
Genome Sequences of the First Phages Infecting Limnohabitans Reveal Their Global Distribution and Metabolic Potential

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Isolation of Limnohabitans and Phages
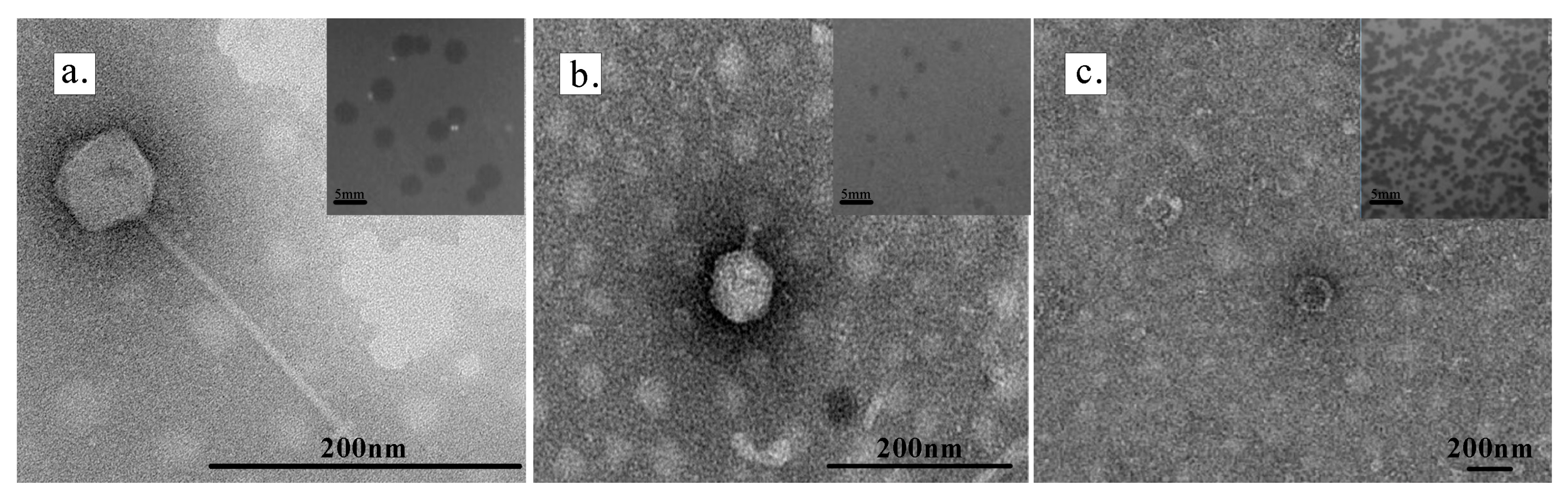
2.2. Transmission Electron Microscopy (TEM)
2.3. Extraction of Bacterial and Phage Genomic DNA
2.4. Phylogenetic Analysis Based on the 16S rRNA Gene
2.5. Phage Genome Sequencing and Bioinformatics Analysis
3. Results
3.1. Host Strains
3.2. Biological Characteristics of Three Limnohabitans Phages
3.3. General Genomic Characteristics of Three Limnohabitans Phages
3.4. Metabolic Potential of Three Limnohabitans Phages
3.5. Globally Distribution of Three Limnohabitans Phages
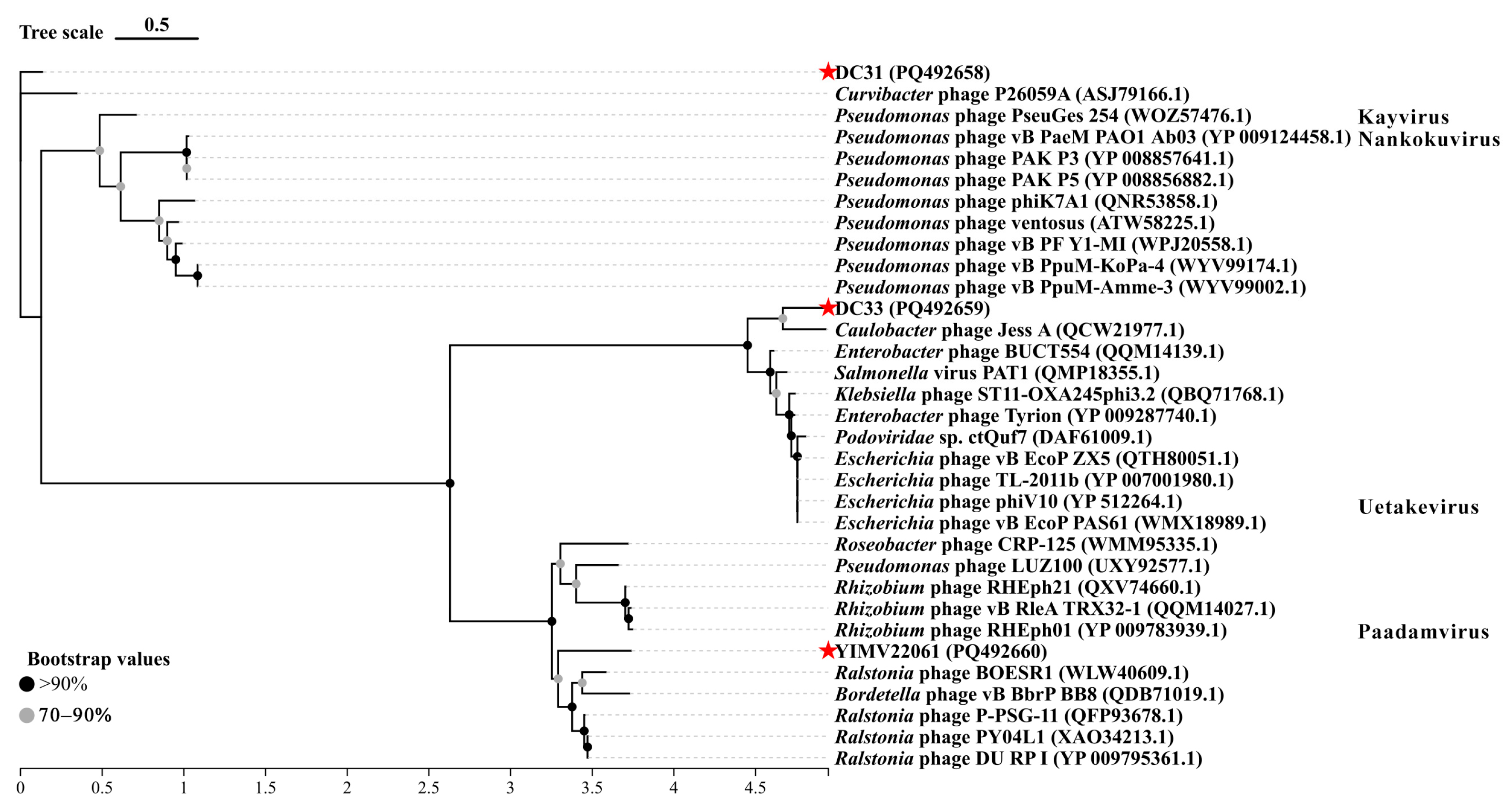
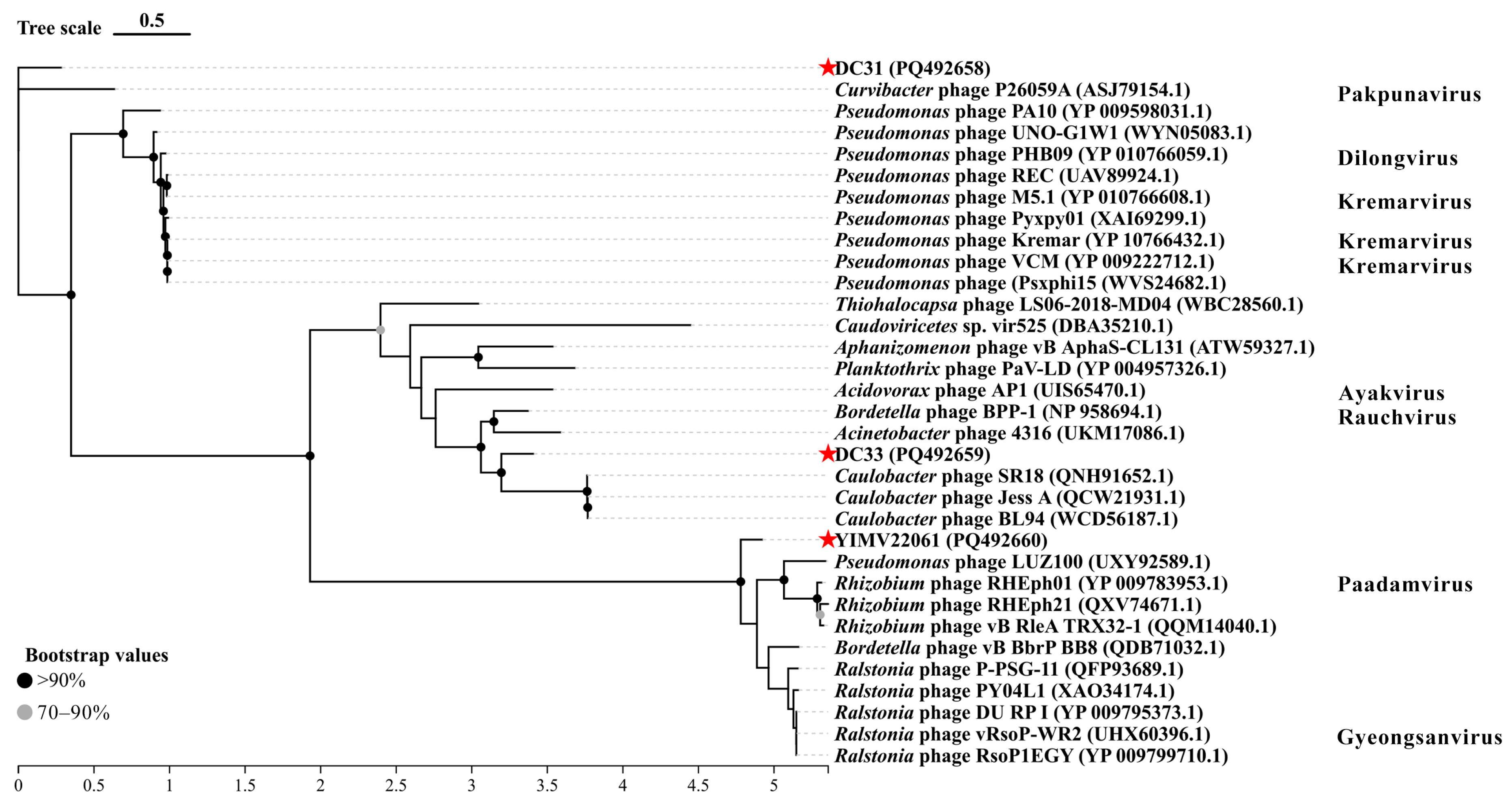
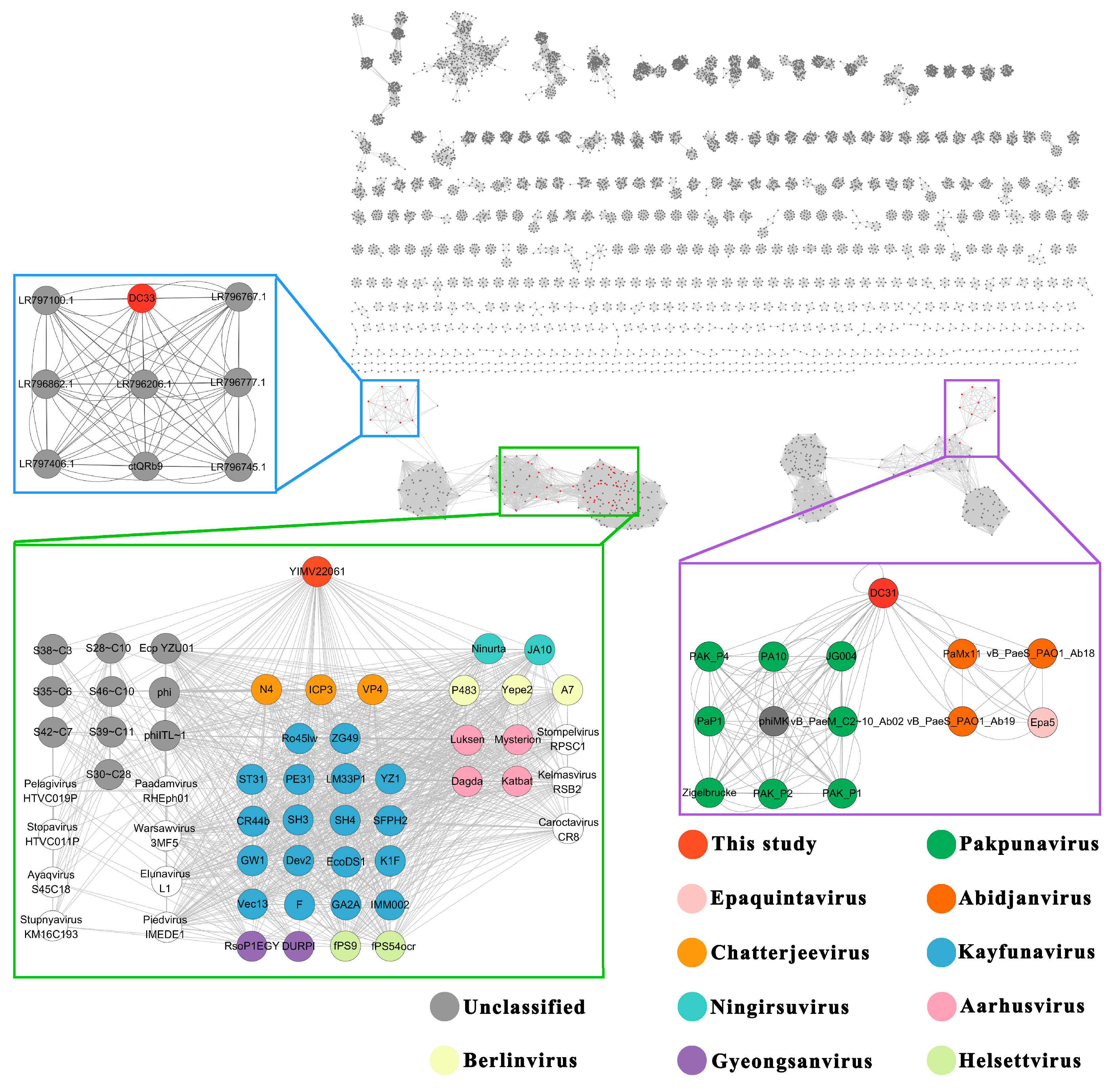
3.6. The First Genome of Phages Infecting Limnohabitans
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AMG | Auxiliary metabolic gene |
| ORF | Open reading frame |
| ATP | Adenosine triphosphate |
| dCMP | Deoxycytidine monophosphate |
| dUMP | Deoxyuridine monophosphate |
| dTMP | Deoxythymidine monophosphate |
| MCP | Major capsid protein |
| TerL | Terminase large subunit |
References
- Sime-Ngando, T. Environmental bacteriophages: Viruses of microbes in aquatic ecosystems. Front. Microbiol. 2014, 5, 355. [Google Scholar] [CrossRef] [PubMed]
- Cobián Güemes, A.G.; Youle, M.; Cantú, V.A.; Felts, B.; Nulton, J.; Rohwer, F. Viruses as winners in the game of life. Annu. Rev. Virol. 2016, 3, 197–214. [Google Scholar] [CrossRef] [PubMed]
- Suttle, C.A. Marine viruses—Major players in the global ecosystem. Nat. Rev. Microbiol. 2007, 5, 801–812. [Google Scholar] [CrossRef]
- Kavagutti, V.S.; Andrei, A.Ş.; Mehrshad, M.; Salcher, M.M.; Ghai, R. Phage-centric ecological interactions in aquatic ecosystems revealed through ultradeep metagenomics. Microbiome 2019, 7, 1–15. [Google Scholar] [CrossRef]
- Zhao, A.; Lu, Y.; Li, Q.; Li, T.; Zhao, J. Metagenomics reveals the diversity and role of surface-water microbes in biogeochemical cycles in lakes at different terrain ladders. Front. Environ. Sci. 2023, 11, 1121775. [Google Scholar] [CrossRef]
- Che, R.; Bai, M.; Xiao, W.; Zhang, S.; Wang, Y.; Cui, X. Nutrient levels and prokaryotes affect viral communities in plateau lakes. Sci. Total. Environ. 2022, 839, 156033. [Google Scholar] [CrossRef]
- Malki, K.; Kula, A.; Bruder, K.; Sible, E.; Hatzopoulos, T.; Steidel, S.; Watkins, S.C.; Putonti, C. Bacteriophages isolated from Lake Michigan demonstrate broad host-range across several bacterial phyla. Virol. J. 2015, 12, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Moon, K.; Kang, I.; Kim, S.; Kim, S.J.; Cho, J.C. Genomic and ecological study of two distinctive freshwater bacteriophages infecting a Comamonadaceae bacterium. Sci. Rep. 2018, 8, 7989. [Google Scholar] [CrossRef]
- Laanto, E.; Oksanen, H.M. Three phages from a boreal lake during ice cover infecting Xylophilus, Caulobacter, and Polaromonas species. Viruses 2023, 15, 307. [Google Scholar] [CrossRef]
- Yang, F.; Jin, H.; Wang, X.Q.; Li, Q.; Zhang, J.T.; Cui, N.; Jiang, Y.L.; Chen, Y.; Wu, Q.F.; Zhou, C.Z.; et al. Genomic analysis of Mic1 reveals a novel freshwater long-tailed cyanophage. Front. Microbiol. 2020, 11, 484. [Google Scholar] [CrossRef]
- Zhang, D.; He, Y.; Gin, K.Y. Genomic characterization of a novel freshwater cyanophage reveals a new lineage of cyanopodovirus. Front. Microbiol. 2022, 12, 768868. [Google Scholar] [CrossRef] [PubMed]
- Simek, K.; Kasalicky, V.; Jezbera, J.; Jezberová, J.; Hejzlar, J.; Hahn, M.W. Broad habitat range of the phylogenetically narrow R-BT065 cluster, representing a core group of the Betaproteobacterial genus Limnohabitans. Appl. Environ. Microbiol. 2010, 76, 631–639. [Google Scholar] [CrossRef]
- Props, R.; Denef, V.J. Temperature and nutrient levels correspond with lineage-specific microdiversification in the ubiquitous and abundant freshwater genus Limnohabitans. Appl. Environ. Microbiol. 2020, 86, e00140-20. [Google Scholar] [CrossRef] [PubMed]
- Kasalický, V.; Zeng, Y.; Piwosz, K.; Šimek, K.; Kratochvilová, H.; Koblížek, M. Aerobic anoxygenic photosynthesis is commonly present within the genus Limnohabitans. Appl. Environ. Microbiol. 2018, 84, e02116-17. [Google Scholar] [CrossRef]
- Rofner, C.; Sommaruga, R.; Pérez, M.T. Differential utilization patterns of dissolved organic phosphorus compounds by heterotrophic bacteria in two mountain lakes. FEMS Microbiol. Ecol. 2016, 92, fiw139. [Google Scholar] [CrossRef]
- Šimek, K.; Kasalický, V.; Zapomělová, E.; Horňák, K. Alga-derived substrates select for distinct betaproteobacterial lineages and contribute to niche separation in Limnohabitans strains. Appl. Environ. Microbiol. 2011, 77, 7307–7315. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Wu, X.; Zhang, Y.; Zheng, B.; Meng, D.; Zhou, H.; Lu, L.; Deng, W.; Shao, Z.; Qin, Y. Management of water quality targets based on river-lake water quality response relationships for lake basins–a case study of Dianchi Lake. Environ. Res. 2020, 186, 109479. [Google Scholar] [CrossRef]
- Lin, Z.; Peng, S. Comparison of multimodel simulations of land use and land cover change considering integrated constraints-A case study of the Fuxian Lake basin. Ecol. Indic. 2022, 142, 109254. [Google Scholar] [CrossRef]
- Fu, C.Q.; Zhao, Q.; Li, Z.Y.; Wang, Y.X.; Zhang, S.Y.; Lai, Y.H.; Xiao, W.; Cui, X.L. A novel Halomonas ventosae-specific virulent halovirus isolated from the Qiaohou salt mine in Yunnan, Southwest China. Extremophiles 2016, 20, 101–110. [Google Scholar] [CrossRef]
- Diao, K.; Li, G.; Sun, X.; Yi, H.; Zhang, S.; Xiao, W. Genomic Characterization of a Halovirus Representing a Novel Siphoviral Cluster. Viruses 2023, 15, 1392. [Google Scholar] [CrossRef]
- Lekunberri, I.; Villagrasa, M.; Balcázar, J.L.; Borrego, C.M. Contribution of bacteriophage and plasmid DNA to the mobilization of antibiotic resistance genes in a river receiving treated wastewater discharges. Sci. Total. Environ. 2017, 601, 206–209. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Chen, Y.; Shi, C.; Huang, Z.; Zhang, Y.; Li, S.; Li, Y.; Ye, J.; Yu, C.; Li, Z.; et al. SOAPnuke: A MapReduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data. Gigascience 2018, 7, gix120. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Li, D.; Liu, C.M.; Luo, R.; Sadakane, K.; Lam, T.W. MEGAHIT: An ultrafast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef] [PubMed]
- Nayfach, S.; Camargo, A.P.; Schulz, F.; Eloe-Fadrosh, E.; Roux, S.; Kyrpides, N.C. CheckV assesses the quality and completeness of metagenome-assembled viral genomes. Nat. Biotechnol. 2021, 39, 578–585. [Google Scholar] [CrossRef]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Marchler-Bauer, A.; Panchenko, A.R.; Shoemaker, B.A.; Thiessen, P.A.; Geer, L.Y.; Bryant, S.H. CDD: A database of conserved domain alignments with links to domain three-dimensional structure. Nucleic Acids Res. 2002, 30, 281–283. [Google Scholar] [CrossRef]
- Jiang, J.Z.; Yuan, W.G.; Shang, J.; Shi, Y.H.; Yang, L.L.; Liu, M.; Zhu, P.; Jin, T.; Sun, Y.; Yuan, L.H. Virus classification for viral genomic fragments using PhaGCN2. Brief. Bioinform. 2023, 24, bbac505. [Google Scholar] [CrossRef]
- Buchfink, B.; Ashkenazy, H.; Reuter, K.; Kennedy, J.A.; Drost, H.G. Sensitive clustering of protein sequences at tree-of-life scale using DIAMOND DeepClust. BioRxiv 2023. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Camargo, A.P.; Nayfach, S.; Chen, I.A.; Palaniappan, K.; Ratner, A.; Chu, K.; Ritter, S.J.; Reddy, T.B.K.; Mukherjee, S.; Schulz, F.; et al. IMG/VR v4: An expanded database of uncultivated virus genomes within a framework of extensive functional, taxonomic, and ecological metadata. Nucleic Acids Res. 2023, 51, D733–D743. [Google Scholar] [CrossRef]
- Xie, J.; Chen, Y.; Cai, G.; Cai, R.; Hu, Z.; Wang, H. Tree Visualization By One Table (tvBOT): A web application for visualizing, modifying and annotating phylogenetic trees. Nucleic Acids Res. 2023, 51, W587–W592. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Göker, M. VICTOR: Genome-based phylogeny and classification of prokaryotic viruses. Bioinformatics 2017, 33, 3396–3404. [Google Scholar] [CrossRef]
- Moraru, C.; Varsani, A.; Kropinski, A.M. VIRIDIC–a novel tool to calculate the intergenomic similarities of prokaryote-infecting viruses. Viruses 2020, 12, 1268. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, S.; Suzuki, Y. Chloramphenicol-, dihydrostreptomycin-, and kanamycin-inactivating enzymes from multiple drug-resistant Escherichia coli carrying episome‘R’. Nature 1965, 208, 1301–1303. [Google Scholar] [CrossRef]
- Lurz, R.; Orlova, E.V.; Günther, D.; Dube, P.; Dröge, A.; Weise, F.; van Heel, M.; Tavares, P. Structural organization of the head-to-tail interface of a bacterial virus. J. Mol. Biol. 2001, 310, 1027–1037. [Google Scholar] [CrossRef]
- Young, R.Y.; Wang, N.; Roof, W.D. Phages will out: Strategies of host cell lysis. Trends Microbiol. 2000, 8, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Cahill, J.; Young, R. Phage lysis: Multiple genes for multiple barriers. Adv. Virus Res. 2019, 103, 33–70. [Google Scholar]
- Hsieh, Y.J.; Wanner, B.L. Global regulation by the seven-component Pi signalling system. Curr. Opin. Microbiol. 2010, 13, 198–203. [Google Scholar] [CrossRef]
- Keller, L.M.; Weber-Ban, E. An emerging class of nucleic acid-sensing regulators in bacteria: WYL domain-containing proteins. Curr. Opin. Microbiol. 2023, 74, 102296. [Google Scholar] [CrossRef] [PubMed]
- Rojas-Pirela, M.; Andrade-Alviárez, D.; Rojas, V.; Kemmerling, U.; Cáceres, A.J.; Michels, P.A.; Concepción, J.L.; Quiñones, W. Phosphoglycerate kinase: Structural aspects and functions, with special emphasis on the enzyme from Kinetoplastea. Open Biol. 2020, 10, 200302. [Google Scholar] [CrossRef]
- Banks, R.D.; Blake, C.C.; Evans, P.R.; Haser, R.; Rice, D.W.; Hardy, G.W.; Merrett, M.; Phillips, A.W. Sequence, structure and activity of phosphoglycerate kinase: A possible hinge-bending enzyme. Nature 1979, 279, 773–777. [Google Scholar] [CrossRef] [PubMed]
- Maley, F.; Maley, G.F. A tale of two enzymes, deoxycytidylate deaminase and thymidylate synthase. Prog. Nucleic Acid Res. Mol. Biol. 1990, 39, 49–80. [Google Scholar]
- Holmgren, A. Thioredoxin and glutaredoxin systems. J. Biol. Chem. 1989, 264, 13963–13966. [Google Scholar] [CrossRef] [PubMed]
- Stewart, E.J.; Åslund, F.; Beckwith, J. Disulfide bond formation in the Escherichia coli cytoplasm: An in vivo role reversal for the thioredoxins. EMBO J. 1998, 17, 5543–5550. [Google Scholar] [CrossRef]
- Rioseras, B.; Yagüe, P.; López-García, M.T.; Gonzalez-Quiñonez, N.; Binda, E.; Marinelli, F.; Manteca, A. Characterization of SCO4439, a D-alanyl-D-alanine carboxypeptidase involved in spore cell wall maturation, resistance and germination in Streptomyces coelicolor. Sci. Rep. 2016, 6, 21659. [Google Scholar] [CrossRef]
- Greve, J.M.; Pinkham, A.M.; Cowan, J.A. Human aspartyl (asparaginyl) hydroxylase. A multifaceted enzyme with broad intra- and extra-cellular activity. Metallomics 2021, 13, mfab044. [Google Scholar] [CrossRef]
- Bollen, M.; Gijsbers, R.; Ceulemans, H.; Stalmans, W.; Stefan, C. Nucleotide pyrophosphatases/phosphodiesterases on the move. Crit. Rev. Biochem. Mol. Biol. 2000, 35, 393–432. [Google Scholar] [CrossRef]
- Iyer, L.M.; Koonin, E.V.; Aravind, L. Classification and evolutionary history of the single-strand annealing proteins, RecT, Redβ, ERF and RAD52. BMC Genom. 2002, 3, 1–11. [Google Scholar] [CrossRef]
- Fleming, J.R.; Hauth, F.; Hartig, J.S.; Mayans, O. Crystal structure of a GCN5-related N-acetyltransferase from Lactobacillus curiae. Acta Crystallogr. F Struct. Biol. Commun. 2023, 79 Pt 8, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Alekshun, M.N.; Levy, S.B. The mar regulon: Multiple resistance to antibiotics and other toxic chemicals. Trends Microbiol. 1999, 7, 410–413. [Google Scholar] [CrossRef] [PubMed]
- Haque, M.M.; Kabir, M.S.; Aini, L.Q.; Hirata, H.; Tsuyumu, S. SlyA, a MarR family transcriptional regulator, is essential for virulence in Dickeya dadantii 3937. J. Bacteriol. 2009, 191, 5409–5418. [Google Scholar] [CrossRef] [PubMed]
- Nazaret, F.; Alloing, G.; Mandon, K.; Frendo, P. MarR family transcriptional regulators and their roles in plant-interacting bacteria. Microorganisms 2023, 11, 1936. [Google Scholar] [CrossRef]
- Holtappels, D.; Alfenas-Zerbini, P.; Koskella, B. Drivers and consequences of bacteriophage host range. FEMS Microbiol. Rev. 2023, 47, fuad038. [Google Scholar] [CrossRef]
- Goldsmith, D.B.; Crosti, G.; Dwivedi, B.; McDaniel, L.D.; Varsani, A.; Suttle, C.A.; Weinbauer, M.G.; Sandaa, R.A.; Breitbart, M. Development of phoH as a novel signature gene for assessing marine phage diversity. Appl. Environ. Microbiol. 2011, 77, 7730–7739. [Google Scholar] [CrossRef]
- Wang, X.; Liu, J.; Yu, Z.; Jin, J.; Liu, X.; Wang, G. Novel groups and unique distribution of phage phoH genes in paddy waters in northeast China. Sci. Rep. 2016, 6, 38428. [Google Scholar] [CrossRef]
- Nilsson, E.; Li, K.; Fridlund, J.; Šulčius, S.; Bunse, C.; Karlsson, C.M.; Lindh, M.; Lundin, D.; Pinhassi, J.; Holmfeldt, K. Genomic and seasonal variations among aquatic phages infecting the Baltic Sea gammaproteobacterium Rheinheimera sp. strain BAL341. Appl. Environ. Microbiol. 2019, 85, e01003-19. [Google Scholar] [CrossRef]
- Kim, S.K.; Makino, K.O.; Amemura, M.I.; Shinagawa, H.I.; Nakata, A.T. Molecular analysis of the phoH gene, belonging to the phosphate regulon in Escherichia coli. J. Bacteriol. 1993, 175, 1316–1324. [Google Scholar] [CrossRef]
- Martiny, A.C.; Coleman, M.L.; Chisholm, S.W. Phosphate acquisition genes in Prochlorococcus ecotypes: Evidence for genome-wide adaptation. Proc. Natl. Acad. Sci. USA 2006, 103, 12552–12557. [Google Scholar] [CrossRef]
- Kazakov, A.E.; Vassieva, O.; Gelfand, M.S.; Osterman, A.; Overbeek, R. Bioinformatics classification and functional analysis of PhoH homologs. In Silico Biol. 2003, 3, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Maley, G.F. Deoxycytidylate deaminase from T2-infected Escherichia coli. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 1978; Volume 51, pp. 412–418. [Google Scholar]
- Dunigan, D.D.; Agarkova, I.V.; Esmael, A.; Alvarez, S.; Van Etten, J.L. Early-Phase Drive to the Precursor Pool: Chloroviruses Dive into the Deep End of Nucleotide Metabolism. Viruses 2023, 15, 911. [Google Scholar] [CrossRef] [PubMed]
- Wikner, J.; Vallino, J.J.; Steward, G.F.; Smith, D.C.; Azam, F. Nucleic acids from the host bacterium as a major source of nucleotides for three marine bacteriophages. FEMS Microbiol. Ecol. 1993, 12, 237–248. [Google Scholar] [CrossRef]
- Zimmerman, A.E.; Howard-Varona, C.; Needham, D.M.; John, S.G.; Worden, A.Z.; Sullivan, M.B.; Waldbauer, J.R.; Coleman, M.L. Metabolic and biogeochemical consequences of viral infection in aquatic ecosystems. Nat. Rev. Microbiol. 2020, 18, 21–34. [Google Scholar] [CrossRef]
- Rihtman, B.; Bowman-Grahl, S.; Millard, A.; Corrigan, R.M.; Clokie, M.R.; Scanlan, D.J. Cyanophage MazG is a pyrophosphohydrolase but unable to hydrolyse magic spot nucleotides. Environ. Microbiol. Rep. 2019, 11, 448–455. [Google Scholar] [CrossRef] [PubMed]
- Shi, K.X.; Wu, Y.K.; Tang, B.K.; Zhao, G.P.; Lyu, L.D. Housecleaning of pyrimidine nucleotide pool coordinates metabolic adaptation of nongrowing Mycobacterium tuberculosis. Emerg. Microbes Infect. 2019, 8, 40–44. [Google Scholar] [CrossRef]
- Ho, P.; Chen, Y.; Biswas, S.; Canfield, E.; Abdolvahabi, A.; Feldman, D.E. Bacteriophage antidefense genes that neutralize TIR and STING immune responses. Cell Rep. 2023, 42, 112305. [Google Scholar] [CrossRef]
- Qian, P.Y.; Cheng, A.; Wang, R.; Zhang, R. Marine biofilms: Diversity, interactions and biofouling. Nat. Rev. Microbiol. 2022, 20, 671–684. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deng, B.; Che, R.; Zhu, P.; Wang, Y.; Li, Z.; Zhang, S.; Xiao, W. Genome Sequences of the First Phages Infecting Limnohabitans Reveal Their Global Distribution and Metabolic Potential. Microorganisms 2025, 13, 1324. https://doi.org/10.3390/microorganisms13061324
Deng B, Che R, Zhu P, Wang Y, Li Z, Zhang S, Xiao W. Genome Sequences of the First Phages Infecting Limnohabitans Reveal Their Global Distribution and Metabolic Potential. Microorganisms. 2025; 13(6):1324. https://doi.org/10.3390/microorganisms13061324
Chicago/Turabian StyleDeng, Boxuan, Raoqiong Che, Pinxin Zhu, Yongxia Wang, Zhiying Li, Shiying Zhang, and Wei Xiao. 2025. "Genome Sequences of the First Phages Infecting Limnohabitans Reveal Their Global Distribution and Metabolic Potential" Microorganisms 13, no. 6: 1324. https://doi.org/10.3390/microorganisms13061324
APA StyleDeng, B., Che, R., Zhu, P., Wang, Y., Li, Z., Zhang, S., & Xiao, W. (2025). Genome Sequences of the First Phages Infecting Limnohabitans Reveal Their Global Distribution and Metabolic Potential. Microorganisms, 13(6), 1324. https://doi.org/10.3390/microorganisms13061324