of Stenotrophomonas was significantly higher in the NC group than in the NN group (p < 0.05). Both Cetobacterium and Pseudomonas were significantly more abundant (p < 0.01) in the CN group than in the purebred group (Figure 5B) and Lactobacillus and Serratia were significantly more abundant in the CN group than in the purebred group (p < 0.05). The majority of microbiota taxa showing significantly higher abundance in the hybrid groups than in the purebred groups belonged to the Proteobacteria and Fusobacteria phyla.
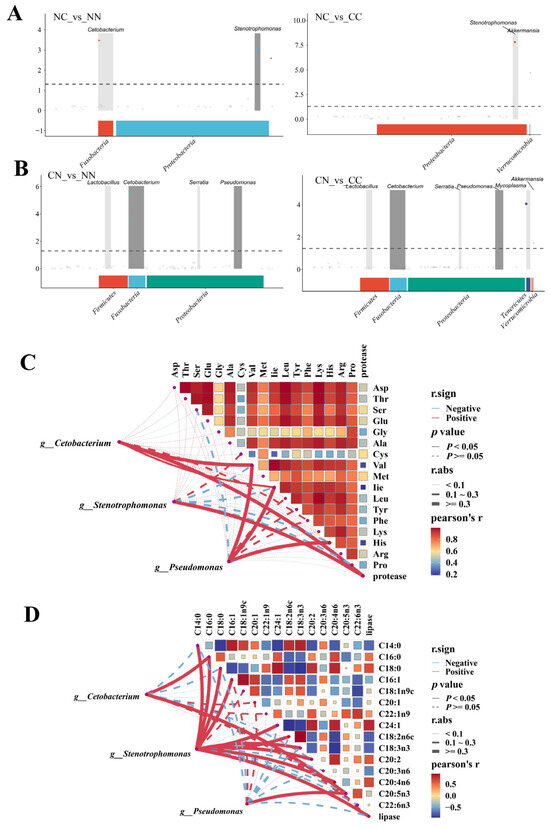
Figure 5.
Associations between microbiome and muscle nutrients. (A) Manhattan map based on MetagenomeSeq analysis between NC group and parental lines. (B) Manhattan map based on MetagenomeSeq analysis between CN group and parental lines. Horizontal axis represents annotation of ASV at phylum level; vertical axis indicates −log10(adj-Pvalue) value. The dashed line denotes threshold for significance level of the difference. Correlation network heatmap represents relationships between genera and (C) amino acids and (D) fatty acids. Correlation analysis of network was conducted using Mantel’s test; correlation analysis of the heat map was conducted by Spearman’s test. Red and blue indicate positive and negative correlations, respectively.
Cetobacterium, Pseudomonas, and Stenotrophomonas were selected to investigate the correlations between microbial genera and the contents of amino acids and fatty acids and the activity of digestive enzymes. For these three microbial taxa, their abundance was significantly associated with the amino acid content in the muscle of largemouth bass (Figure 5C). Specifically, the abundance of Cetobacterium was significantly correlated with Val content, while the abundance of Pseudomonas was significantly correlated with the contents of Val, Ile, and His (p < 0.05). Both Cetobacterium and Stenotrophomonas were significantly correlated with protease activity (p < 0.05). The abundance of Stenotrophomonas exhibited significant correlations with multiple fatty acids (p < 0.05, Figure 5D), including C14:0, C16:0, C18:0, C16:1, C24:1, C18:2n6c, C18:3n3, C20:2, C14:3, C14:2, and C20:4n6, as well as lipase activity. Both Cetobacterium and Pseudomonas were significantly associated with C16:0 and C20:5n3 (p < 0.05). All of the detected correlations were positive. In addition, in pairwise comparisons, most amino acids were strongly positively correlated with each other, whereas fatty acids showed relatively weak correlations.
4. Discussion
Hybridization allows for the combination of alleles from parents with different genotypes, thereby enhancing the genetic diversity of the crossbred progeny, and allowing them to inherit superior traits from both parents or exhibit traits superior to those of both parents [11]. The evaluation of the heterosis rate of target traits and analyses of the underlying genetic mechanisms have biological significance and are also relevant for production. Heterosis has been exploited to enhance economically important traits, such as growth performance and nutritional quality. The heterosis rate serves as a valuable metric for assessing phenotypic heterosis [10,35]. In this study, the H% values of the NC group and CN group reached statistically significant levels in terms of body length, body thickness, and body height. These findings suggest that the hybrid fish in this study exhibited positive heterosis.
The muscle nutrient profile is an important indicator of fish product quality [36]. Previous studies have demonstrated that the muscle nutrient composition of aquacultured animals can be improved through hybridization [37,38]. In this study, the lipid content of muscle was significantly higher in the hybrid groups than in the purebred groups. Based on the results of our previous study, we propose that the increased muscle lipid content in the hybrid groups may enhance the flavor of the meat [22]. The amino acid profiles of muscle were similar between the NC group and NN group, but these two groups showed significantly higher total amino acid and essential amino acid contents compared with the CN group and the CC group. Compared with the parental lines, the hybrid groups exhibited significantly lower polyunsaturated fatty acid contents in muscle. Additionally, the level of monounsaturated fatty acids in muscle was significantly lower in the NC group than in the other groups. Therefore, the NC group in this study inherited the superior amino acid composition from the paternal line and exhibited a more favorable amino acid profile compared with that of the maternal line. However, in the CN group, the amino acid and fatty acid contents in muscle generally demonstrated negative heterosis, a phenomenon that has been reported in other studies [39,40]. It is important to understand the mechanisms of positive heterosis, but equally important to understand those of negative heterosis. Considering the critical role of gut microbiota in nutrient digestion and absorption, we expected that gut microbial functions may differ among the various groups, and these differences may help to explain the heterosis of muscle nutrient profiles.
Because the gut microbiota is influenced by factors such as diet and environment, its composition and structure exhibit significant variations among populations. The host’s genetic information is one of the critical factors influencing the structure of the gut microbial community [14,15]. Previous studies have demonstrated that hybrid aquatic species, including hybrid abalone (Haliotis laevigata × Haliotis rubra) [41], hybrid Chinook salmon (Oncorhynchus tshawytscha) [42], and whitefish (Coregonus clupeaformis) [43], display markedly different gut microbial communities compared with those of their parental lines. In this study, there was no significant difference in the α-diversity of the gut microbial community among the four groups, suggesting that the gut microbiota of largemouth bass is more stable than those of other aquatic animals. In another study, the gut microbes of fish subjected to stress conditions displayed relatively stable richness and evenness [34], a finding that aligns with this study. The β diversity is a key indicator of the composition and structure of gut microbiota [44]. Based on the consistency of the water environment maintained in all groups, we conclude that despite the uniformity in microbial abundance observed across all groups, the composition of the gut microbial community differed between the hybrid groups and the purebred groups. Regarding the influence of the water environment on the composition and structure of the gut microbiota in largemouth bass, we will develop another research protocol to conduct a comprehensive and systematic analysis. Notably, digestive enzyme activity was significantly higher in the hybrid groups than in the purebred groups. Digestive enzymes play critical roles in the absorption and utilization of nutrients [45]. We found that certain microbial taxa exhibited significant positive correlations with digestive enzyme activity, suggesting that changes in the abundance of particular gut microbes in the hybrid groups may underlie the increased digestive enzyme activity and the changes in muscle nutrient composition.
Non-additive inheritance plays a crucial role in the formation of heterosis [46,47]. To determine its contribution to heterosis of largemouth bass, we categorized the bacterial genera showing significant differences in abundance between the hybrid and purebred groups into 12 genetic inheritance patterns, in three classes: additive, non-additive (dominant), and nonadditive (over-dominant). The majority of microbial taxa in the two hybrid groups showed non-additive inheritance patterns, consistent with the findings of Mai et al. [10] and Huang et al. [20]. The non-additive inheritance of gut microbiota in largemouth bass may be associated with the development of heterosis. Notably, the proportion of bacteria showing non-additive inheritance was lower in the NC group (2.9%) than in the CN group (4.8%). Accordingly, compared with the CN group, the NC group showed marginally superior growth performance and muscle nutrient composition. This shows that non-additive inheritance of gut microbiota may significantly affect the growth and muscle nutrient composition of hybrid largemouth bass [48,49]. Functional annotation analyses showed that a large number of microbial taxa potentially contribute to the synthesis and metabolism of amino acids and fatty acids, with significant differences observed in their relative abundance across groups. Thus, the differences in microbial abundance in the hybrid groups compared with the purebred groups may serve as a critical factor contributing to the differences in muscle nutrient composition between these groups.
Proteobacteria and Fusobacteria are significant components of the gut microbiota in bony fish [50,51]. In this study, these two phyla were identified as the dominant microbial groups among the four groups of largemouth bass, with their combined relative abundance exceeding 70%. Tan et al. [52] found that Proteobacteria was the predominant microbial group in largemouth bass. Similarly, Jin et al. [53] found that the relative abundance of Proteobacteria reached over 90% in the microbiota of largemouth bass. These findings underscore the absolute dominance of Proteobacteria and Fusobacteria in the gut microbiota of largemouth bass. Proteobacteria include numerous genera or strains that are pathogenic or opportunistic pathogens, which are generally considered as unfavorable for fish survival [54,55]. Proteobacteria are thought to negatively affect their hosts because members of this phylum contain a large number of genes encoding virulence factors and antibiotic resistance factors [56]. However, this conclusion warrants further scrutiny. The physiological roles and functions of Proteobacteria in fish have not yet been comprehensively investigated. Other studies have demonstrated a direct proportional relationship between the abundance of Proteobacteria and the amino acid and fatty acid contents in fish [57]; and a relationship between the high abundance of Proteobacteria in short-intestinal fish and enhanced metabolism of amino acids and lipids [58]. These findings suggest that Proteobacteria may play a role in metabolic processes related to energy and material metabolism in fish. Fusobacteria have been demonstrated to exhibit a positive correlation with the levels of lipids, amino acids, and other nutrients in fish [59], and may potentially contribute to the bioaccumulation of toxic substances [52]. In this study, the Proteobacteria and Fusobacteria in the gut microbiota of largemouth bass were likely involved in the biological processes associated with amino acid and fatty acid synthesis and metabolism. Notably, previous studies have shown that the abundance of Proteobacteria and Fusobacteria is generally negatively correlated [52,54,60]. Li et al. [61] proposed that Proteobacteria and Fusobacteria/Firmicutes/Bacteroides can be categorized into two distinct functional groups, which exhibit functional differences in carbohydrate utilization, short-chain fatty acid production, virulence factors, and antibiotic resistance. Furthermore, Li et al. [61] suggested that under non-stress conditions, the ratio of functional group 2 (Fusobacteria, Firmicutes, Bacteroides) to functional group 1 (Proteobacteria) is positively correlated with fish growth. These findings suggest that there are interactions between Proteobacteria and Fusobacteria. In this study, hybrid populations with a higher relative abundance of Fusobacteria exhibited similar characteristics.
We also conducted an in-depth analysis of the differences in microbiome composition at the genus level between the hybrid and purebred groups. We detected significantly higher abundance of Cetobacterium, Stenotrophomonas, Pseudomonas, Lactobacillus, and Serratia in the hybrid groups than in the purebred groups. These microbial taxa may be associated with heterosis. Previous studies have demonstrated that Cetobacterium is able to synthesize essential amino acids, including Leu, Ile, Val, and Gly. Furthermore, it can utilize carbohydrates for polysaccharide biosynthesis and improve carbohydrate utilization in fish [62,63]. Stenotrophomonas, which was significantly enriched in the NC group, may have promoted the growth and development of fish in this group. Although some studies have suggested that Stenotrophomonas may be a pathogen associated with certain fish diseases [64,65], there is probably a more complex relationship at play. In fact, a positive correlation was detected between the abundance of Stenotrophomonas and the growth rate of fish [66]. Mondal et al. [67] demonstrated that the abundance of Stenotrophomonas increased significantly in infected rohu (Labeo rohita). Furthermore, Stenotrophomonas quickly became a dominant genus in the gut of sheepshead minnow (Cyprinodon variegatus) during exposure to environmental stress [68]. These findings indicate that Stenotrophomonas may enhance the host’s growth and/or play a role in its stress response. Similarly to Stenotrophomonas, Pseudomonas is commonly recognized as a pathogen affecting aquacultured animals [69]. However, Pseudomonas has also been shown to exhibit probiotic properties [70].
The mechanisms by which particular microbes influence the growth and metabolism of fish warrant further investigation. In this study, Cetobacterium, Stenotrophomonas, and Pseudomonas exhibited positive correlations with the contents of multiple amino acids and fatty acids. Notably, the abundance of Stenotrophomonas was significantly positively correlated with the levels of various fatty acids. The relationship between the gut microbiota and muscle nutrient profiles has been well established in previous studies [71]. Our results indicate that these microbial taxa, particularly Stenotrophomonas, may play crucial roles in determining the composition of amino acids and fatty acids in the muscle of largemouth bass. Interestingly, these three microbial taxa belong to Proteobacteria and Fusobacteria, emphasizing the important role of these phyla in promoting the growth of largemouth bass and in determining the amino acid and fatty acid profiles of its muscle tissue. However, the precise mechanisms by which the aforementioned microbial taxa influence heterosis remain unclear. In our subsequent research, we will use the findings of this study to investigate the roles of Proteobacteria and Firmicutes in heterosis, exploring potential pathways for addressing heterosis-related challenges via gut microbiota.
5. Conclusions
Crossbred progeny derived from two geographically isolated parental lines exhibited significant heterosis in several phenotypic traits, including body length, body thickness, and body height. Compared with the purebred groups, the crossbred progeny demonstrated enhanced digestive enzyme activity, altered muscle nutrient composition, and differences in the abundance of gut microbes, primarily following a non-additive pattern of inheritance. These changes were significantly different from those in the purebred groups, and were significantly correlated with variations in phenotype and muscle nutrient profiles. The relative abundance of microorganisms harboring genes enriched in KEGG pathways associated with amino acid and fatty acid synthesis and metabolism exhibited significant differences between the hybrid and purebred groups, which contributed to the differences in muscle nutrient profiles between these groups. Proteobacteria and Fusobacteria were identified as the dominant bacterial phyla in largemouth bass. These phyla are crucial for various physiological and biochemical processes, including growth and metabolism. The abundance of Cetobacterium, Stenotrophomonas, and Pseudomonas exhibited significant correlations with amino acids and fatty acids, highlighting their importance in promoting heterosis of hybrid largemouth bass. However, the mechanisms by which the aforementioned microbial taxa influence heterosis require further in-depth investigation.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/microorganisms13071449/s1.
Author Contributions
J.H. (Jixiang Hua): Writing—original draft, Software, Investigation. Q.W.: Writing—review and editing, Software, Investigation. Y.T.: Validation, Supervision. H.S.: Investigation, Formal analysis. S.L.: Software, Methodology, Y.Z.: Validation. W.C.: Supervision. K.L. and J.H. (Jie He): Funding acquisition, Project administration. J.Q.: Writing—review and editing, Validation, Formal analysis, Resources, Project administration, Funding acquisition, Data curation, Conceptualization. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported by The Project of Seed Industry Revitalization in Jiangsu Province, China (JBGS [2021] 130), Major Technology Collaborative Promotion Plan for Largemouth Bass Industry in Jiangsu Province (2022-ZYXT-07), The Provincial Key Research and Development Program of Anhui (No. 202423l10050003), Central Public-interest Scientific Institution Basal Research Fund, CAFS (NO. 2023TD11), The key research and development plan of Guangxi (AB241484006).
Institutional Review Board Statement
The study was conducted according to the guidelines of the Declaration of Helsinki and was approved by the Bioethical Committee of the Freshwater Fisheries Research Center (FFRC), Chinese Academy of Fishery Sciences (2013863BCE, 6 August 2013).
Informed Consent Statement
Not applicable.
Data Availability Statement
The original contributions presented in this study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Conflicts of Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Liu, Q.; Long, Y.; Li, B.; Zhao, L.; Luo, J.; Xu, L.; Luo, W.; Du, Z.; Zhou, J.; Yang, S. Rice-shrimp culture: A better intestinal microbiota, immune enzymatic activities, and muscle relish of crayfish (Procambarus clarkii) in Sichuan Province. Appl. Microbiol. Biotechnol. 2020, 104, 9413–9420. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Sun, Y.; Ni, A.; Li, Y.; Yuan, J.; Ma, H.; Wang, P.; Shi, L.; Zong, Y.; Zhao, J.; et al. Research Note: Heterosis for egg production and oviposition pattern in reciprocal crossbreeds of indigenous and elite laying chickens. Poult. Sci. 2022, 101, 102201. [Google Scholar] [CrossRef] [PubMed]
- Qiao, J.; Li, K.; Miao, N.; Xu, F.; Han, P.; Dai, X.; Abdelkarim, O.F.; Zhu, M.; Zhao, Y. Additive and Dominance Genome-Wide Association Studies Reveal the Genetic Basis of Heterosis Related to Growth Traits of Duhua Hybrid Pigs. Animals 2024, 14, 1944. [Google Scholar] [CrossRef]
- Yu, D.; Gu, X.; Zhang, S.; Dong, S.; Miao, H.; Gebretsadik, K.; Bo, K. Molecular basis of heterosis and related breeding strategies reveal its importance in vegetable breeding. Hortic. Res. 2021, 8, 120. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Sugi, N.; Nojima, Y.; Entani, T.; Shiba, H. Synchronization of Arabidopsis flowering time and vegetative growth stage via FT overexpression can reveal inherent heterosis due to heterozygosity in intraspecific hybrids. Biosci. Biotechnol. Biochem. 2023, 87, 877–882. [Google Scholar] [CrossRef]
- Zhang, S.; Huang, X.; Han, B. Understanding the genetic basis of rice heterosis: Advances and prospects. Crop. J. 2021, 9, 688–692. [Google Scholar] [CrossRef]
- Xiao, Q.; Huang, Z.; Shen, Y.; Gan, Y.; Wang, Y.; Gong, S.; Lu, Y.; Luo, X.; You, W.; Ke, C. Transcriptome analysis reveals the molecular mechanisms of heterosis on thermal resistance in hybrid abalone. BMC Genom. 2021, 22, 650. [Google Scholar] [CrossRef]
- Yang, H.; Li, Q. The DNA methylation level is associated with the superior growth of the hybrid crosses in the Pacific oyster Crassostrea gigas. Aquaculture 2022, 547, 737421. [Google Scholar] [CrossRef]
- Liang, Y.; Zhang, G.; Jiang, G.; Hu, Y.; Fang, J.; Chi, Y.; Xu, C.; Liu, W.; Liu, H.; Li, Q. Hybridization between “Haida No. 1” and Orange-shell line of the Pacific oyster reveals high heterosis in survival. Aquaculture 2022, 551, 737945. [Google Scholar] [CrossRef]
- Mai, C.; Wen, C.; Xu, Z.; Xu, G.; Chen, S.; Zheng, J.; Sun, C.; Yang, N. Genetic basis of negative heterosis for growth traits in chickens revealed by genome-wide gene expression pattern analysis. J. Anim. Sci. Biotechnol. 2021, 12, 52. [Google Scholar] [CrossRef]
- Charlesworth, D.; Willis, J.H. The genetics of inbreeding depression. Nat. Rev. Genet. 2009, 10, 783–796. [Google Scholar] [CrossRef] [PubMed]
- Moore, S.; Lukens, L. An Evaluation of Arabidopsis thaliana Hybrid Traits and Their Genetic Control. G3 Genes|Genomes|Genetics 2011, 1, 571–579. [Google Scholar] [CrossRef] [PubMed]
- McFall-Ngai, M.; Hadfield, M.G.; Bosch, T.C.G.; Carey, H.V.; Domazet-Lošo, T.; Douglas, A.E.; Dubilier, N.; Eberl, G.; Fukami, T.; Gilbert, S.F.; et al. Animals in a bacterial world, a new imperative for the life sciences. Proc. Natl. Acad. Sci. USA 2013, 110, 3229–3236. [Google Scholar] [CrossRef] [PubMed]
- Youngblut, N.D.; Reischer, G.H.; Walters, W.; Schuster, N.; Walzer, C.; Stalder, G.; Ley, R.E.; Farnleitner, A.H. Host diet and evolutionary history explain different aspects of gut microbiome diversity among vertebrate clades. Nat. Commun. 2019, 10, 2200. [Google Scholar] [CrossRef]
- Cui, X.; Zhang, Q.; Zhang, Q.; Zhang, Y.; Chen, H.; Liu, G.; Zhu, L. Research Progress of the Gut Microbiome in Hybrid Fish. Microorganisms 2022, 10, 891. [Google Scholar] [CrossRef]
- Hembrom, P.S.; Barik, S.; Deepthi, M.; Kannoth, S.; Grace, T. Influence of gut microbiome on health and development of penaeid shrimps. Aquat. Sci. 2023, 86, 4. [Google Scholar] [CrossRef]
- Brucker, R.M.; Bordenstein, S.R. Speciation by symbiosis. Trends Ecol. Evol. 2012, 27, 443–451. [Google Scholar] [CrossRef]
- Gebiola, M.; Kelly, S.E.; Hammerstein, P.; Giorgini, M.; Hunter, M.S. “Darwin’s corollary” and cytoplasmic incompatibility induced by Cardinium may contribute to speciation in Encarsia wasps (Hymenoptera: Aphelinidae). Evolution 2016, 70, 2447–2458. [Google Scholar] [CrossRef]
- Li, Z.; Wright, A.G.; Si, H.; Wang, X.; Qian, W.; Zhang, Z.; Li, G. Changes in the rumen microbiome and metabolites reveal the effect of host genetics on hybrid crosses. Environ. Microbiol. Rep. 2016, 8, 1016–1023. [Google Scholar] [CrossRef]
- Huang, Q.; Wen, C.; Gu, S.; Jie, Y.; Li, G.; Yan, Y.; Tian, C.; Wu, G.; Yang, N. Synergy of gut microbiota and host genome in driving heterosis expression of chickens. J. Genet. Genom. 2024, 51, 1121–1134. [Google Scholar] [CrossRef]
- Ahmed, I.; Jan, K.; Fatma, S.; Dawood, M.A.O. Muscle proximate composition of various food fish species and their nutritional significance: A review. J. Anim. Physiol. Anim. Nutr. 2022, 106, 690–719. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Lu, S.; Tao, Y.; Hua, J.; Zhuge, Y.; Chen, W.; Qiang, J. Characteristic Muscle Quality Parameters of Male Largemouth Bass (Micropterus salmoides) Distinguished from Female and Physiological Variations Revealed by Transcriptome Profiling. Biology 2024, 13, 1029. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.-N.; Wang, A.-L.; Bao, L.; Wang, J.; Liu, Y.; Sun, R.-Y. Changes of protein-bound and free amino acids in the muscle of the freshwater prawn Macrobrachium nipponense in different salinities. Aquaculture 2004, 233, 561–571. [Google Scholar] [CrossRef]
- Kasiga, T.; White, B.M.; Bruce, T.J.; Brown, M.L. Effect of fish meal replacement with Carinata Brassica carinata in low animal protein diets of rainbow trout Oncorhynchus mykiss (Walbaum) on trypsin activity, protein and amino acid digestibility and bioavailability. Aquac. Res. 2020, 51, 2134–2149. [Google Scholar] [CrossRef]
- Lai, X.; Xiao, Q.; Ji, H.; Huang, Z.; Huang, H.; You, W.; Luo, X.; Ke, C. Comparative analysis of the growth and biochemical composition of backcrosses and their parents in abalone. Aquaculture 2023, 570, 739445. [Google Scholar] [CrossRef]
- Zhang, W.; Song, Q.; Wu, F.; Zhang, J.; Xu, M.; Li, H.; Han, Z.; Gao, H.; Xu, N. Evaluation of the four breeds in synthetic line of Jiaxing Black Pigs and Berkshire for meat quality traits, carcass characteristics, and flavor substances. Anim. Sci. J. 2019, 90, 574–582. [Google Scholar] [CrossRef]
- Slaughter, J.E.; Wright, R.A.; DeVries, D.R. Latitudinal Influence on First-Year Growth and Survival of Largemouth Bass. N. Am. J. Fish. Manag. 2008, 28, 993–1000. [Google Scholar] [CrossRef]
- Du, J.; Li, S.; Shao, J.; Song, H.; Jiang, P.; Lei, C.; Bai, J.; Han, L. Genetic diversity analysis and development of molecular markers for the identification of largemouth bass (Micropterus salmoides L.) based on whole-genome re-sequencing. Front. Genet. 2022, 13, 936610. [Google Scholar] [CrossRef]
- Hargrove, J.S.; Weyl, O.L.F.; Zhao, H.; Peatman, E.; Austin, J.D. Using species-diagnostic SNPs to detail the distribution and dynamics of hybridized black bass populations in southern Africa. Biol. Invasions 2019, 21, 1499–1509. [Google Scholar] [CrossRef]
- Lutz-Carrillo, D.J.; Schlechte, W.; Norman, J.; Bennett, D.L. Lineage and hybridization effects on size potential in the Largemouth Bass complex. Trans. Am. Fish. Soc. 2023, 152, 145–168. [Google Scholar] [CrossRef]
- Li, W.; Liu, J.; Tan, H.; Yang, C.; Ren, L.; Liu, Q.; Wang, S.; Hu, F.; Xiao, J.; Zhao, R.; et al. Genetic Effects on the Gut Microbiota Assemblages of Hybrid Fish from Parents with Different Feeding Habits. Front. Microbiol. 2018, 9, 2972. [Google Scholar] [CrossRef] [PubMed]
- A Bryden, C.; Heath, J.W.; Heath, D.D. Performance and heterosis in farmed and wild Chinook salmon (Oncorhynchus tshawytscha) hybrid and purebred crosses. Aquaculture 2004, 235, 249–261. [Google Scholar] [CrossRef]
- Perriman, B.; Bentzen, P.; Wringe, B.; Duffy, S.; Islam, S.; Fleming, I.; Solberg, M.; Bradbury, I. Morphological consequences of hybridization between farm and wild Atlantic salmon Salmo salar under both wild and experimental conditions. Aquac. Environ. Interactions 2022, 14, 85–96. [Google Scholar] [CrossRef]
- Wang, Q.; Ye, W.; Tao, Y.; Li, Y.; Lu, S.; Xu, P.; Qiang, J. Transport Stress Induces Oxidative Stress and Immune Response in Juvenile Largemouth Bass (Micropterus salmoides): Analysis of Oxidative and Immunological Parameters and the Gut Microbiome. Antioxidants 2023, 12, 157. [Google Scholar] [CrossRef]
- Hou, W.; Yi, Z. Heterosis for biomass yield and quality traits in a reciprocal cross population between Miscanthus sinensis and Miscanthus lutarioriparius. Ind. Crop. Prod. 2023, 205, 117451. [Google Scholar] [CrossRef]
- Hansen, A.-C.; Hemre, G.-I. Effects of replacing fish meal and oil with plant resources in on-growing diets for Atlantic cod Gadus morhua L. Aquac. Nutr. 2013, 19, 641–650. [Google Scholar] [CrossRef]
- Xie, Q.; Liu, Y. Comparative analysis of the nutrient components in the muscle and skin tissues of hybrid sturgeon (Acipenser baerii ♀ × Acipenser schrenckii ♂) of different sizes. Aquac. Res. 2022, 53, 6124–6134. [Google Scholar] [CrossRef]
- Ali, A.; Wang, J.; Khan, I.; Wei, S.; Sun, Q.; Xia, Q.; Wang, Z.; Han, Z.; Liu, S. Physicochemical parameters and nutritional profile of back and abdomen muscle of fresh golden pompano (Trachinotus ovatus) and hybrid grouper (Epinephelus lanceolatus × Epinephelus fuscoguttatus). Food Sci. Nutr. 2023, 11, 1024–1039. [Google Scholar] [CrossRef]
- Lu, X.; Luan, S.; Cao, B.; Sui, J.; Dai, P.; Meng, X.; Luo, K.; Kong, J. Heterosis and heritability estimates for the survival of the Pacific white shrimp (Litopenaeus vannamei) under the commercial scale ponds. Acta Oceanol. Sin. 2016, 36, 62–68. [Google Scholar] [CrossRef]
- Yuan, J.; Zhao, J.; Sun, Y.; Wang, Y.; Li, Y.; Ni, A.; Zong, Y.; Ma, H.; Wang, P.; Shi, L.; et al. The mRNA-lncRNA landscape of multiple tissues uncovers key regulators and molecular pathways that underlie heterosis for feed intake and efficiency in laying chickens. Genet. Sel. Evol. 2023, 55, 69. [Google Scholar] [CrossRef]
- Danckert, N.P.; Wilson, N.; Phan-Thien, K.-Y.; Stone, D.A. The intestinal microbiome of Australian abalone, Haliotis laevigata and Haliotis laevigata × Haliotis rubra, over a 1-year period in aquaculture. Aquaculture 2021, 534, 736245. [Google Scholar] [CrossRef]
- Ziab, M.; Chaganti, S.R.; Heath, D.D. The effects of host quantitative genetic architecture on the gut microbiota composition of Chinook salmon (Oncorhynchus tshawytscha). Heredity 2023, 131, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Sevellec, M.; Laporte, M.; Bernatchez, A.; Derome, N.; Bernatchez, L. Evidence for host effect on the intestinal microbiota of whitefish (Coregonus sp.) species pairs and their hybrids. Ecol. Evol. 2019, 9, 11762–11774. [Google Scholar] [CrossRef] [PubMed]
- Reese, A.T.; Dunn, R.R. Drivers of Microbiome Biodiversity: A Review of General Rules, Feces, and Ignorance. mBio 2018, 9, e01294-18. [Google Scholar] [CrossRef]
- Martínez, R.; Santos, R.; Mascaró, M.; Canseco, L.; Caamal-Monsreal, C.; Rosas, C. Digestive dynamics during chyme formation of Octopus maya (Mollusca, Cephalopoda). Aquac. Res. 2012, 43, 1119–1126. [Google Scholar] [CrossRef]
- Wu, X.; Li, R.; Li, Q.; Bao, H.; Wu, C. Comparative transcriptome analysis among parental inbred and crosses reveals the role of dominance gene expression in heterosis in Drosophila melanogaster. Sci. Rep. 2016, 6, 21124. [Google Scholar] [CrossRef]
- Mai, C.; Wen, C.; Sun, C.; Xu, Z.; Chen, S.; Yang, N. Implications of Gene Inheritance Patterns on the Heterosis of Abdominal Fat Deposition in Chickens. Genes 2019, 10, 824. [Google Scholar] [CrossRef]
- Fu, J.; Zhang, Y.; Yan, T.; Li, Y.; Jiang, N.; Zhou, Y.; Zhou, Q.; Qin, P.; Fu, C.; Lin, H.; et al. Transcriptome profiling of two super hybrid rice provides insights into the genetic basis of heterosis. BMC Plant Biol. 2022, 22, 314. [Google Scholar] [CrossRef]
- Zhang, G.; Li, J.; Zhang, J.; Liang, X.; Zhang, X.; Wang, T.; Yin, S. Integrated Analysis of Transcriptomic, miRNA and Proteomic Changes of a Novel Hybrid Yellow Catfish Uncovers Key Roles for miRNAs in Heterosis. Mol. Cell. Proteom. 2019, 18, 1437–1453. [Google Scholar] [CrossRef]
- Perry, W.B.; Lindsay, E.; Payne, C.J.; Brodie, C.; Kazlauskaite, R. The role of the gut microbiome in sustainable teleost aquaculture. Proc. R. Soc. B Biol. Sci. 2020, 287, 20200184. [Google Scholar] [CrossRef]
- Johny, T.K.; Puthusseri, R.M.; Bhat, S.G. A primer on metagenomics and next-generation sequencing in fish gut microbiome research. Aquac. Res. 2021, 52, 4574–4600. [Google Scholar] [CrossRef]
- Tan, S.; Xu, X.; Cheng, H.; Wang, J.; Wang, X. The alteration of gut microbiome community play an important role in mercury biotransformation in largemouth bass. Environ. Res. 2022, 204, 112026. [Google Scholar] [CrossRef] [PubMed]
- Jin, W.; Jiang, L.; Hu, S.; Zhu, A. Metabolite features of serum and intestinal microbiota response of largemouth bass (Micropterus salmoides) after Aeromonas hydrophila challenge. Comp. Biochem. Physiol. Part C Toxicol. Pharmacol. 2023, 263. [Google Scholar] [CrossRef]
- Song, Z.; Ye, W.; Tao, Y.; Zheng, T.; Qiang, J.; Li, Y.; Liu, W.; Xu, P. Transcriptome and 16S rRNA Analyses Reveal That Hypoxic Stress Affects the Antioxidant Capacity of Largemouth Bass (Micropterus salmoides), Resulting in Intestinal Tissue Damage and Structural Changes in Microflora. Antioxidants 2022, 12, 1. [Google Scholar] [CrossRef]
- Shin, N.-R.; Whon, T.W.; Bae, J.-W. Proteobacteria: Microbial signature of dysbiosis in gut microbiota. Trends Biotechnol. 2015, 33, 496–503. [Google Scholar] [CrossRef]
- Litvak, Y.; Byndloss, M.X.; Bäumler, A.J. Colonocyte metabolism shapes the gut microbiota. Science 2018, 362, eaat9076. [Google Scholar] [CrossRef]
- Chen, X.; Chen, H.; Liu, Q.; Ni, K.; Ding, R.; Wang, J.; Wang, C. High Plasticity of the Gut Microbiome and Muscle Metabolome of Chinese Mitten Crab (Eriocheir sinensis) in Diverse Environments. J. Microbiol. Biotechnol. 2021, 31, 240–249. [Google Scholar] [CrossRef]
- Liu, Y.; Li, X.; Li, Y.; Li, J.; Zhu, S. Gut microbiomes of cyprinid fish exhibit host-species symbiosis along gut trait and diet. Front. Microbiol. 2022, 13, 936601. [Google Scholar] [CrossRef]
- Huang, X.; Fang, S.; Yang, H.; Gao, J.; He, M.; Ke, S.; Zhao, Y.; Chen, C.; Huang, L. Evaluating the contribution of gut microbiome to the variance of porcine serum glucose and lipid concentration. Sci. Rep. 2017, 7, 14928. [Google Scholar] [CrossRef]
- Yang, J.H.; Park, J.W.; Kim, H.S.; Lee, S.; Yerke, A.M.; Jaiswal, Y.S.; Williams, L.L.; Hwang, S.; Moon, K.H. Effects of Antibiotic Residues on Fish Gut Microbiome Dysbiosis and Mucosal Barrier-Related Pathogen Susceptibility in Zebrafish Experimental Model. Antibiotics 2024, 13, 82. [Google Scholar] [CrossRef]
- Li, M.; Liang, H.; Yang, H.; Ding, Q.; Xia, R.; Chen, J.; Zhou, W.; Yang, Y.; Zhang, Z.; Yao, Y.; et al. Deciphering the gut microbiome of grass carp through multi-omics approach. Microbiome 2024, 12, 2. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Fan, Z.; Yi, M.; Liu, Z.; Ke, X.; Gao, F.; Cao, J.; Wang, M.; Chen, G.; Lu, M. Characterization of the core gut microbiota of Nile tilapia (Oreochromis niloticus): Indication of a putative novel Cetobacterium species and analysis of its potential function on nutrition. Arch. Microbiol. 2022, 204, 690. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Zhang, Z.; Ding, Q.; Yang, Y.; Bindelle, J.; Ran, C.; Zhou, Z. Intestinal Cetobacterium and acetate modify glucose homeostasis via parasympathetic activation in zebrafish. Gut Microbes 2021, 13, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Abraham, T.J.; Paul, P.; Adikesavalu, H.; Patra, A.; Banerjee, S. Stenotrophomonas maltophilia as an opportunistic pathogen in cultured African catfish Clarias gariepinus (Burchell, 1822). Aquaculture 2016, 450, 168–172. [Google Scholar] [CrossRef]
- Yang, Q.; Guo, K.; Zhou, X.; Tang, X.; Yu, X.; Yao, W.; Wu, Z. Histopathology, antioxidant responses, transcriptome and gene expression analysis in triangle sail mussel Hyriopsis cumingii after bacterial infection. Dev. Comp. Immunol. 2021, 124, 104175. [Google Scholar] [CrossRef]
- Zhao, R.; Symonds, J.E.; Walker, S.P.; Steiner, K.; Carter, C.G.; Bowman, J.P.; Nowak, B.F. Relationship between gut microbiota and Chinook salmon (Oncorhynchus tshawytscha) health and growth performance in freshwater recirculating aquaculture systems. Front. Microbiol. 2023, 14, 1065823. [Google Scholar] [CrossRef]
- Mondal, H.K.; Maji, U.J.; Mohanty, S.; Sahoo, P.K.; Maiti, N.K. Alteration of gut microbiota composition and function of Indian major carp, rohu (Labeo rohita) infected with Argulus siamensis. Microb. Pathog. 2022, 164, 105420. [Google Scholar] [CrossRef]
- Wigren, M.A.; Johnson, T.A.; Griffitt, R.J.; Hay, A.G.; Knott, J.A.; Sepúlveda, M.S. Limited impact of weathered residues from the Deepwater Horizon oil spill on the gut-microbiome and foraging behavior of sheepshead minnows (Cyprinodon variegatus). J. Toxicol. Environ. Health Part A 2024, 87, 1–21. [Google Scholar] [CrossRef]
- Sumithra, T.; Gayathri, S.; Sharma, S.K.; Ebeneezar, S.; Anikuttan, K.; Sajina, K.; Narasimapallavan, G.I.; Reshma, K.; Vishnu, R.; Tamilmani, G.; et al. Metagenomic signatures of transportation stress in the early life stages of cobia (Rachycentron canadum) to aid in mitigation strategies. Aquaculture 2022, 559, 738407. [Google Scholar] [CrossRef]
- Fu, L.; Zhang, X.; Wang, Y.; Peng, L.; Li, W. Nitrogen removal characteristics of Pseudomonas stutzeri F11 and its application in grass carp culture. Fish. Sci. 2016, 83, 89–98. [Google Scholar] [CrossRef]
- Wen, C.; Wang, Q.; Gu, S.; Jin, J.; Yang, N. Emerging perspectives in the gut–muscle axis: The gut microbiota and its metabolites as important modulators of meat quality. Microb. Biotechnol. 2024, 17, e14361. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).