
Salinity Effect on Soil Bacterial and Archaeal Diversity and Assembly in Phragmites australis Salt Marshes in the Qaidam Basin, China

 , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Soil Sampling and Physicochemical Analysis
2.2. DNA Extraction and Microbial Community Sequencing
2.3. Co-Occurrence Network Construction
2.4. Statistical Analysis
3. Results
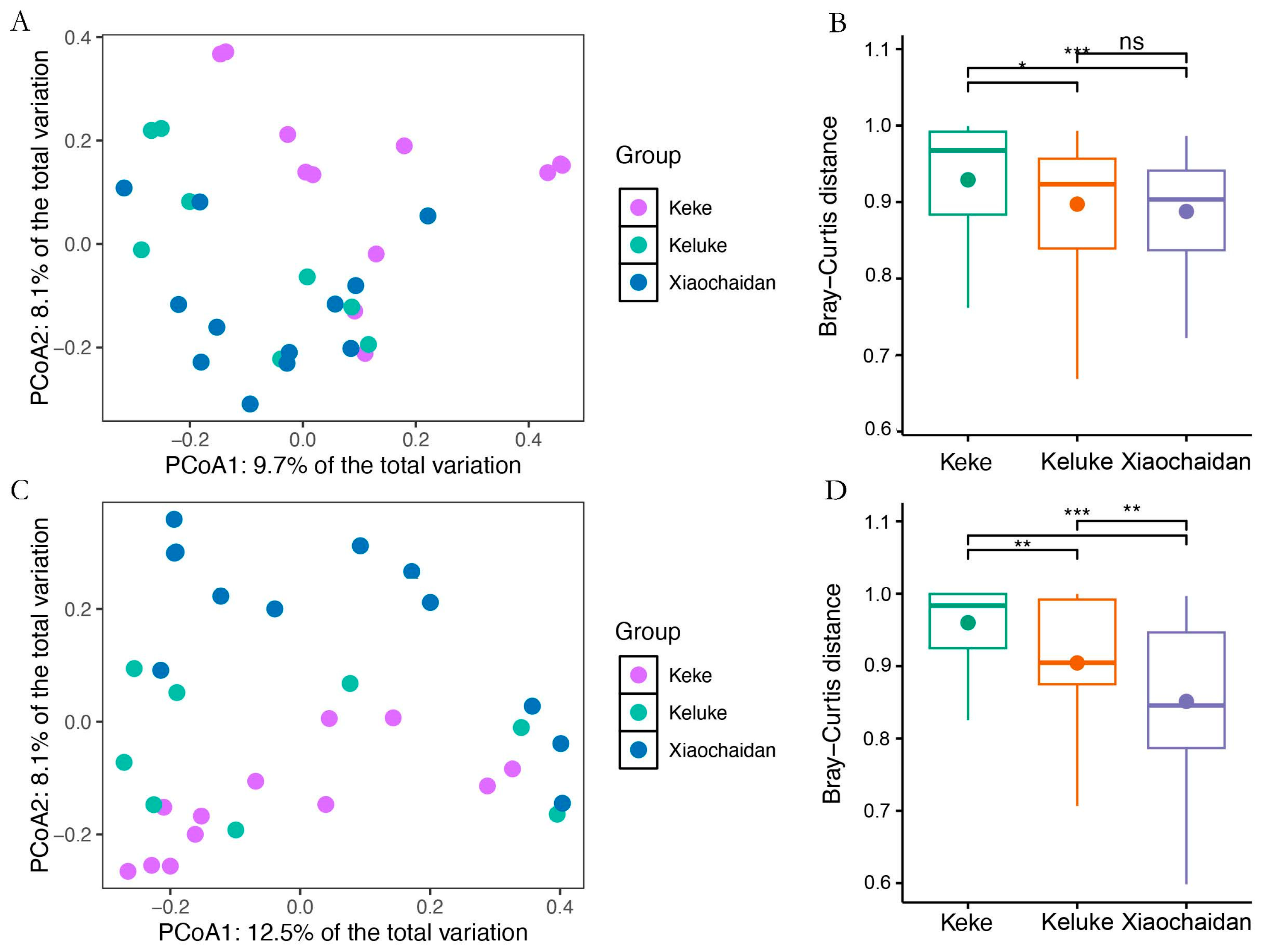
3.1. Microbial Community Composition
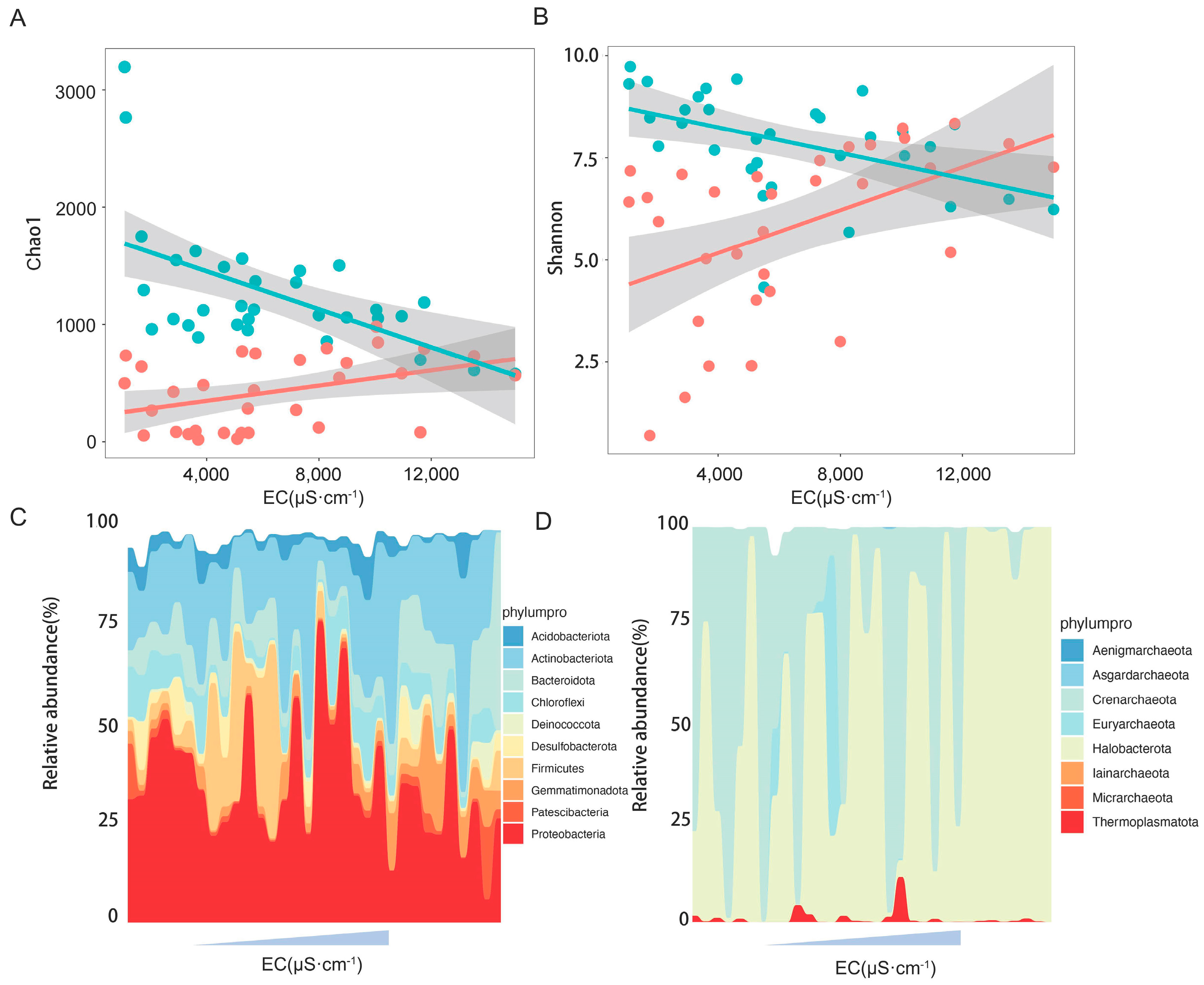
3.2. Effect of Salinity on Microbial Community
3.3. Microbial Community Assembly Process
3.4. Ecological Function of Microbial Community
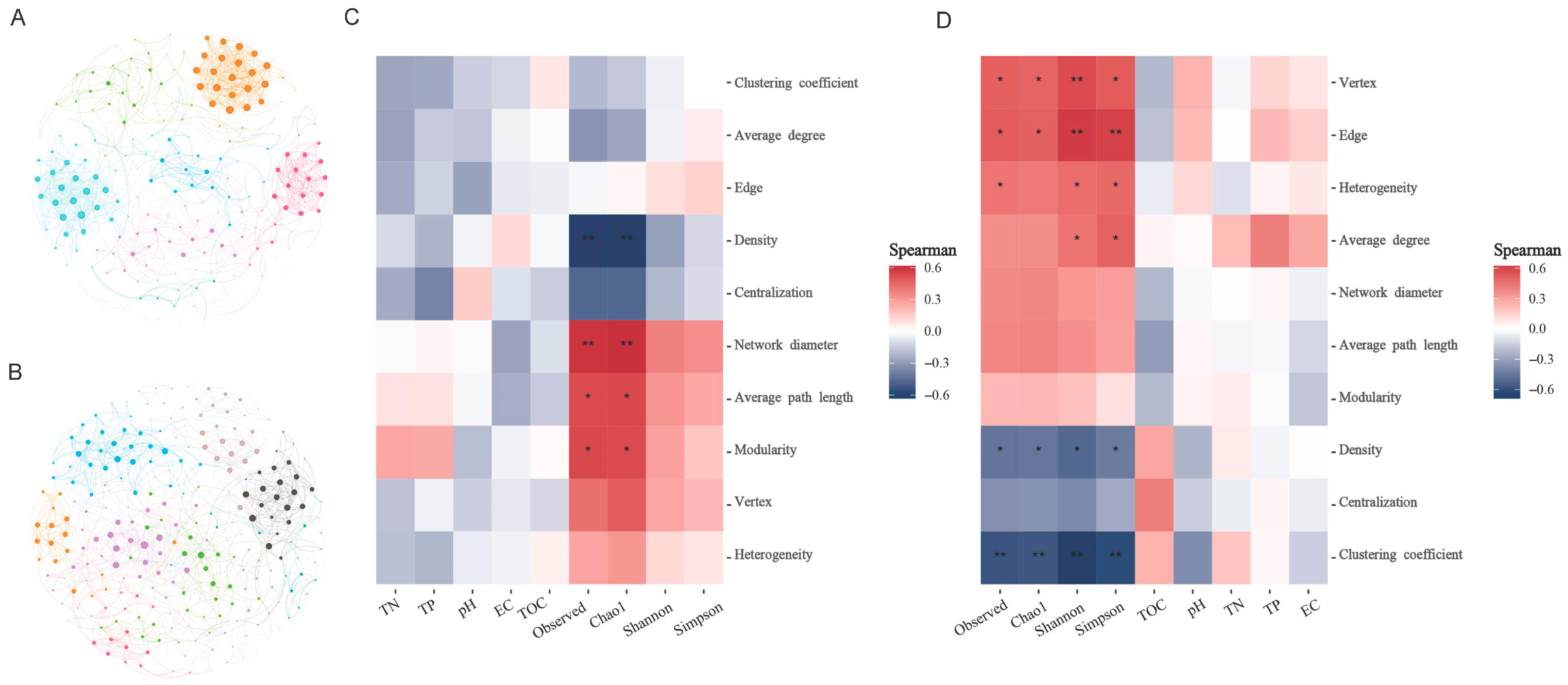
3.5. Co-Occurrence Network of Microbial Community
4. Discussion
4.1. Salinity Is a Key Environmental Factor Affecting Microbial Communities of Salt Marshes
4.2. Stochastic Process Driving Microbial Community Assembly
4.3. Alpha Diversity Links to Microbial Co-Occurrence Network Complexity
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Messager, M.L.; Lehner, B.; Grill, G.; Nedeva, I.; Schmitt, O. Estimating the volume and age of water stored in global lakes using a geo-statistical approach. Nat. Commun. 2016, 7, 13603. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Tan, E.; Wang, B.; Gan, Z.; Yang, J.; Han, J.; Zhang, X.; Kao, S.-J.; King, G.; Dong, H.; et al. Salinity change induces distinct climate feedbacks of nitrogen removal in saline lakes. Water Res. 2023, 245, 120668. [Google Scholar] [CrossRef]
- Edwardson, C.F.; Hollibaugh, J.T. Composition and activity of microbial communities along the redox gradient of an alkaline, hypersaline, lake. Front. Microbiol. 2018, 9, 14. [Google Scholar] [CrossRef]
- Middleton, B.A.; Boudell, J. Salinification of coastal wetlands and freshwater management to support resilience. Ecosyst. Health Sustain. 2023, 9, 83. [Google Scholar] [CrossRef]
- Wurtsbaugh, W.A.; Miller, C.; Null, S.E.; Derose, R.J.; Wilcock, P.; Hahnenberger, M.; Howe, F.; Moore, J. Decline of the world’s saline lakes. Nat. Geosci. 2017, 10, 816–821. [Google Scholar] [CrossRef]
- Li, X.; Wang, A.; Wan, W.; Luo, X.; Zheng, L.; He, G.; Huang, D.; Chen, W.; Huang, Q. High salinity inhibits soil bacterial community mediating nitrogen cycling. Appl. Environ. Microbiol. 2021, 87, e0136621. [Google Scholar] [CrossRef] [PubMed]
- Capooci, M.; Barba, J.; Seyfferth, A.L.; Vargas, R. Experimental influence of storm-surge salinity on soil greenhouse gas emissions from a tidal salt marsh. Sci. Total Environ. 2019, 686, 1164–1172. [Google Scholar] [CrossRef]
- Coban, O.; De Deyn, G.B.; van der Ploeg, M. Soil microbiota as game-changers in restoration of degraded lands. Science 2022, 375, eabe0725. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Du, N.; Eller, F.; Ye, S.; Li, X.; Wei, J.; Guo, Y.; Brix, H.; Guo, W. Ecological mechanisms of carbon sequestration in vegetated coastal wetland ecosystem: Exploring the roles of biodiversity and environmental changes. J. Mar. Environ. Eng. 2025, 12, 35–47. [Google Scholar] [CrossRef]
- Fierer, N. Embracing the unknown: Disentangling the complexities of the soil microbiome. Nat. Rev. Microbiol. 2017, 15, 579–590. [Google Scholar] [CrossRef]
- Rath, K.M.; Rousk, J. Salt effects on the soil microbial decomposer community and their role in organic carbon cycling: A review. Soil Biol. Biochem. 2015, 81, 108–123. [Google Scholar] [CrossRef]
- Gao, Y.; Liu, L.; Zhu, P.; Yang, S.; Guo, W.; Yu, X. Patterns and dynamics of the soil microbial community with gradual vegetation succession in the Yellow River Delta, China. Wetlands 2021, 41, 9. [Google Scholar] [CrossRef]
- Wang, L.; Qin, L.; Sun, X.; Zhao, S.; Yu, L.; Wang, M.; Chen, S. Linking bacterial growth responses to soil salinity with cd availability. Bull. Environ. Contam. Toxicol. 2022, 109, 286–297. [Google Scholar] [CrossRef]
- Zhao, Q.; Bai, J.; Gao, Y.; Zhao, H.; Zhang, G.; Cui, B. Shifts in the soil bacterial community along a salinity gradient in the Yellow River Delta. Land Degrad. Dev. 2020, 31, 2255–2267. [Google Scholar] [CrossRef]
- Zhang, G.; Bai, J.; Tebbe, C.C.; Zhao, Q.; Jia, J.; Wang, W.; Wang, X.; Yu, L. Salinity controls soil microbial community structure and function in coastal estuarine wetlands. Environ. Microbiol. 2021, 23, 1020–1037. [Google Scholar] [CrossRef]
- Zhang, X.; Qi, L.; Li, W.; Hu, B.X.; Dai, Z. Bacterial community variations with salinity in the saltwater-intruded estuarine aquifer. Sci. Total Environ. 2021, 755, 142423. [Google Scholar] [CrossRef]
- Zhao, Q.; Zhao, H.; Gao, Y.; Zheng, L.; Wang, J.; Bai, J. Alterations of bacterial and archaeal communities by freshwater input in coastal wetlands of the Yellow River Delta, China. Appl. Soil Ecol. 2020, 153, 103581. [Google Scholar] [CrossRef]
- Li, C.; Jin, L.; Zhang, C.; Li, S.; Zhou, T.; Hua, Z.; Wang, L.; Ji, S.; Wang, Y.; Gan, Y.; et al. Destabilized microbial networks with distinct performances of abundant and rare biospheres in maintaining networks under increasing salinity stress. iMeta 2023, 2, e79. [Google Scholar] [CrossRef]
- Zhu, P.; Liu, C.; Wei, W.; Wu, Y.; Sardar, M.F.; Yu, X.; Guo, W. Stochastic processes limit the effect of organic fertilizer application on soil bacterial community composition in salt marsh Suaeda salsa. J. Clean. Prod. 2024, 441, 141034. [Google Scholar] [CrossRef]
- Gong, X.; Chen, Z.; Deng, Y.; Zhao, D.; Gao, P.; Zhang, L.; Tu, Q.; Qu, L.; Zheng, L.; Zhang, Y.; et al. Contrasting archaeal and bacterial community assembly processes and the importance of rare taxa along a depth gradient in shallow coastal sediments. Sci. Total Environ. 2022, 852, 158411. [Google Scholar] [CrossRef]
- Milke, F.; Wagner-Doebler, I.; Wienhausen, G.; Simon, M. Selection, drift and community interactions shape microbial biogeographic patterns in the Pacific Ocean. ISME J. 2022, 16, 2653–2665. [Google Scholar] [CrossRef] [PubMed]
- Dini-Andreote, F.; Stegen, J.C.; van Elsas, J.D.; Salles, J.F. Disentangling mechanisms that mediate the balance between stochastic and deterministic processes in microbial succession. Proc. Natl. Acad. Sci. USA 2015, 112, E1326–E1332. [Google Scholar] [CrossRef] [PubMed]
- Stegen, J.C.; Lin, X.; Fredrickson, J.K.; Chen, X.; Kennedy, D.W.; Murray, C.J.; Rockhold, M.L.; Konopka, A. Quantifying community assembly processes and identifying features that impose them. ISME J. 2013, 7, 2069–2079. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.-F.; Pan, J.; Pan, Y.-P.; Li, M. Biogeography, assembly patterns, driving factors, and interactions of archaeal community in mangrove sediments. mSystems 2021, 6, e0138120. [Google Scholar] [CrossRef]
- Wagg, C.; Schlaeppi, K.; Banerjee, S.; Kuramae, E.E.; Van Der Heijden, M.G.A. Fungal-bacterial diversity and microbiome complexity predict ecosystem functioning. Nat. Commun. 2019, 10, 4841. [Google Scholar] [CrossRef]
- Yang, G.; Wagg, C.; Veresoglou, S.D.; Hempel, S.; Rillig, M.C. How soil biota drive ecosystem stability. Trends Plant Sci. 2018, 23, 1057–1067. [Google Scholar] [CrossRef]
- Zhang, H.; Dong, L.; Yao, X.; Wang, W. Soil fertility shifts the relative importance of saprotrophic and mycorrhizal fungi for maintaining ecosystem stability. Glob. Change Biol. 2023, 29, 1206–1216. [Google Scholar] [CrossRef]
- Yuan, M.M.; Guo, X.; Wu, L.; Zhang, Y.; Xiao, N.; Ning, D.; Shi, Z.; Zhou, X.; Wu, L.; Yang, Y.; et al. Climate warming enhances microbial network complexity and stability. Nat. Clim. Chang. 2021, 11, 343–348. [Google Scholar] [CrossRef]
- Lima-Mendez, G.; Faust, K.; Henry, N.; Decelle, J.; Colin, S.; Carcillo, F.; Chaffron, S.; Ignacio-Espinosa, J.C.; Roux, S.; Vincent, F.; et al. Determinants of community structure in the global plankton interactome. Science 2025, 348, 1262073. [Google Scholar] [CrossRef]
- Li, X.; Liu, Q.; Yu, X.; Zhang, C.; Liu, M.; Zhou, X.; Gu, C.; Wang, M.; Shao, H.; Li, J.; et al. Spatial pattern and co-occurrence network of microbial community in response to extreme environment of salt lakes on the Qinghai-Tibet Plateau. Environ. Sci. Pollut. Res. 2023, 30, 20615–20630. [Google Scholar] [CrossRef]
- Callahan, B.J.; Mcmurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Liu, C.; Cui, Y.; Li, X.; Yao, M. microeco: An R package for data mining in microbial community ecology. FEMS Microbiol. Ecol. 2021, 97, fiaa255. [Google Scholar] [CrossRef]
- Louca, S.; Parfrey, L.W.; Doebeli, M. Decoupling function and taxonomy in the global ocean microbiome. Science 2016, 353, 1272–1277. [Google Scholar] [CrossRef] [PubMed]
- Bacci, G.; Cerri, M.; Lastrucci, L.; Ferranti, F.; Ferri, V.; Foggi, B.; Gigante, D.; Venanzoni, R.; Viciani, D.; Mengoni, A.; et al. Applying predictive models to decipher rhizobacterial modifications in common reed die-back affected populations. Sci. Total Environ. 2018, 642, 708–722. [Google Scholar] [CrossRef] [PubMed]
- Xia, S.; Song, Z.; Van Zwieten, L.; Guo, L.; Yu, C.; Wang, W.; Li, Q.; Hartley, I.P.; Yang, Y.; Liu, H.; et al. Storage, patterns and influencing factors for soil organic carbon in coastal wetlands of China. Glob. Change Biol. 2022, 28, 6065–6085. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Wu, Y.; Yin, M.; Ma, X.; Yu, X.; Guo, X.; Du, N.; Eller, F.; Guo, W. Soil salinity, not plant genotype or geographical distance, shapes soil microbial community of a reed wetland at a fine scale in the Yellow River Delta. Sci. Total Environ. 2023, 856 Pt 2, 159136. [Google Scholar] [CrossRef]
- Liu, Y.; Priscu, J.C.; Xiong, J.; Conrad, R.; Vick-Majors, T.; Chu, H.; Hou, J. Salinity drives archaeal distribution patterns in high altitude lake sediments on the Tibetan Plateau. FEMS Microbiol. Ecol. 2016, 92, fiw033. [Google Scholar] [CrossRef]
- van Wolferen, M.; Pulschen, A.A.; Baum, B.; Gribaldo, S.; Albers, S.-V. The cell biology of archaea. Nat. Microbiol. 2022, 7, 1744–1755. [Google Scholar] [CrossRef]
- Medina-Chávez, N.O.; Travisano, M. Archaeal Communities: The Microbial Phylogenomic Frontier. Front. Genet. 2022, 12, 693193. [Google Scholar] [CrossRef]
- Li, M.; Mi, T.; He, H.; Chen, Y.; Zhen, Y.; Yu, Z. Active bacterial and archaeal communities in coastal sediments: Biogeography pattern, assembly process and co-occurrence relationship. Sci. Total Environ. 2021, 750, 142252. [Google Scholar] [CrossRef]
- Zhu, P.; Yang, S.; Wu, Y.; Ru, Y.; Yu, X.; Wang, L.; Guo, W. Shifts in soil microbial community composition, function, and co-occurrence network of phragmites australis in the Yellow River Delta. Front. Microbiol. 2022, 13, 858125. [Google Scholar] [CrossRef]
- Mukhopadhya, I.; Hansen, R.; El-Omar, E.M.; Hold, G.L. IBD—What role do Proteobacteria play? Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Dai, X.; Zhu, Y.; Yang, J.; Jiang, H.; Dong, H.; Huang, L. (Meta)Genomic analysis reveals diverse energy conservation strategies employed by globally distributed Gemmatimonadota. mSystems 2022, 7, e0022822. [Google Scholar] [CrossRef] [PubMed]
- Hazzouri, K.M.; Sudalaimuthuasari, N.; Saeed, E.E.; Kundu, B.; Al-Maskari, R.S.; Nelson, D.; AlShehhi, A.A.; Aldhuhoori, M.A.; Almutawa, D.S.; Alshehhi, F.R.; et al. Salt flat microbial diversity and dynamics across salinity gradient. Sci. Rep. 2022, 12, 11293. [Google Scholar] [CrossRef] [PubMed]
- Eichler, J. Halobacterium salinarum: Life with more than a grain of salt. Microbiology 2023, 169, 001327. [Google Scholar] [CrossRef]
- Santos, A.; Bruna, P.; Martinez-Urtaza, J.; Solís, F.; Valenzuela, B.; Zamorano, P.; Barrientos, L. Two archaeal metagenome-assembled genomes from El tatio provide new insights into the Crenarchaeota phylum. Genes 2021, 12, 391. [Google Scholar] [CrossRef]
- Zhang, N.-C.; Hong, Z.-F.; Qiu, R.-L.; Chao, Y.-Q.; Yu, Y.-F. Removal pathway quantification and co-metabolic mechanism evaluation of alkylphenols from synthetic wastewater by phenolic root exudates in the rhizosphere of Phragmites australis. J. Hazard. Mater. 2022, 424 Pt A, 127269. [Google Scholar] [CrossRef]
- Niu, Y.; Zheng, Y.; Hou, L.; Gao, D.; Chen, F.; Pei, C.; Dong, H.; Liang, X.; Liu, M. Microbial dynamics and activity of denitrifying anaerobic methane oxidizers in China’s estuarine and coastal wetlands. Sci. Total Environ. 2022, 806 Pt 1, 150425. [Google Scholar] [CrossRef]
- Gao, D.; Liu, F.; Li, L.; Chen, C.; Liang, H. Diversity and community structure of ammonia oxidizers in a marsh wetland of the northeast China. Appl. Microbiol. Biotechnol. 2018, 102, 8561–8571. [Google Scholar] [CrossRef]
- Höfferle, Š.; Nicol, G.W.; Pal, L.; Hacin, J.; Prosser, J.I.; Mandić-Mulec, I. Ammonium supply rate influences archaeal and bacterial ammonia oxidizers in a wetland soil vertical profile. FEMS Microbiol. Ecol. 2010, 74, 302–315. [Google Scholar] [CrossRef]
- He, R.; Zeng, J.; Zhao, D.; Wang, S.; Wu, Q.L. Decreased spatial variation and deterministic processes of bacterial community assembly in the rhizosphere of Phragmites australis across the Middle–Lower Yangtze plain. Mol. Ecol. 2022, 31, 1180–1195. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Gu, S.; Zhang, Z.; Li, S.; Zhou, Y.; Zhang, Z.; Zhang, Q.; Wang, L.; Ju, Z.; Yan, C.; et al. Spatial distribution patterns across multiple microbial taxonomic groups. Environ. Res. 2023, 223, 115470. [Google Scholar] [CrossRef]
- Duan, Y.; Wang, X.; Wang, L.; Lian, J.; Wang, W.; Wu, F.; Li, Y.; Li, Y. Biogeographic patterns of soil microbe communities in the deserts of the Hexi Corridor, northern China. Catena 2022, 211, 106026. [Google Scholar] [CrossRef]
- Clark, D.R.; Underwood, G.J.; McGenity, T.J.; Dumbrell, A.J. What drives study-dependent differences in distance–decay relationships of microbial communities? Glob. Ecol. Biogeogr. 2021, 30, 811–825. [Google Scholar] [CrossRef]
- Zhou, J.; Ning, D. Stochastic community assembly: Does it matter in microbial ecology? Microbiol. Mol. Biol. Rev. 2017, 81, e00002-17. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Wang, X.; Song, Y.; Zeng, Q.; Zhang, Y.; Luo, H. Prochlorococcus have low global mutation rate and small effective population size. Nat. Ecol. Evol. 2022, 6, 183–194. [Google Scholar] [CrossRef]
- Wang, Z.-B.; Sun, Y.-Y.; Li, Y.; Chen, X.-L.; Wang, P.; Ding, H.-T.; Chen, B.; Zhang, X.-Y.; Song, X.-Y.; Wang, M.; et al. Significant bacterial distance-decay relationship in continuous, well-connected southern ocean surface water. Microb. Ecol. 2020, 80, 73–80. [Google Scholar] [CrossRef]
- Qiu, L.; Kong, W.; Zhu, H.; Zhang, Q.; Banerjee, S.; Ishii, S.; Sadowsky, M.J.; Gao, J.; Feng, C.; Wang, J.; et al. Halophytes increase rhizosphere microbial diversity, network complexity and function in inland saline ecosystem. Sci. Total Environ. 2022, 831, 154944. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Keke | Keluke | Xiaochaidan | ||||
|---|---|---|---|---|---|---|
| Bacteria | Archaea | Bacteria | Archaea | Bacteria | Archaea | |
| Variable Selection | 36.36 | 24.24 | 0 | 7.14 | 16.67 | 0 |
| Homogeneous Selection | 0 | 0 | 7.14 | 0 | 1.51 | 0 |
| Dispersal Limitation | 16.67 | 3.03 | 21.43 | 21.43 | 19.70 | 27.27 |
| Homogeneous Dispersal | 12.12 | 15.15 | 7.14 | 14.29 | 1.52 | 0 |
| Drift | 34.85 | 57.58 | 64.29 | 57.14 | 60.61 | 72.73 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, P.; Wang, Y.; Sheng, W.; Yu, M.; Wei, W.; Sun, W.; Gao, J.; Xu, Z.; Cao, M.; Wang, Y.; et al. Salinity Effect on Soil Bacterial and Archaeal Diversity and Assembly in Phragmites australis Salt Marshes in the Qaidam Basin, China. Microorganisms 2025, 13, 1253. https://doi.org/10.3390/microorganisms13061253
Zhu P, Wang Y, Sheng W, Yu M, Wei W, Sun W, Gao J, Xu Z, Cao M, Wang Y, et al. Salinity Effect on Soil Bacterial and Archaeal Diversity and Assembly in Phragmites australis Salt Marshes in the Qaidam Basin, China. Microorganisms. 2025; 13(6):1253. https://doi.org/10.3390/microorganisms13061253
Chicago/Turabian StyleZhu, Pengcheng, Yuhui Wang, Wenyi Sheng, Mingyang Yu, Wei Wei, Wenlong Sun, Jian Gao, Zhenwei Xu, Ming Cao, Yuzhi Wang, and et al. 2025. "Salinity Effect on Soil Bacterial and Archaeal Diversity and Assembly in Phragmites australis Salt Marshes in the Qaidam Basin, China" Microorganisms 13, no. 6: 1253. https://doi.org/10.3390/microorganisms13061253
APA StyleZhu, P., Wang, Y., Sheng, W., Yu, M., Wei, W., Sun, W., Gao, J., Xu, Z., Cao, M., Wang, Y., Liu, L., & Guo, W. (2025). Salinity Effect on Soil Bacterial and Archaeal Diversity and Assembly in Phragmites australis Salt Marshes in the Qaidam Basin, China. Microorganisms, 13(6), 1253. https://doi.org/10.3390/microorganisms13061253