

Transitions of the Bacteria–Fungi Microbiomes Associated with Different Life Cycle Stages of Dinoflagellate Scrippsiella acuminata

 ,
,  ,
,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Establishment of an Algal Culture and Maintenance, and Resting Cysts’ Production
2.2. Sample Types and Their Preparations
2.3. DNA Extraction and PCR Amplification of 16S and ITS rRNA Genes
2.4. Sequence Processing and Bioinformatic Analyses
2.5. Identification of the Core Bacterial and Fungal Taxa
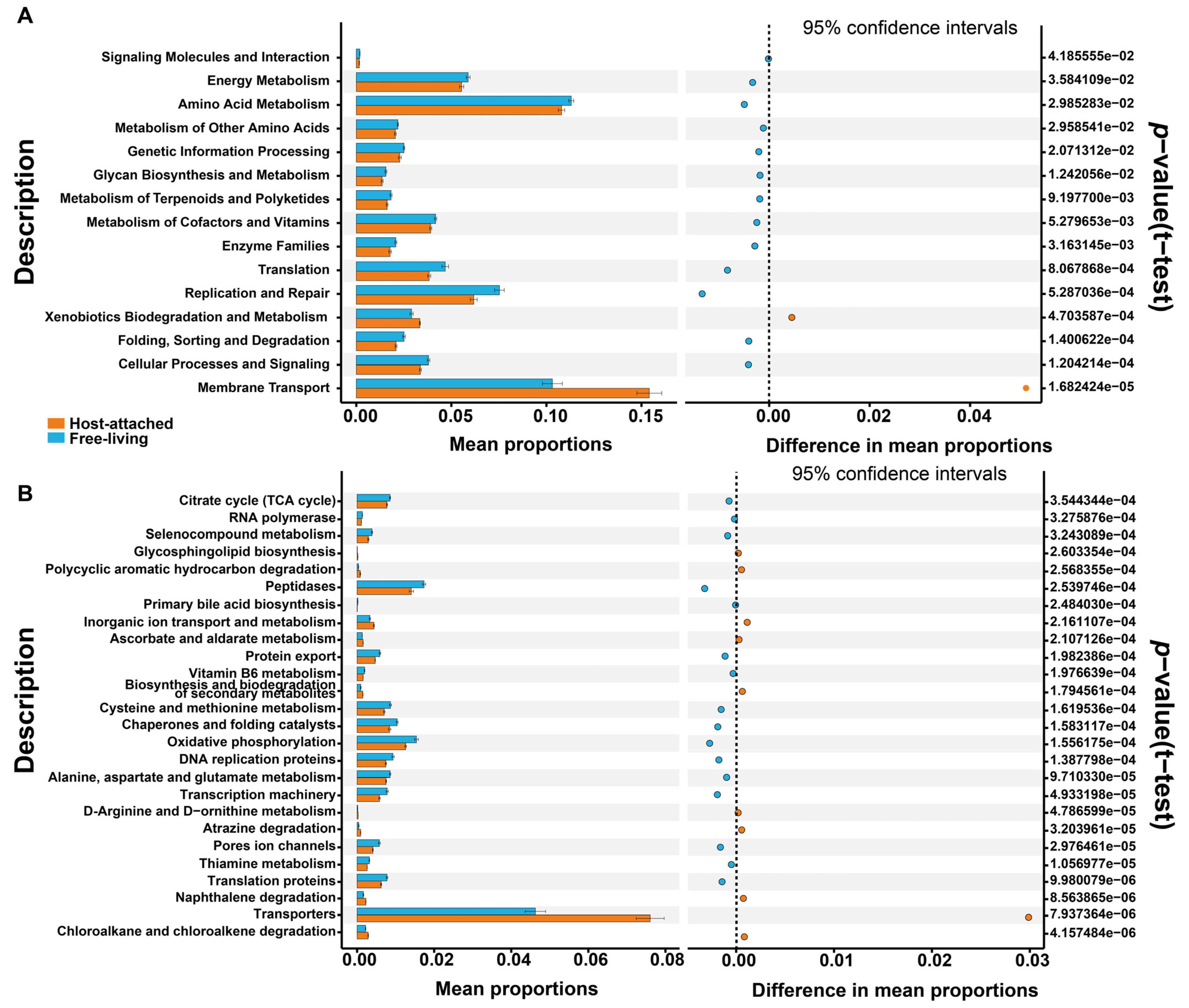
2.6. Prediction of the Functional Potential of the Bacterial and Fungal Microbiomes
3. Results
3.1. General Descriptions of Bacterial and Fungal Diversity Associated with Scrippsiella acuminata
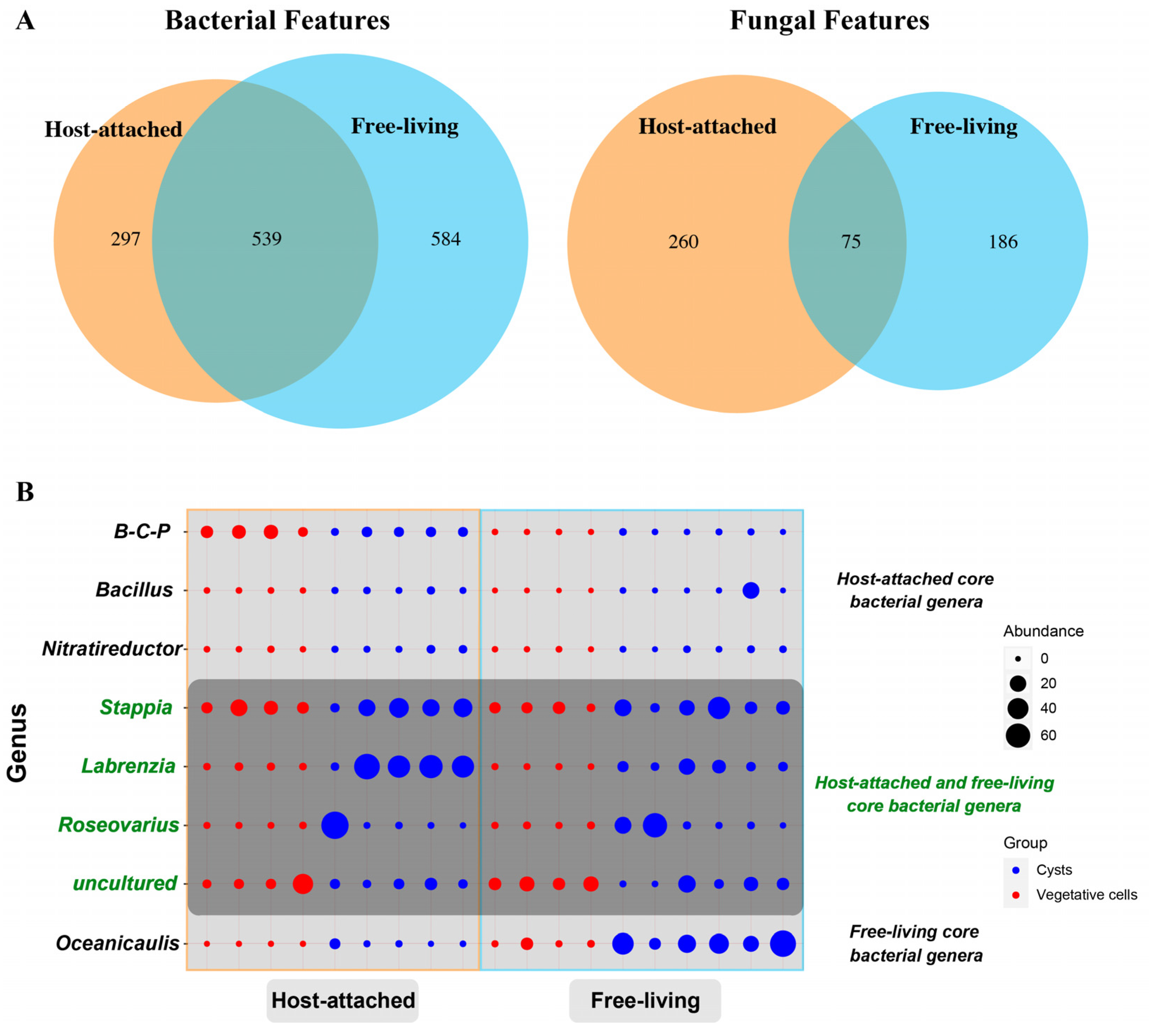
3.2. Common Species Shared by the Attached and Free-Living Groups and Core Genera Determination
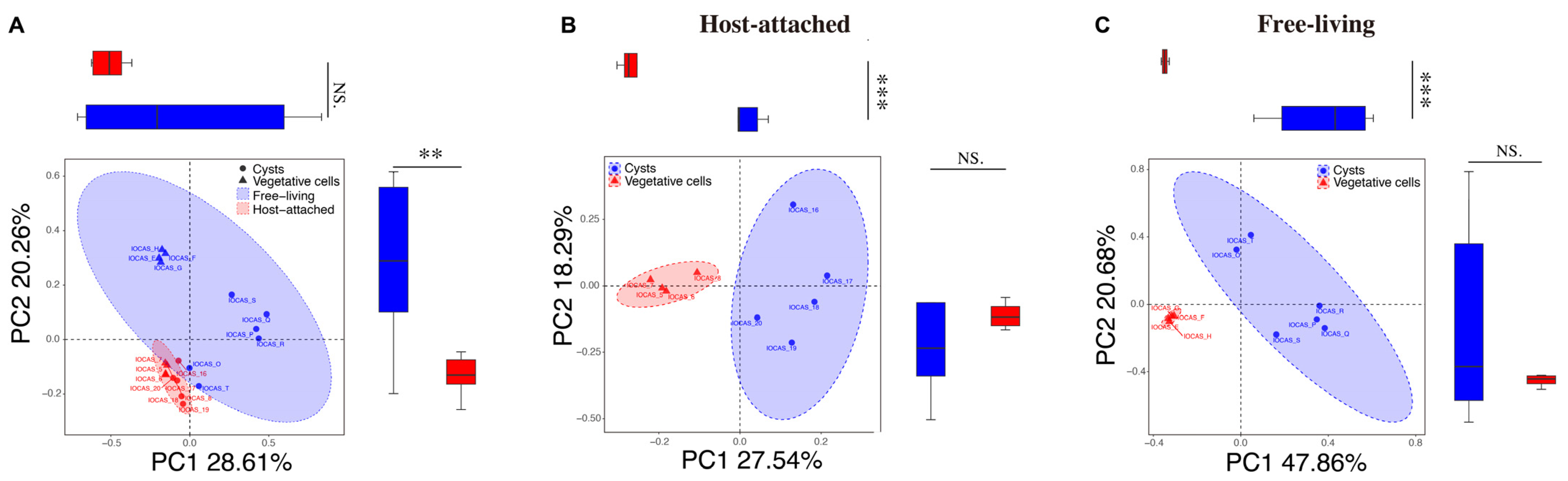
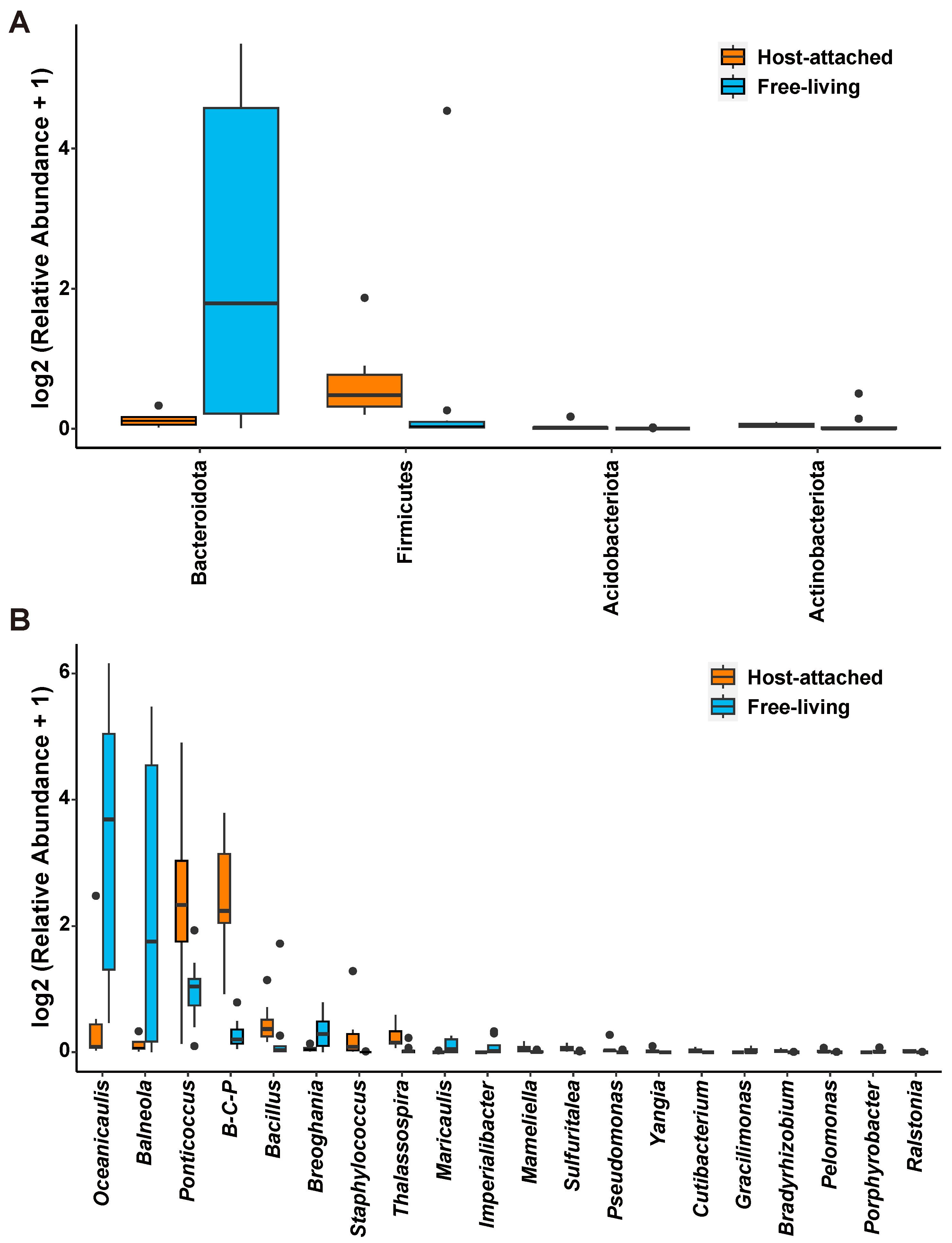
3.3. Comparison Between the Host-Attached and Free-Living Microbiome Communities
3.4. Comparisons of the Bacterial and Fungal Communities Among Different Life Cycle Stages
4. Discussion
4.1. Core Bacteria–Fungi Genera Stably Co-Existed with S. acuminata
4.2. Host-Attached and Free-Living Microbiomes Showed Different Nutritional Preferences
4.3. Dinoflagellate Resting Cysts in Dormancy Still Interact with Bacteria and Fungi
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Correction Statement
References
- Cole, J.J. Interactions between bacteria and algae in aquatic ecosystems. Annu. Rev. Ecol. Syst. 1982, 13, 291–314. [Google Scholar] [CrossRef]
- Seymour, J.R.; Amin, S.A.; Raina, J.B.; Stocker, R. Zooming in on the phycosphere: The ecological interface for phytoplankton-bacteria relationships. Nat. Microbiol. 2017, 2, 17065. [Google Scholar] [CrossRef] [PubMed]
- Rolland, J.l.; Stien, D.; Sanchez-Ferandin, S.; Lami, R. Quorum sensing and quorum quenching in the phycosphere of phytoplankton: A case of chemical interactions in ecology. J. Chem. Ecol. 2016, 42, 1201–1211. [Google Scholar] [CrossRef]
- Waring, B.G.; Averill, C.; Hawkes, C.V. Differences in fungal and bacterial physiology alter soil carbon and nitrogen cycling: Insights from meta-analysis and theoretical models. Ecol. Lett. 2013, 16, 887–894. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.H.; Sun, C.; Guo, Z.H.; Liu, L.Y.; Zhang, X.T.; Sun, K.; Zheng, Y.F.; Gates, A.J.; Todd, J.D.; Zhang, X.H. An S-methyltransferase that produces the climate-active gas dimethylsulfide is widespread across diverse marine bacteria. Nat. Microbiol. 2024, 9, 2614–2625. [Google Scholar] [CrossRef] [PubMed]
- Grossart, H.-P.; Van den Wyngaert, S.; Kagami, M.; Wurzbacher, C.; Cunliffe, M.; Rojas-Jimenz, K. Fungi in aquatic ecosystems. Nat. Rev. Microbiol. 2019, 17, 339–354. [Google Scholar] [CrossRef]
- Amend, A.; Burgaud, G.; Cunliffe, M.; Edgcomb, V.P.; Ettinger, C.L.; Gutierrez, M.H.; Heitman, J.; Hom, E.F.Y.; Ianiri, G.; Jones, A.C.; et al. Fungi in the Marine Environment: Open Questions and Unsolved Problems. Mbio 2019, 10, e01189-18. [Google Scholar] [CrossRef]
- Droop, M.R. Vitamins, phytoplankton and bacteria: Symbiosis or scavenging? J. Plankton Res. 2007, 29, 107–113. [Google Scholar] [CrossRef]
- Cirri, E.; Pohnert, G. Algae-bacteria interactions that balance the planktonic microbiome. New Phytol. 2019, 223, 100–106. [Google Scholar] [CrossRef]
- Tang, Y.Z.; Koch, F.; Gobler, C.J. Most harmful algal bloom species are vitamin B1 and B12 auxotrophs. Proc. Natl. Acad. Sci. USA 2010, 107, 20756–20761. [Google Scholar] [CrossRef]
- Croft, M.T.; Lawrence, A.D.; Raux-Deery, E.; Warren, M.J.; Smith, A.G. Algae acquire vitamin B-12 through a symbiotic relationship with bacteria. Nature 2005, 438, 90–93. [Google Scholar] [CrossRef] [PubMed]
- Kouzuma, A.; Watanabe, K. Exploring the potential of algae/bacteria interactions. Curr. Opin. Biotechnol. 2015, 33, 125–129. [Google Scholar] [CrossRef] [PubMed]
- Prieto, A.; Barber-Lluch, E.; Hernandez-Ruiz, M.; Martinez-Garcia, S.; Fernandez, E.; Teira, E. Assessing the role of phytoplankton-bacterioplankton coupling in the response of microbial plankton to nutrient additions. J. Plankton Res. 2016, 38, 55–63. [Google Scholar] [CrossRef]
- Geng, H.F.; Belas, R. Molecular mechanisms underlying roseobacter-phytoplankton symbioses. Curr. Opin. Biotechnol. 2010, 21, 332–338. [Google Scholar] [CrossRef]
- Fortin, S.G.; Song, B.; Anderson, I.C.; Reece, K.S. Blooms of the harmful algae Margalefidinium polykrikoides and Alexandrium monilatum alter the York River Estuary microbiome. Harmful Algae 2022, 114, 102216. [Google Scholar] [CrossRef]
- Flynn, K.J.; Stoecker, D.K.; Mitra, A.; Raven, J.A.; Glibert, P.M.; Hansen, P.J.; Graneli, E.; Burkholder, J.M. Misuse of the phytoplanktonzooplankton dichotomy: The need to assign organisms as mixotrophs within plankton functional types. J. Plankton Res. 2013, 35, 3–11. [Google Scholar] [CrossRef]
- Lin, S.; Cheng, S.; Song, B.; Zhong, X.; Lin, X.; Li, W.; Li, L.; Zhang, Y.; Zhang, H.; Ji, Z.; et al. The Symbiodinium kawagutii genome illuminates dinoflagellate gene expression and coral symbiosis. Science 2015, 350, 691–694. [Google Scholar] [CrossRef]
- Lin, S. Genomic understanding of dinoflagellates. Res. Microbiol. 2011, 162, 551–569. [Google Scholar] [CrossRef]
- Hackett, J.D.; Anderson, D.M.; Erdner, D.L.; Bhattacharya, D. Dinoflagellates: A remarkable evolutionary experiment. Am. J. Bot. 2004, 91, 1523–1534. [Google Scholar] [CrossRef]
- Li, A.S.; Stoecker, D.K.; Coats, D.W. Mixotrophy in Gyrodinium galatheanum (Dinophyceae): Grazing responses to light intensity and inorganic nutrients. J. Phycol. 2000, 36, 33–45. [Google Scholar] [CrossRef]
- Adolf, J.E.; Bachvaroff, T.R.; Krupatkina, D.N.; Nonogaki, H.; Brown, P.J.P.; Lewitus, A.J.; Harvey, H.R.; Place, A.R. Species specificity and potential roles of Karlodinium micrum toxin. Afr. J. Mar. Sci. 2006, 28, 415–419. [Google Scholar] [CrossRef]
- Tang, Y.Z.; Hu, Z.; Deng, Y. Characteristical life history (resting cyst) provides a mechanism for recurrence and geographic expansion of harmful algal blooms of dinoflagellates: A review. Stud. Mar. Sin. 2016, 51, 132–154. [Google Scholar] [CrossRef]
- Taylor, F.J.R.; Hoppenrath, M.; Saldarriaga, J.F. Dinoflagellate diversity and distribution. Biodivers. Conserv. 2008, 17, 407–418. [Google Scholar] [CrossRef]
- Bravo, I.; Figueroa, R.I. Towards an ecological understanding of dinoflagellate cyst functions. Microorganisms 2014, 2, 11–32. [Google Scholar] [CrossRef]
- Smayda, T. Harmful algal blooms: Their ecophysiology and general relevance to phytoplankton blooms in the sea. Limnol. Oceanogr. 1997, 42, 1137–1153. [Google Scholar] [CrossRef]
- Jeong, H.J.; Kang, H.C.; Lim, A.S.; Jang, S.H.; Lee, K.; Lee, S.Y.; Ok, J.H.; You, J.H.; Kim, J.H.; Lee, K.H.; et al. Feeding diverse prey as an excellent strategy of mixotrophic dinoflagellates for global dominance. Sci. Adv. 2021, 7, eabe4214. [Google Scholar] [CrossRef]
- Needham, D.M.; Fuhrman, J.A. Pronounced daily succession of phytoplankton, archaea and bacteria following a spring bloom. Nat. Microbiol. 2016, 1, 16005. [Google Scholar] [CrossRef]
- Doucette, G.J. Interactions between bacteria and harmful algae: A review. Nat. Toxins 1995, 3, 65–74. [Google Scholar] [CrossRef]
- Soria-Dengg, S.; Reissbrodt, R.; Horstmann, U. Siderophores in marine, coastal waters and their relevance for iron uptake by phytoplankton: Experiments with the diatom Phaeodactylum tricornutum. Mar. Ecol. Prog. Ser. 2001, 220, 73–82. [Google Scholar] [CrossRef]
- Lin, S.; Hu, Z.; Song, X.; Gobler, C.J.; Tang, Y.Z. Vitamin B12-auxotrophy in dinoflagellates caused by incomplete or absent cobalamin-independent methionine synthase genes (metE). Fundam. Res. 2022, 2, 727–737. [Google Scholar] [CrossRef]
- Teeling, H.; Fuchs, B.M.; Becher, D.; Klockow, C.; Gardebrecht, A.; Bennke, C.M.; Kassabgy, M.; Huang, S.; Mann, A.J.; Waldmann, J.; et al. Substrate-controlled succession of marine bacterioplankton populations induced by a phytoplankton bloom. Science 2012, 336, 608–611. [Google Scholar] [CrossRef] [PubMed]
- Buchan, A.; LeCleir, G.R.; Gulvik, C.A.; González, J.M. Master recyclers: Features and functions of bacteria associated with phytoplankton blooms. Nat. Rev. Microbiol. 2014, 12, 686–698. [Google Scholar] [CrossRef]
- Mayali, X.; Franks, P.J.S.; Burton, R.S. Temporal attachment dynamics by distinct bacterial taxa during a dinoflagellate bloom. Aquat. Microb. Ecol. 2011, 63, 111–122. [Google Scholar] [CrossRef]
- Yang, C.; Li, Y.; Zhou, B.; Zhou, Y.; Zheng, W.; Tian, Y.; Van Nostrand, J.D.; Wu, L.; He, Z.; Zhou, J.; et al. Illumina sequencing-based analysis of free-living bacterial community dynamics during an Akashiwo sanguine bloom in Xiamen sea, China. Sci. Rep. 2015, 5, 8476. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Wang, K.; Hu, Z.; Hu, Q.; Tang, Y.Z. Identification and implications of a core bacterial microbiome in 19 clonal cultures laboratory-reared for months to years of the cosmopolitan dinoflagellate Karlodinium veneficum. Front. Microbiol. 2022, 13, 967610. [Google Scholar] [CrossRef]
- Deng, Y.Y.; Wang, K.; Hu, Z.X.; Tang, Y.Z. Abundant Species Diversity and Essential Functions of Bacterial Communities Associated with Dinoflagellates as Revealed from Metabarcoding Sequencing for Laboratory-Raised Clonal Cultures. Int. J. Environ. Res. Public Health 2022, 19, 4446. [Google Scholar] [CrossRef] [PubMed]
- Bolch, C.J.S.; Bejoy, T.A.; Green, D.H. Bacterial Associates Modify Growth Dynamics of the Dinoflagellate Gymnodinium catenatum. Front. Microbiol. 2017, 8, 670. [Google Scholar] [CrossRef]
- Park, B.S.; Guo, R.; Lim, W.A.; Ki, J.S. Importance of free-living and particle-associated bacteria for the growth of the harmful dinoflagellate Prorocentrum minimum: Evidence in culture stages. Mar. Freshw. Res. 2018, 69, 290–299. [Google Scholar] [CrossRef]
- Shin, H.; Lee, E.; Shin, J.; Ko, S.-R.; Oh, H.-S.; Ahn, C.-Y.; Oh, H.-M.; Cho, B.-K.; Cho, S. Elucidation of the bacterial communities associated with the harmful microalgae Alexandrium tamarense and Cochlodinium polykrikoides using nanopore sequencing. Sci. Rep. 2018, 8, 5323. [Google Scholar] [CrossRef]
- Li, S.; Chen, M.; Chen, Y.; Tong, J.; Wang, L.; Xu, Y.; Hu, Z.; Chen, H. Epibiotic bacterial community composition in red-tide dinoflagellate Akashiwo sanguinea culture under various growth conditions. FEMS Microbiol. Ecol. 2019, 95, fiz057. [Google Scholar] [CrossRef]
- Guo, R.; Wang, P.; Lu, D.; Dai, X. Comparison of bacterial communities associated with Prorocentrum donghaiense and Karenia mikimotoistrains from Chinese coastal waters. Mar. Freshw. Res. 2020, 71, 1662–1671. [Google Scholar] [CrossRef]
- Tarazona-Janampa, U.I.; Cembella, A.D.; Pelayo-Zarate, M.C.; Pajares, S.; Marquez-Valdelamar, L.M.; Okolodkov, Y.B.; Tebben, J.; Krock, B.; Duran-Riveroll, L.M. Associated Bacteria and Their Effects on Growth and Toxigenicity of the Dinoflagellate Prorocentrum lima Species Complex From Epibenthic Substrates Along Mexican Coasts. Front. Mar. Sci. 2020, 7, 569. [Google Scholar] [CrossRef]
- Sun, J.Y.; Song, Y.; Ma, Z.P.; Zhang, H.J.; Yang, Z.D.; Cai, Z.H.; Zhou, J. Fungal community dynamics during a marine dinoflagellate (Noctiluca scintillans) bloom. Mar. Environ. Res. 2017, 131, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Riemann, L.; Steward, G.F.; Azam, F. Dynamics of cacterial community composition and activity during a mesocosm diatom bloom. Appl. Environ. Microciol. 2000, 66, 578–587. [Google Scholar] [CrossRef] [PubMed]
- Kan, J.J.; Chen, F. Co-monitoning bacterial and dinoflagellates communities by denaturing gradient gel electrophoresis (DGGE) and SSU rRNA sequencing during a dinoflagellates bloom. Acta Oceanol. Sin. 2004, 23, 483–492. [Google Scholar]
- Jones, K.L.; Mikulski, C.M.; Barnhorst, A.; Doucette, G.J. Comparative analysis of bacterioplankton assemblages from Karenia brevis bloom and nonbloom water on the west Florida shelf (Gulf of Mexico, USA) using 16S rRNA gene clone libraries. FEMS Microbiol. Ecol. 2010, 73, 468–485. [Google Scholar] [CrossRef]
- Theroux, S.; Huang, Y.; Amaral Zettler, L. Comparative Molecular Microbial Ecology of the Spring Haptophyte Bloom in a Greenland Arctic Oligosaline Lake. Front. Microbiol. 2012, 3, 33594. [Google Scholar] [CrossRef]
- Zhou, J.; Zhang, B.Y.; Yu, K.; Du, X.P.; Zhu, J.M.; Zeng, Y.H.; Cai, Z.H. Functional profiles of phycospheric microorganisms during a marine dinoflagellate bloom. Water Res. 2020, 173, 115554. [Google Scholar] [CrossRef]
- Foysal, M.J.; Timms, V.; Neilan, B.A. Dynamics of the benthic and planktic microbiomes in a Planktothrix-dominated toxic cyanobacterial bloom in Australia. Water Res. 2024, 249, 120980. [Google Scholar] [CrossRef]
- Coyne, K.J.; Wang, Y.; Johnson, G. Algicidal Bacteria: A Review of Current Knowledge and Applications to Control Harmful Algal Blooms. Front. Microbiol. 2022, 13, 871177. [Google Scholar] [CrossRef]
- Wang, N.; Mark, N.; Launer, N.; Hirtler, A.; Weston, C.; Cleckner, L.; Faehndrich, C.; LaGorga, L.; Xia, L.; Pyrek, D.; et al. Harmful algal blooms in Cayuga lake, NY: From microbiome analysis to eDNA monitoring. J. Environ. Manag. 2024, 354, 120128. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Song, Q.S.; Li, D.M.; Wang, X.Y.; Liu, H.; Wu, C.C.; Shen, P.P. Structural and functional diversity of the microbiome during the Phaeocystis globosa winter bloom in the southern Yellow Sea of China. Int. Biodeterior. Biodegrad. 2023, 181, 105617. [Google Scholar] [CrossRef]
- Grossart, H.P.; Levold, F.; Allgaier, M.; Simon, M.; Brinkhoff, T. Marine diatom species harbour distinct bacterial communities. Environ. Microbiol. 2005, 7, 860–873. [Google Scholar] [CrossRef] [PubMed]
- Lupette, J.; Lami, R.; Krasovec, M.; Grimsley, N.; Moreau, H.; Piganeau, G.; Sanchez-Ferandin, S. Marinobacter dominates the bacterial community of the Ostreococcus tauri phycosphere in culture. Front. Microbiol. 2016, 7, 1414. [Google Scholar] [CrossRef]
- Maire, J.; Girvan, S.K.; Barkla, S.E.; Perez-Gonzalez, A.; Suggett, D.J.; Blackall, L.L.; van Oppen, M.J.H. Intracellular bacteria are common and taxonomically diverse in cultured and in hospite algal endosymbionts of coral reefs. ISME J. 2021, 15, 2028–2042. [Google Scholar] [CrossRef]
- Brisbin, M.M.; Mitarai, S.; Saito, M.A.; Alexander, H. Microbiomes of bloom-forming Phaeocystis algae are stable and consistently recruited, with both symbiotic and opportunistic modes. ISME J. 2022, 16, 2255–2264. [Google Scholar] [CrossRef] [PubMed]
- Dang, H.; Lovell, C.R. Microbial surface colonization and biofilm development in marine environments. Microbiol. Mol. Biol. Rev. 2016, 80, 91–138. [Google Scholar] [CrossRef]
- Tang, Y.Z.; Gu, H.; Wang, Z.; Liu, D.; Wang, Y.; Lu, D.; Hu, Z.; Deng, Y.; Shang, L.; Qi, Y. Exploration of resting cysts (stages) and their relevance for possibly HABs-causing species in China. Harmful Algae 2021, 107, 102050. [Google Scholar] [CrossRef]
- Ellegaard, M.; Ribeiro, S. The long-term persistence of phytoplankton resting stages in aquatic ‘seed banks’. Biol. Rev. 2018, 93, 166–183. [Google Scholar] [CrossRef]
- Adachi, M.; Kanno, T.; Matsubara, T.; Nishijima, T.; Itakura, S.; Yamaguchi, M. Promotion of cyst formation in the toxic dinoflagellate Alexandrium (Dinophyceae) by natural bacterial assemblages from Hiroshima Bay, Japan. Mar. Ecol. Prog. Ser. 1999, 191, 175–185. [Google Scholar] [CrossRef]
- Adachi, M.; Matsubara, T.; Okamoto, R.; Nishijima, T.; Itakura, S.; Yamaguchi, M. Inhibition of cyst formation in the toxic dinoflagellate Alexandrium (Dinophyceae) by bacteria from Hiroshima Bay, Japan. Aquat. Microb. Ecol. 2002, 26, 223–233. [Google Scholar] [CrossRef]
- Adachi, M.; Kanno, T.; Okamoto, R.; Itakura, S.; Yamaguchi, M.; Nishijima, T. Population structure of Alexandrium (Dinophyceae) cyst formation-promoting bacteria in Hiroshima Bay, Japan. Appl. Environ. Microbiol. 2003, 69, 6560–6568. [Google Scholar] [CrossRef] [PubMed]
- Mayali, X.; Franks, P.J.S.; Azam, F. Bacterial induction of temporary cyst formation by the dinoflagellate Lingulodinium polyedrum. Aquat. Microb. Ecol. 2007, 50, 51–62. [Google Scholar] [CrossRef]
- Head, M.J.; Mertens, K.N.; Fensome, R.A. Dual nomenclature in organic-walled dinoflagellate cysts I: Concepts, methods and applications. Palynology 2024, 48, 2290200. [Google Scholar] [CrossRef]
- Kretschmann, J.; Elbraezchter, M.; Zinssmeister, C.; Soehner, S.; Kirsch, M.; Kusber, W.-H.; Gottschling, M. Taxonomic clarification of the dinophyte Peridinium acuminatum Ehrenb., Scrippsiella acuminata, comb. nov. (Thoracosphaeraceae, Peridiniales). Phytotaxa 2015, 220, 239–256. [Google Scholar] [CrossRef]
- Tang, Y.Z.; Gobler, C.J. Lethal effects of Northwest Atlantic Ocean isolates of the dinoflagellate, Scrippsiella trochoidea, on Eastern oyster (Crassostrea virginica) and Northern quahog (Mercenaria mercenaria) larvae. Mar. Biol. 2012, 159, 199–210. [Google Scholar] [CrossRef]
- Kim, Y.O.; Han, M.S. Seasonal relationships between cyst germination and vegetative population of Scrippsiella trochoidea (Dinophyceae). Mar. Ecol. Prog. Ser. 2000, 204, 111–118. [Google Scholar] [CrossRef]
- Ishikawa, A.; Taniguchi, A. The role of cysts on population dynamics of Scrippsiella spp. (Dinophyceae) in Onagawa Bay, northeast Japan. Mar. Biol. 1994, 119, 39–44. [Google Scholar] [CrossRef]
- Qi, Y.; Zheng, L.; Wang, R. The life cycle of Scrippsiella trochoidea and its physiol-ecological control. Oceanol. Limnol. Sin. 1997, 28, 588–593. [Google Scholar]
- Binder, B.J.; Anderson, D.M. Biochemical-composition and metabolic-activity of Scrippsiella Trochoidea (Dinophyceae) resting cysts. J. Phycol. 1990, 26, 289–298. [Google Scholar] [CrossRef]
- Lirdwitayaprasit, T.; Okaichi, T.; Montani, S.; Ochi, T.; Anderson, D.M. Changes in Cell Chemical-Composition during the Life Cycle of Scrippsiella Trochoidea (Dinophyceae). J. Phycol. 1990, 26, 299–306. [Google Scholar] [CrossRef]
- Deng, Y.; Hu, Z.; Shang, L.; Peng, Q.; Tang, Y.Z. Transcriptomic analyses of Scrippsiella trochoidea reveals processes regulating encystment and dormancy in the life cycle of a dinoflagellate, with a particular attention to the role of abscisic acid. Front. Microbiol. 2017, 8, 2450. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.Y.; Yue, C.X.; Yang, H.J.; Li, F.T.; Hu, Z.X.; Shang, L.X.; Chai, Z.Y.; Lin, S.J.; Tang, Y.Z. Broad active metabolic pathways, autophagy, and antagonistic hormones regulate dinoflagellate cyst dormancy in marine sediments. Sci. Adv. 2025, 11, eads7789. [Google Scholar] [CrossRef]
- Li, F.T.; Yue, C.X.; Deng, Y.Y.; Tang, Y.Z. Full-length transcriptome analysis of a bloom-forming dinoflagellate Scrippsiella acuminata (Dinophyceae). Sci. Data 2025, 12, 352. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Li, F.; Shang, L.; Hu, Z.; Yue, C.; Tang, Y.Z. The resting cyst of dinoflagellate Scrippsiella acuminata host bacterial microbiomes with more diverse trophic strategies under conditions typically observed in marine sediments. Front. Microbiol. 2024, 15, 1407459. [Google Scholar] [CrossRef]
- Smith, W.L.; Chanley, M.H. Culture of Marine Invertebrate Animals; Springer: New York, NY, USA, 1975; pp. 29–60. [Google Scholar]
- Yue, C.; Chai, Z.; Hu, Z.; Shang, L.; Deng, Y.; Tang, Y.Z. Deficiency of nitrogen but not phosphorus triggers the life cycle transition of the dinoflagellate Scrippsiella acuminata from vegetative growth to resting cyst formation. Harmful Algae 2022, 118, 102312. [Google Scholar] [CrossRef]
- Kreyling, J.; Schweiger, A.H.; Bahn, M.; Ineson, P.; Migliavacca, M.; Morel-Journel, T.; Christiansen, J.R.; Schtickzelle, N.; Larsen, K.S. To replicate, or not to replicate—That is the question: How to tackle nonlinear responses in ecological experiments. Ecol. Lett. 2018, 21, 1629–1638. [Google Scholar] [CrossRef]
- Su, J.; Yang, X.; Zheng, T.; Hong, H. An efficient method to obtain axenic cultures of Alexandrium tamarense—A PSP-producing dinoflagellate. J. Microbiol. Methods 2007, 69, 425–430. [Google Scholar] [CrossRef]
- Bodenhausen, N.; Horton, M.W.; Bergelson, J. Bacterial Communities Associated with the Leaves and the Roots of Arabidopsis thaliana. PLoS ONE 2013, 8, e56329. [Google Scholar] [CrossRef]
- Jarvis, S.G.; Woodward, S.; Taylor, A.F.S. Strong altitudinal partitioning in the distributions of ectomycorrhizal fungi along a short (300 m) elevation gradient. New Phytol. 2015, 206, 1145–1155. [Google Scholar] [CrossRef]
- Deng, Y.Y.; Wang, K.; Hu, Z.X.; Hu, Q.; Tang, Y.Z. Different geographic strains of dinoflagellate Karlodinium veneficum host highly diverse fungal community and potentially serve as possible niche for colonization of fungal endophytes. Int. J. Mol. Sci. 2023, 24, 1672. [Google Scholar] [CrossRef]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahe, F. VSEARCH: A versatile open source tool for metagenomics. Peerj 2016, 4, e2584. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2 (vol 37, pg 852, 2019). Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Cole, J.R.; Wang, Q.; Cardenas, E.; Fish, J.; Chai, B.; Farris, R.J.; Kulam-Syed-Mohideen, A.S.; McGarrell, D.M.; Marsh, T.; Garrity, G.M.; et al. The Ribosomal Database Project: Improved alignments and new tools for rRNA analysis. Nucleic Acids Res. 2009, 37, D141–D145. [Google Scholar] [CrossRef] [PubMed]
- Abarenkov, K.; Nilsson, R.H.; Larsson, K.H.; Alexander, I.J.; Eberhardt, U.; Erland, S.; Hoiland, K.; Kjoller, R.; Larsson, E.; Pennanen, T.; et al. The UNITE database for molecular identification of fungi—Recent updates and future perspectives. New Phytol. 2010, 186, 281–285. [Google Scholar] [CrossRef]
- Lindahl, B.D.; Nilsson, R.H.; Tedersoo, L.; Abarenkov, K.; Carlsen, T.; Kjoller, R.; Koljalg, U.; Pennanen, T.; Rosendahl, S.; Stenlid, J.; et al. Fungal community analysis by high-throughput sequencing of amplified markers—A user’s guide. New Phytol. 2013, 199, 288–299. [Google Scholar] [CrossRef] [PubMed]
- Lawson, C.A.; Raina, J.B.; Kahlke, T.; Seymour, J.R.; Suggett, D.J. Defining the core microbiome of the symbiotic dinoflagellate, Symbiodinium. Environ. Microbiol. Rep. 2018, 10, 7–11. [Google Scholar] [CrossRef]
- Hattenrath-Lehmann, T.K.; Jankowiak, J.; Koch, F.; Gobler, C.J. Prokaryotic and eukaryotic microbiomes associated with blooms of the ichthyotoxic dinoflagellate Cochlodinium (Margalefidinium) polykrikoides in New York, USA, estuaries. PLoS ONE 2019, 14, e0223067. [Google Scholar] [CrossRef]
- Sorenson, E.; Bertos-Fortis, M.; Farnelid, H.; Kremp, A.; Krueger, K.; Lindehoff, E.; Legrand, C. Consistency in microbiomes in cultures of Alexandrium species isolated from brackish and marine waters. Environ. Microbiol. Rep. 2019, 11, 425–433. [Google Scholar] [CrossRef]
- Wu, Z.; Lee, W.H.; Liu, Z.; Lin, S.; Lam, P.K.S. Microbiome Associated With Gambierdiscus balechii Cultures Under Different Toxicity Conditions. Front. Mar. Sci. 2022, 9, 760553. [Google Scholar] [CrossRef]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef] [PubMed]
- Caspi, R.; Billington, R.; Keseler, I.M.; Kothari, A.; Krummenacker, M.; Midford, P.E.; Ong, W.K.; Paley, S.; Subhraveti, P.; Karp, P.D. The MetaCyc database of metabolic pathways and enzymes—A 2019 update. Nucleic Acids Res. 2020, 48, D445–D453. [Google Scholar] [CrossRef] [PubMed]
- Parks, D.H.; Tyson, G.W.; Hugenholtz, P.; Beiko, R.G. STAMP: Statistical analysis of taxonomic and functional profiles. Bioinformatics 2014, 30, 3123–3124. [Google Scholar] [CrossRef] [PubMed]
- Astudillo Garcia, C.; Bell, J.J.; Webster, N.S.; Glasl, B.; Jompa, J.; Montoya, J.M.; Taylor, M.W. Evaluating the core microbiota in complex communities: A systematic investigation. Environ. Microbiol. 2017, 19, 1450–1462. [Google Scholar] [CrossRef]
- Kim, B.C.; Park, J.R.; Bae, J.W.; Rhee, S.K.; Kim, K.H.; Oh, J.W.; Park, Y.H. Stappia marina sp nov., a marine bacterium isolated from the Yellow Sea. Int. J. Syst. Evol. Microbiol. 2006, 56, 75–79. [Google Scholar] [CrossRef]
- Weber, C.F.; King, G.M. Physiological, ecological, and phylogenetic characterization of Stappia, a marine CO-oxidizing bacterial genus. Appl. Environ. Microbiol. 2007, 73, 1266–1276. [Google Scholar] [CrossRef]
- Trang Thi, V.; Kwon, B.R.; Eom, J.I.; Shin, B.K.; Kim, S.M. Interaction between marine bacterium Stappia sp. K01 and diatom Phaeodactylum tricornutum through extracellular fatty acids. J. Appl. Phycol. 2020, 32, 71–82. [Google Scholar] [CrossRef]
- Biebl, H.; Pukall, R.; Luensdorf, H.; Schulz, S.; Allgaier, M.; Tindall, B.J.; Wagner-Doebler, I. Description of Labrenzia alexandrii gen. nov., sp nov., a novel alphaproteobacterium containing bacterlochlorophyll a, and a proposal for reclassification of Stappia aggregata as Labrenzia aggregata comb. nov., of Stappia marina as Labrenzia marina comb. nov and of Stappia alba as Labrenzia alba comb. nov., and emended descriptions of the genera Pannonibacter, Stappia and Roseibium, and of the species Roseibium denhamense and Roseibium hamelinense. Int. J. Syst. Evol. Microbiol. 2007, 57, 1095–1107. [Google Scholar] [CrossRef]
- Denaro, R.; Crisafi, F.; Smedile, F.; Soprano, V.; Rossi, R.; Zingone, A.; Acosta, F.; Giacobbe, M.G. Microbiomes associated with cultures of Gambierdiscus australes and Ostreopsis cf. ovata, two epibenthic dinoflagellates from the NE Atlantic Ocean (Las Palmas, Gran Canaria). Mar. Ecol.-Evol. Persp. 2022, 43, e12699. [Google Scholar] [CrossRef]
- Shiba, T. Roseobacter litoralis gen. nov., sp. nov., and Roseobacter denitrificans sp. nov., Aerobic Pink-Pigmented Bacteria which Contain Bacteriochlorophyll a. Syst. Appl. Microbiol. 1991, 14, 140–145. [Google Scholar] [CrossRef]
- Csotonyi, J.T.; Maltman, C.; Swiderski, J.; Stackebrandt, E.; Yurkov, V. Extremely ’vanadiphilic’ multiply metal-resistant and halophilic aerobic anoxygenic phototrophs, strains EG13 and EG8, from hypersaline springs in Canada. Extremophiles 2015, 19, 127–134. [Google Scholar] [CrossRef]
- Gonzalez, J.M.; Kiene, R.P.; Moran, M.A. Transformation of sulfur compounds by an abundant lineage of marine bacteria in the alpha-subclass of the class Proteobacteria. Appl. Environ. Microbiol. 1999, 65, 3810–3819. [Google Scholar] [CrossRef] [PubMed]
- Kieft, B.; Li, Z.; Bryson, S.; Hettich, R.L.; Pan, C.L.; Mayali, X.; Mueller, R.S. Phytoplankton exudates and lysates support distinct microbial consortia with specialized metabolic and ecophysiological traits. Proc. Natl. Acad. Sci. USA 2021, 118, e2101178118. [Google Scholar] [CrossRef] [PubMed]
- Cooksey, K.E.; Wigglesworthcooksey, B. Adhesion of bacteria and diatoms to surface in the sea—A review Aquat. Microb. Ecol. 1995, 9, 87–96. [Google Scholar] [CrossRef]
- Vanloosdrecht, M.C.M.; Lyklema, J.; Norde, W.; Zehnder, A.J.B. Influence of interactions on microbial acitivity. Microbiol. Rev. 1990, 54, 75–87. [Google Scholar] [CrossRef]
- DeLong, E.F.; Franks, D.G.; Alldredge, A.L. Phylogenetic diversity of aggregate-attached vs. free-living marine bacterial assemblages. Limnol. Oceanogr. 1993, 38, 924–934. [Google Scholar] [CrossRef]
- Park, B.S.; Guo, R.Y.; Lim, W.A.; Ki, J.S. Pyrosequencing reveals specific associations of bacterial clades Roseobacter and Flavobacterium with the harmful dinoflagellate Cochlodinium polykrikoides growing in culture. Mar. Ecol.-Evol. Persp. 2017, 38, e12474. [Google Scholar] [CrossRef]
- Martínez-Mercado, M.A.; Cembella, A.D.; Sánchez-Castrejón, E.; Saavedra-Flores, A.; Galindo-Sánchez, C.E.; Durán-Riveroll, L.M. Functional diversity of bacterial microbiota associated with the toxigenic benthic dinoflagellate Prorocentrum. PLoS ONE 2024, 19, e0306108. [Google Scholar] [CrossRef]
- Prince, R.; Gramain, A.; McGenity, T. Handbook of Hydrocarbon and Lipid Microbiology; Springer: Berlin/Heidelberg, Germany, 2010; pp. 1669–1692. [Google Scholar]
- Gutierrez, T.; Nichols, P.D.; Whitman, W.B.; Aitken, M.D. Porticoccus hydrocarbonoclasticus sp. nov., an aromatic hydrocarbon-degrading bacterium identified in laboratory cultures of marine phytoplankton. Appl. Environ. Microbiol. 2012, 78, 628–637. [Google Scholar] [CrossRef]
- Parales, R.E.; Ditty, J.L.; Harwood, C.S. Toluene-degrading bacteria are chemotactic towards the environmental pollutants benzene, toluene, and trichloroethylene. Appl. Environ. Microbiol. 2000, 66, 4098–4104. [Google Scholar] [CrossRef]
- Saadoun, I.; Al-Akhras, M.A.; Abu-Ashour, J. Bacterial degradation of hydrocarbons as evidenced by respirometric analysis. Microbios 1999, 100, 19–25. [Google Scholar]
- Li, G.; He, Z.; An, T.; Zeng, X.; Sheng, G.; Fu, J. Comparative study of the elimination of toluene vapours in twin biotrickling filters using two microorganisms Bacillus cereus S1 and S2. J. Chem. Technol. Biotechnol. 2008, 83, 1019–1026. [Google Scholar] [CrossRef]
- Antonio, T. Marine Enzymes for Biocatalysis; Woodhead Publishing: Sawston, UK, 2013; pp. 373–402. [Google Scholar]
- Berry, D.; Gutierrez, T. Evaluating the Detection of Hydrocarbon-Degrading Bacteria in 16S rRNA Gene Sequencing Surveys. Front. Microbiol. 2017, 8, 896. [Google Scholar] [CrossRef] [PubMed]
- Lueders, T.; Kindler, R.; Miltner, A.; Friedrich, M.W.; Kaestner, M. Identification of bacterial micropredators distinctively active in a soil microbial food web. Appl. Environ. Microbiol. 2006, 72, 5342–5348. [Google Scholar] [CrossRef]
- Qi, Q.S.; Rehm, B.H.A. Polyhydroxybutyrate biosynthesis in Caulobacter crescentus: Molecular characterization of the polyhydroxybutyrate synthase. Microbiology 2001, 147, 3353–3358. [Google Scholar] [CrossRef] [PubMed]
- McAdams, H.H. Bacterial stalks are nutrient-scavenging antennas. Proc. Natl. Acad. Sci. USA 2006, 103, 11435–11436. [Google Scholar] [CrossRef]
- Gallego, S.; Vila, J.; Maria Nieto, J.; Urdiain, M.; Rossello-Mora, R.; Grifolla, M. Breoghania corrubedonensis gen. nov sp nov., a novel alphaproteobacterium isolated from a Galician beach (NW Spain) after the Prestige fuel oil spill, and emended description of the family Cohaesibacteraceae and the species Cohaesibacter gelatinilyticus. Syst. Appl. Microbiol. 2010, 33, 316–321. [Google Scholar] [CrossRef]
- Oh, H.-M.; Kang, I.; Vergin, K.L.; Lee, K.; Giovannoni, S.J.; Cho, J.-C. Genome Sequence of Oceanicaulis sp. Strain HTCC2633, Isolated from the Western Sargasso Sea. J. Bacteriol. 2011, 193, 317–318. [Google Scholar] [CrossRef]
- He, C.; Zheng, L.; Ding, J.; Gao, W.; Chi, W.; Ding, Y. Complete Genome Sequence of an N-Acyl Homoserine Lactone Producer, Breoghania sp. Strain L-A4, Isolated from Rhizosphere of Phragmites australis in a Coastal Wetland. Microbiol. Resour. Ann. 2019, 8, e01539-18. [Google Scholar] [CrossRef]
- Kong, X.; Li, C.; Wang, P.; Huang, G.; Li, Z.; Han, Z. Soil pollution characteristics and microbial responses in a vertical profile with long-term tannery sludge contamination in Hebei, China. Int. J. Environ. Res. Public Health 2019, 16, 563. [Google Scholar] [CrossRef]
- Zhang, X.L.; Qi, M.; Li, Q.H.; Cui, Z.D.; Yang, Q. Maricaulis alexandrii sp. nov., a novel active bioflocculants-bearing and dimorphic prosthecate bacterium isolated from marine phycosphere. Anton. Leeuw. Int. J. G. 2021, 114, 1195–1203. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Dong, X.; Luo, C.; Ma, S.; Xu, J.; Cui, Y. Nitrogen enrichment reduces the diversity of bacteria and alters their nutrient strategies in intertidal zones. Front. Mar. Sci. 2022, 9, 942074. [Google Scholar] [CrossRef]
- Uetake, J.; Yoshimura, Y.; Nagatsuka, N.; Kanda, H. Isolation of oligotrophic yeasts from supraglacial environments of different altitude on the Gulkana Glacier (Alaska). FEMS Microbiol. Ecol. 2012, 82, 279–286. [Google Scholar] [CrossRef]
- Wang, W.; Li, D.; Li, Y.; Hua, H.; Ma, E.; Li, Z. Caryophyllene Sesquiterpenes from the Marine-Derived Fungus Ascotricha sp ZJ-M-5 by the One Strain-Many Compounds Strategy. J. Nat. Prod. 2014, 77, 1367–1371. [Google Scholar] [CrossRef] [PubMed]
- Costa, F.d.F.; da Silva, N.M.; Voidaleski, M.F.; Weiss, V.A.; Moreno, L.F.; Schneider, G.X.; Najafzadeh, M.J.; Sun, J.; Gomes, R.R.; Raittz, R.T.; et al. Environmental prospecting of black yeast-like agents of human disease using culture-independent methodology. Sci. Rep. 2020, 10, 14229. [Google Scholar] [CrossRef]
- Liu, M.; Liu, L.; Chen, H.; Yu, Z.; Yang, J.R.; Xue, Y.; Huang, B.; Yang, J. Community dynamics of free-living and particle-attached bacteria following a reservoir Microcystis bloom. Sci. Total Environ. 2019, 660, 501–511. [Google Scholar] [CrossRef]
- Ortega-Retuerta, E.; Joux, F.; Jeffrey, W.H.; Ghiglione, J.F. Spatial variability of particle-attached and free-living bacterial diversity in surface waters from the Mackenzie River to the Beaufort Sea (Canadian Arctic). Biogeosciences 2013, 10, 2747–2759. [Google Scholar] [CrossRef]
- Xu, H.; Zhao, D.; Huang, R.; Cao, X.; Zeng, J.; Yu, Z.; Hooker, K.V.; Hambright, K.D.; Wu, Q.L. Contrasting Network Features between Free-Living and Particle-Attached Bacterial Communities in Taihu Lake. Microb. Ecol. 2018, 76, 303–313. [Google Scholar] [CrossRef]
- Hu, Y.; Xie, G.; Jiang, X.; Shao, K.; Tang, X.; Gao, G. The relationships between the free-living and particle-attached bacterial communities in response to elevated eutrophication. Front. Microbiol. 2020, 11, 423. [Google Scholar] [CrossRef]
- Larsson, U.; Hagstrom, A. Phytoplankton exudate release as an energy source for the growth of pelagic bacteria. Mar. Biol. 1979, 52, 199–206. [Google Scholar] [CrossRef]
- Biddanda, B.A.; Pomeroy, L.R. Microbial aggregation and degradation of phytoplankton-derived detritus in seawater. I. microbial succession. Mar. Ecol. Prog. Ser. 1988, 42, 79–88. [Google Scholar] [CrossRef]
- Pikovskaya, R.I. Mobilization of phosphorus in soil in connection with the vital activity of some microbial species. Mikrobiologya 1948, 7, 362–370. [Google Scholar]
- Marra, L.M.; Fonseca Sousa Soares, C.R.; de Oliveira, S.M.; Avelar Ferreira, P.A.; Soares, B.L.; Carvalho, R.d.F.; de Lima, J.M.; de Souza Moreira, F.M. Biological nitrogen fixation and phosphate solubilization by bacteria isolated from tropical soils. Plant Soil 2012, 357, 289–307. [Google Scholar] [CrossRef]
- Zeng, Q.G.; Luo, F.; Zhang, Z.B.; Yan, R.M.; Zhu, D. Phosphate solubilizing rhizospherebacterial T21 isolated from Dongxiang wild rice species promotes cultivated rice growth. Appl. Mech. Mater. 2012, 108, 167–175. [Google Scholar] [CrossRef]
- Lu, C.; Liu, H.; Jiang, D.; Wang, L.; Jiang, Y.; Tang, S.; Hou, X.; Han, X.; Liu, Z.; Zhang, M.; et al. Paecilomyces variotii extracts (ZNC) enhance plant immunity and promote plant growth. Plant Soil 2019, 441, 383–397. [Google Scholar] [CrossRef]
- Constantin, M.; Raut, I.; Gurban, A.-M.; Doni, M.; Radu, N.; Alexandrescu, E.; Jecu, L. Exploring the Potential Applications of Paecilomyces lilacinus 112. Appl. Sci. 2022, 12, 7572. [Google Scholar] [CrossRef]
- Ahuja, A.; D’Souza, S.F. Bioprocess for Solubilization of Rock Phosphate on Starch Based Medium by Paecilomyces marquandii Immobilized on Polyurethane Foam. Appl. Biochem. Biotechnol. 2009, 152, 1–5. [Google Scholar] [CrossRef]
- Wang, C.; Lin, X.; Li, L.; Lin, L.; Lin, S. Glyphosate Shapes a Dinoflagellate-Associated Bacterial Community While Supporting Algal Growth as Sole Phosphorus Source. Front. Microbiol. 2017, 8, 2530. [Google Scholar] [CrossRef]
- Guidi, F.; Pezzolesi, L.; Vanucci, S. Microbial dynamics during harmful dinoflagellate Ostreopsis cf. ovata growth: Bacterial succession and viral abundance pattern. Microbiologyopen 2018, 7, e00584. [Google Scholar] [CrossRef]
- Nampoothiri, K.M.; Tomes, G.J.; Roopesh, K.; Szakacs, G.; Nagy, V.; Soccol, C.R.; Pankey, A. Thermostable phytase production by Thermoascus aurantiacus in submerged fermentation. Appl. Biochem. Biotechnol. 2004, 118, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Asan Ozusaglam, M.; Ozcan, N. Cloning of Phytase Gene in Probiotic Bacterium Bacillus coagulans. Adv. Stud. Biol. 2008, 1, 15–24. [Google Scholar]
- Ellwood, N.T.W.; Pasella, M.M.; Totti, C.; Accoroni, S. Growth and phosphatase activities of Ostreopsis cf. ovata biofilms supplied with diverse dissolved organic phosphorus (DOP) compounds. Aquat. Microb. Ecol. 2020, 85, 155–166. [Google Scholar] [CrossRef]
- Singh, B.; Boukhris, I.; Pragya; Kumar, V.; Yadav, A.N.; Farhat-Khemakhem, A.; Kumar, A.; Singh, D.; Blibech, M.; Chouayekh, H.; et al. Contribution of microbial phytases to the improvement of plant growth and nutrition: A review. Pedosphere 2020, 30, 295–313. [Google Scholar] [CrossRef]
- Paytan, A.; McLaughlin, K. The oceanic phosphorus cycle. Chem. Rev. 2007, 107, 563–576. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Litaker, R.W.; Sunda, W.G. Phosphorus physiological ecology and molecular mechanisms in marine phytoplankton. J. Phycol. 2016, 52, 10–36. [Google Scholar] [CrossRef]
- Ou, L.; Wang, D.; Huang, B.; Hong, H.; Qi, Y.; Lu, S. Comparative study of phosphorus strategies of three typical harmful algae in Chinese coastal waters. J. Plankton Res. 2008, 30, 1007–1017. [Google Scholar] [CrossRef]
- Jauzein, C.; Labry, C.; Youenou, A.; Quere, J.; Delmas, D.; Collos, Y. Growth and phosphorus uptake by the toxic dinoflagellate Alexandrium Catenella (Dinophyceae) in response to phosphate limitation. J. Phycol. 2010, 46, 926–936. [Google Scholar] [CrossRef]
- Rengefors, K.; Anderson, D.M.; Pettersson, K. Phosphorus uptake by resting cysts of the marine dinoflagellate Scrippsiella trochoidea. J. Plankton Res. 1996, 18, 1753–1765. [Google Scholar] [CrossRef]
- Rengefors, K.; McCall, R.D.; Heaney, S.I. Quantitative X-ray microanalysis as a method for measuring phosphorus in dinoflagellate resting cysts. Eur. J. Phycol. 1999, 34, 171–177. [Google Scholar] [CrossRef]
- Li, F.; Yue, C.; Deng, Y.; Tang, Y.Z. Characterizing the Status of Energetic Metabolism of Dinoflagellate Resting Cysts under Mock Conditions of Marine Sediments via Physiological and Transcriptional Measurements. Int. J. Mol. Sci. 2022, 23, 15033. [Google Scholar] [CrossRef] [PubMed]
- Lundholm, N.; Ribeiro, S.; Andersen, T.J.; Koch, T.; Godhe, A.; Ekelund, F.; Ellegaard, M. Buried alive—Germination of up to a century-old marine protist resting stages. Phycologia 2011, 50, 629–640. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID | Sample Name | Association with Scrippsiella acuminata Cells | Life Cycle Stage | Temperature | Incubation Time |
|---|---|---|---|---|---|
| IOCAS_5 | V7T15-A | Attached | Vegetative cells | 15 °C | 7 d |
| IOCAS_6 | V20T15-A | Attached | Vegetative cells | 15 °C | 20 d |
| IOCAS_7 | V7T21-A | Attached | Vegetative cells | 21 °C | 7 d |
| IOCAS_8 | V20T21-A | Attached | Vegetative cells | 21 °C | 20 d |
| IOCAS_15 | C30T4-A | Attached | Cysts | 4 °C | 30 d |
| IOCAS_16 | C45T4-A | Attached | Cysts | 4 °C | 45 d |
| IOCAS_17 | C30T15-A | Attached | Cysts | 15 °C | 30 d |
| IOCAS_18 | C45T15-A | Attached | Cysts | 15 °C | 45 d |
| IOCAS_19 | C30T21-A | Attached | Cysts | 21 °C | 30 d |
| IOCAS_20 | C45T21-A | Attached | Cysts | 21 °C | 45 d |
| IOCAS_E | V7T15-FL | Free-living | Vegetative cells | 15 °C | 7 d |
| IOCAS_F | V20T15-FL | Free-living | Vegetative cells | 15 °C | 20 d |
| IOCAS_G | V7T21-FL | Free-living | Vegetative cells | 21 °C | 7 d |
| IOCAS_H | V20T21-FL | Free-living | Vegetative cells | 21 °C | 20 d |
| IOCAS_O | C30T4-FL | Free-living | Cysts | 4 °C | 30 d |
| IOCAS_P | C45T4-FL | Free-living | Cysts | 4 °C | 45 d |
| IOCAS_Q | C30T15-FL | Free-living | Cysts | 15 °C | 30 d |
| IOCAS_R | C45T15-FL | Free-living | Cysts | 15 °C | 45 d |
| IOCAS_S | C30T21-FL | Free-living | Cysts | 21 °C | 30 d |
| IOCAS_T | C45T21-FL | Free-living | Cysts | 21 °C | 45 d |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yue, C.; Chai, Z.; Li, F.; Shang, L.; Hu, Z.; Deng, Y.; Tang, Y.-Z. Transitions of the Bacteria–Fungi Microbiomes Associated with Different Life Cycle Stages of Dinoflagellate Scrippsiella acuminata. Microorganisms 2025, 13, 1340. https://doi.org/10.3390/microorganisms13061340
Yue C, Chai Z, Li F, Shang L, Hu Z, Deng Y, Tang Y-Z. Transitions of the Bacteria–Fungi Microbiomes Associated with Different Life Cycle Stages of Dinoflagellate Scrippsiella acuminata. Microorganisms. 2025; 13(6):1340. https://doi.org/10.3390/microorganisms13061340
Chicago/Turabian StyleYue, Caixia, Zhaoyang Chai, Fengting Li, Lixia Shang, Zhangxi Hu, Yunyan Deng, and Ying-Zhong Tang. 2025. "Transitions of the Bacteria–Fungi Microbiomes Associated with Different Life Cycle Stages of Dinoflagellate Scrippsiella acuminata" Microorganisms 13, no. 6: 1340. https://doi.org/10.3390/microorganisms13061340
APA StyleYue, C., Chai, Z., Li, F., Shang, L., Hu, Z., Deng, Y., & Tang, Y.-Z. (2025). Transitions of the Bacteria–Fungi Microbiomes Associated with Different Life Cycle Stages of Dinoflagellate Scrippsiella acuminata. Microorganisms, 13(6), 1340. https://doi.org/10.3390/microorganisms13061340