
High Genetic Diversity Among Bacillus cereus Isolates Contaminating Donated Milk at a Canadian Human Milk Bank

 , , ,
, , ,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Sampling and Culturing-Dependent Screening
2.2. DNA Extraction and Whole-Genome Sequencing
2.3. Bioinformatics Analysis and Statistical Analysis
3. Results
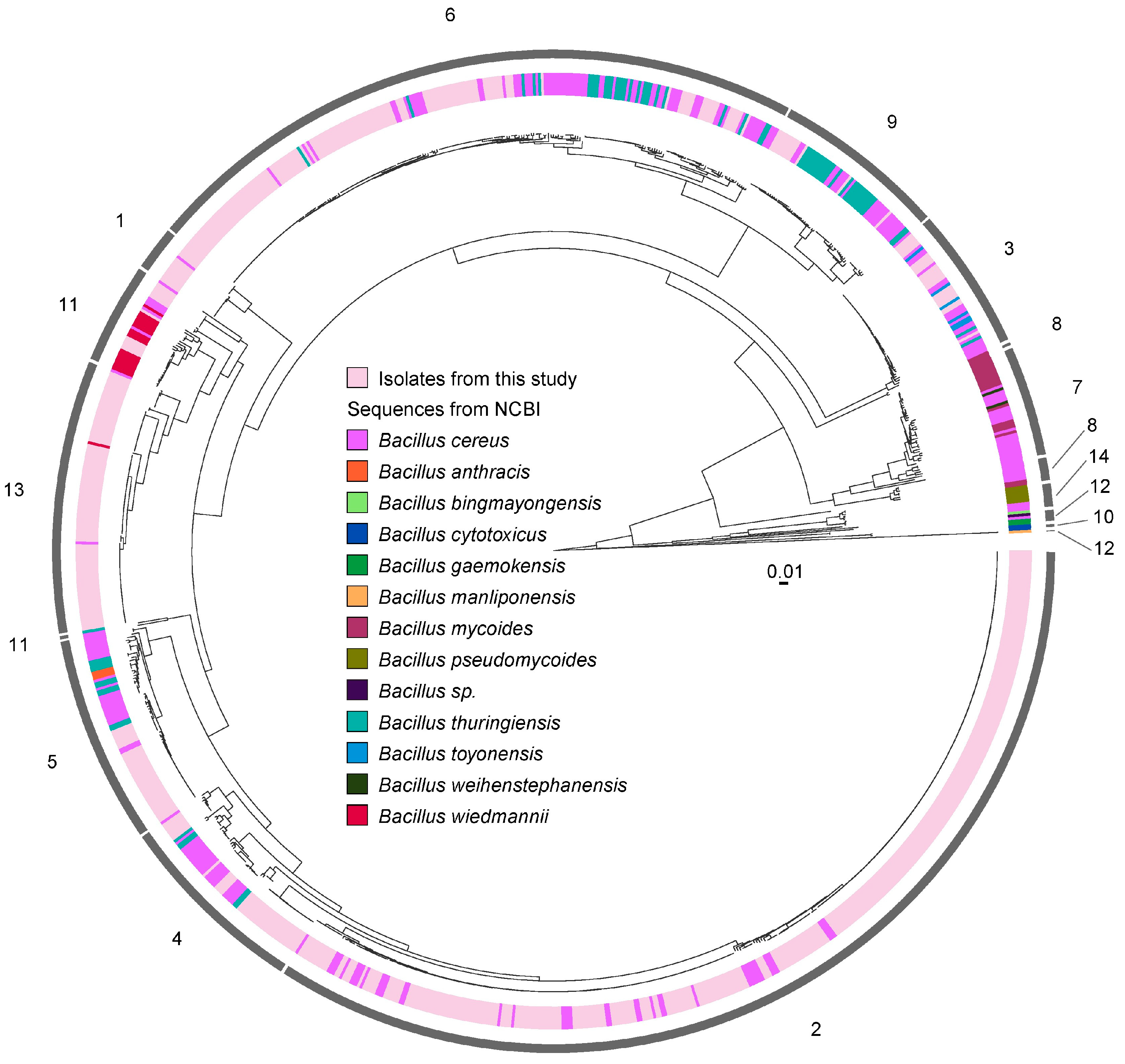
3.1. Species Typing of B. cereus Group Isolates
3.2. Core and Pangenome of the B. cereus Collection
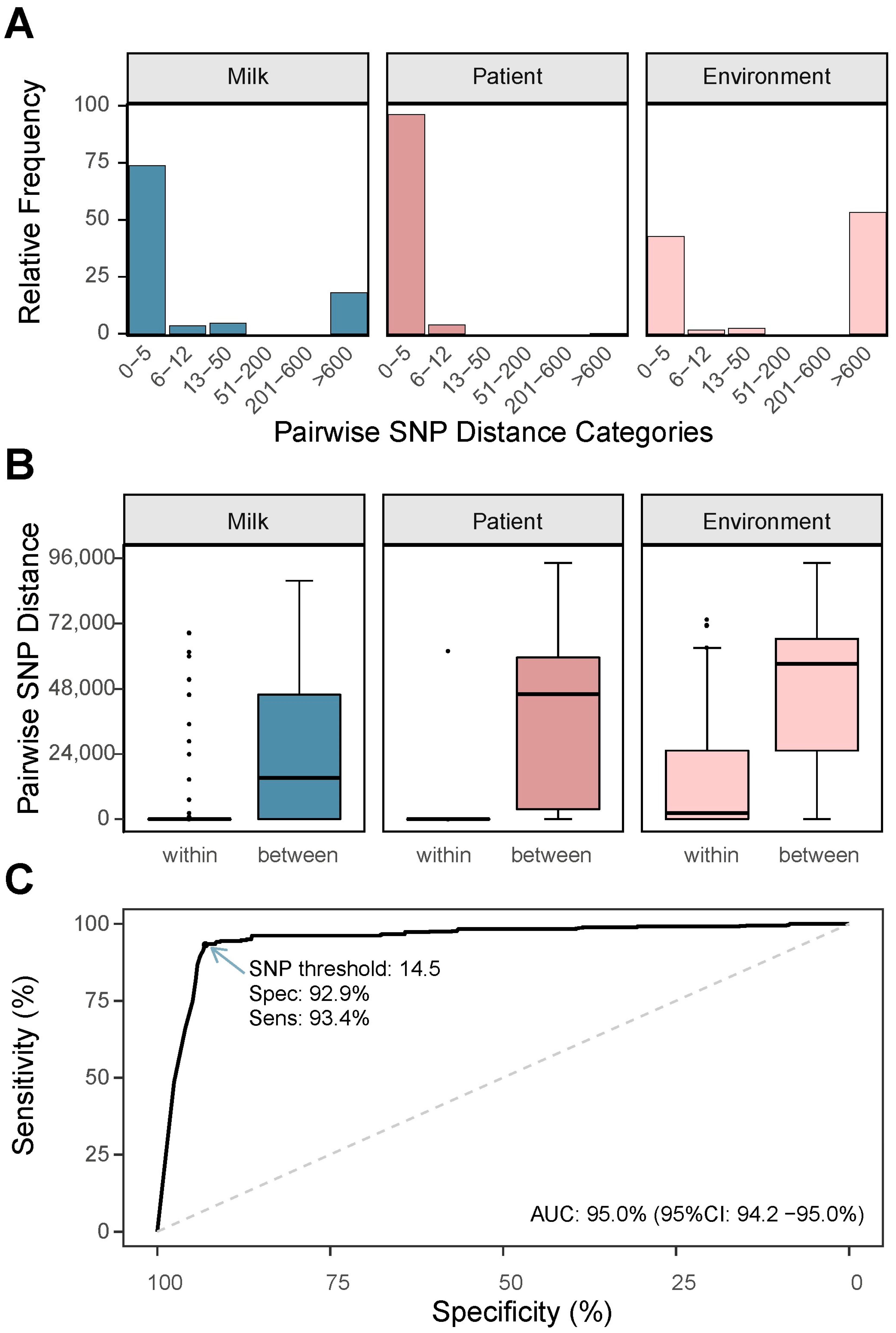
3.3. Analysis of Core Genome SNP Distances Between Isolates
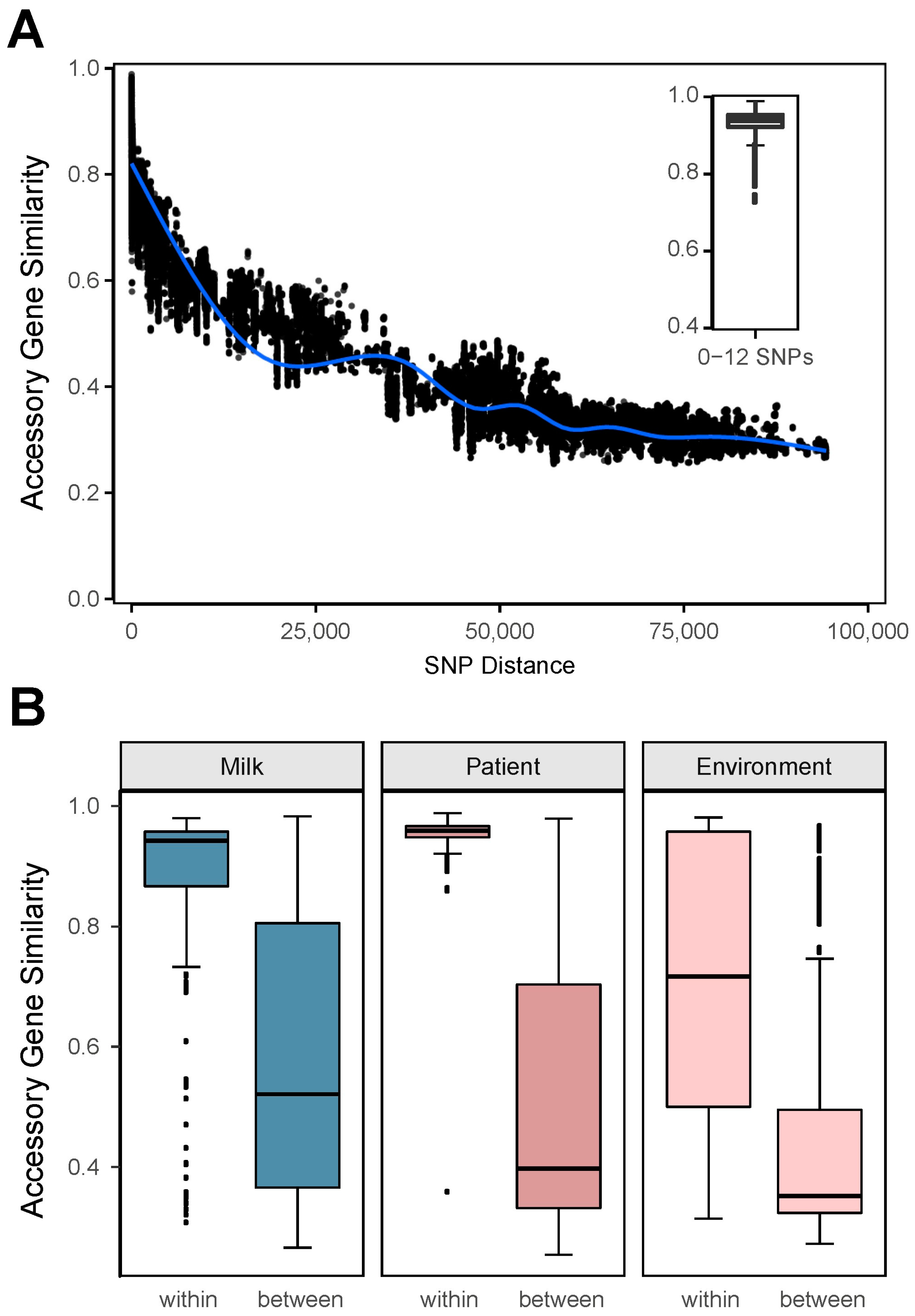
3.4. Accessory Gene Content Similarity and Its Relationship with Core Genome SNPs
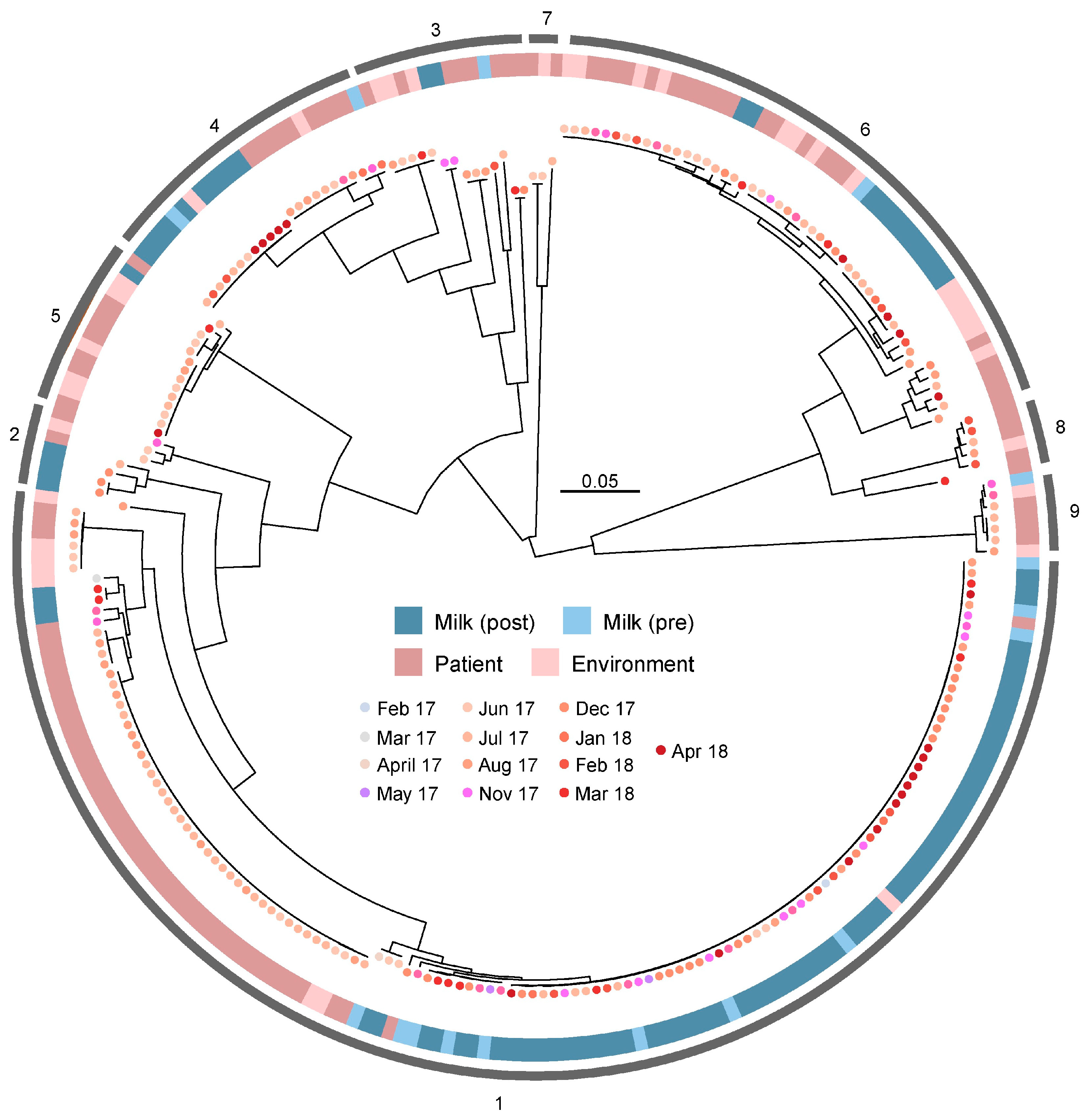
3.5. Absence of Source-Specific Clustering Among B. cereus Isolates
3.6. B. cereus Isolates from Toronto Show Genetic Diversity Comparable to a Global Collection of Isolates
3.7. Identifying Genetic Determinants of Pasteurization Survival in B. cereus Isolates
4. Discussion
4.1. Genomic Diversity and Taxonomic Complexity of B. cereus in the HMB Setting
4.2. B. cereus Clonality and Plate-Level Heterogeneity
4.3. Multifactorial Sources of B. cereus Contamination and Practical Mitigation
4.4. Potential Genetic Determinants for B. cereus Survival in Post-Pasteurization Milk
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AUC | Area under the curve |
| BAPS | Bayesian analysis of population structure |
| CDS | Coding DNA Sequence |
| DNA | Deoxyribonucleic acid |
| GWAS | Genome-wide association study |
| HGT | Horizontal gene transfer |
| HMB | Human milk bank |
| HoP | Holder pasteurization |
| IQR | Interquartile range |
| MAF | Minor allele frequency |
| MALDI-TOF MS | Matrix-assisted laser desorption/ionization-time of flight mass spectrometry |
| MGE | Mobile genetic elements |
| MSH | Mount Sinai Hospital |
| NCBI | National Center for Biotechnology Information |
| NICU | Neonatal intensive care unit |
| ROC | Receiver operating characteristic |
| SD | Standard deviation |
| SNP | Single-nucleotide polymorphism |
| WGS | Whole-genome sequencing |
References
- Statistics Canada. The Proportion of Births at Less Than 37 Weeks of Gestation, 1974 to 2023. Available online: https://www150.statcan.gc.ca/n1/daily-quotidien/240925/cg-c001-eng.htm (accessed on 31 March 2025).
- Pound, C.; Unger, S.; Blair, B. Pasteurized and unpasteurized donor human milk. Paediatr. Child Health 2020, 25, 549. [Google Scholar] [CrossRef]
- Kim, J.; Unger, S. Human milk banking. Paediatr. Child Health 2010, 15, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Bottone, E.J. Bacillus cereus, a volatile human pathogen. Clin. Microbiol. Rev. 2010, 23, 382–398. [Google Scholar] [CrossRef] [PubMed]
- Landers, S.; Updegrove, K. Bacteriological screening of donor human milk before and after Holder pasteurization. Breastfeed. Med. 2010, 5, 117–121. [Google Scholar] [CrossRef]
- Batt, C.A. BACILLUS|Bacillus cereus. In Encyclopedia of Food Microbiology; Academic Press: Cambridge, MA, USA, 2014; pp. 124–128. [Google Scholar] [CrossRef]
- Gopal, N.; Hill, C.; Ross, P.R.; Beresford, T.P.; Fenelon, M.A.; Cotter, P.D. The Prevalence and Control of Bacillus and Related Spore-Forming Bacteria in the Dairy Industry. Front. Microbiol. 2015, 6, 1418. [Google Scholar] [CrossRef]
- Okinaka, R.T.; Keim, P. The Phylogeny of Bacillus cereus sensu lato. Microbiol. Spectr. 2016, 4, 237–251. [Google Scholar] [CrossRef] [PubMed]
- Tourasse, N.J.; Helgason, E.; Okstad, O.A.; Hegna, I.K.; Kolsto, A.B. The Bacillus cereus group: Novel aspects of population structure and genome dynamics. J. Appl. Microbiol. 2006, 101, 579–593. [Google Scholar] [CrossRef]
- Ehling-Schulz, M.; Lereclus, D.; Koehler, T.M.; Fischetti, V.A.; Novick, R.P.; Ferretti, J.J.; Portnoy, D.A.; Braunstein, M.; Rood, J.I. The Bacillus cereus Group: Bacillus Species with Pathogenic Potential. Microbiol. Spectr. 2019, 7, 1–35. [Google Scholar] [CrossRef]
- Tritt, A.; Eisen, J.A.; Facciotti, M.T.; Darling, A.E. An integrated pipeline for de novo assembly of microbial genomes. PLoS ONE 2012, 7, e42304. [Google Scholar] [CrossRef]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Wood, D.E.; Salzberg, S.L. Kraken: Ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 2014, 15, R46. [Google Scholar] [CrossRef] [PubMed]
- Page, A.J.; Cummins, C.A.; Hunt, M.; Wong, V.K.; Reuter, S.; Holden, M.T.; Fookes, M.; Falush, D.; Keane, J.A.; Parkhill, J. Roary: Rapid large-scale prokaryote pan genome analysis. Bioinformatics 2015, 31, 3691–3693. [Google Scholar] [CrossRef]
- Capella-Gutierrez, S.; Silla-Martinez, J.M.; Gabaldon, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef]
- Croucher, N.J.; Page, A.J.; Connor, T.R.; Delaney, A.J.; Keane, J.A.; Bentley, S.D.; Parkhill, J.; Harris, S.R. Rapid phylogenetic analysis of large samples of recombinant bacterial whole genome sequences using Gubbins. Nucleic Acids Res. 2015, 43, e15. [Google Scholar] [CrossRef] [PubMed]
- Seemann, T.; Klötzl, F.; Page, A.J. Snp-Dists: Convert a FASTA Alignment to SNP Distance Matrix. Available online: https://github.com/tseemann/snp-dists (accessed on 30 March 2025).
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2--approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Yu, G.; Lam, T.T.; Zhu, H.; Guan, Y. Two Methods for Mapping and Visualizing Associated Data on Phylogeny Using Ggtree. Mol. Biol. Evol. 2018, 35, 3041–3043. [Google Scholar] [CrossRef] [PubMed]
- Tonkin-Hill, G.; Lees, J.A.; Bentley, S.D.; Frost, S.D.W.; Corander, J. RhierBAPS: An R implementation of the population clustering algorithm hierBAPS. Wellcome Open Res. 2018, 3, 93. [Google Scholar] [CrossRef] [PubMed]
- Lees, J.A.; Galardini, M.; Bentley, S.D.; Weiser, J.N.; Corander, J.; Stegle, O. pyseer: A comprehensive tool for microbial pangenome-wide association studies. Bioinformatics 2018, 34, 4310–4312. [Google Scholar] [CrossRef]
- Jaillard, M.; Lima, L.; Tournoud, M.; Mahé, P.; van Belkum, A.; Lacroix, V.; Jacob, L. A fast and agnostic method for bacterial genome-wide association studies: Bridging the gap between k-mers and genetic events. PLoS Genet. 2018, 14, e1007758. [Google Scholar] [CrossRef]
- R Core Team. A Language and Environment for Statiscal Computing, R Foundation for Statistical Computing: 2024. Available online: https://www.r-project.org/ (accessed on 30 March 2025).
- Jouzani, G.S.; Valijanian, E.; Sharafi, R. Bacillus thuringiensis: A successful insecticide with new environmental features and tidings. Appl. Microbiol. Biotechnol. 2017, 101, 2691–2711. [Google Scholar] [CrossRef]
- Miller, R.A.; Beno, S.M.; Kent, D.J.; Carroll, L.M.; Martin, N.H.; Boor, K.J.; Kovac, J. Bacillus wiedmannii sp. nov., a psychrotolerant and cytotoxic Bacillus cereus group species isolated from dairy foods and dairy environments. Int. J. Syst. Evol. Microbiol. 2016, 66, 4744–4753. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Gao, Q.; Liu, L.; Liu, H.; Wang, Y.; Peng, D.; Ruan, L.; Raymond, B.; Sun, M. Comparative Genomics of Bacillus thuringiensis Reveals a Path to Specialized Exploitation of Multiple Invertebrate Hosts. mBio 2017, 8, e00822-17. [Google Scholar] [CrossRef]
- Carroll, L.M.; Wiedmann, M.; Kovac, J. Proposal of a Taxonomic Nomenclature for the Bacillus cereus Group Which Reconciles Genomic Definitions of Bacterial Species with Clinical and Industrial Phenotypes. mBio 2020, 11, e00034-20. [Google Scholar] [CrossRef] [PubMed]
- Bazinet, A.L. Pan-genome and phylogeny of Bacillus cereus sensu lato. BMC Evol. Biol. 2017, 17, 176. [Google Scholar] [CrossRef] [PubMed]
- Jandova, M.; Mericka, P.; Fiserova, M.; Landfeld, A.; Paterova, P.; Hobzova, L.; Jarkovska, E.; Kacerovsky, M.; Houska, M. Bacillus cereus as a Major Cause of Discarded Pasteurized Human Banked Milk: A Single Human Milk Bank Experience. Foods 2021, 10, 2955. [Google Scholar] [CrossRef]
- Froh, E.B.; Vanderpool, J.; Spatz, D.L. Best Practices to Limit Contamination of Donor Milk in a Milk Bank. J. Obstet. Gynecol. Neonatal Nurs. 2018, 47, 547–555. [Google Scholar] [CrossRef]
- Rigourd, V.; Barnier, J.P.; Ferroni, A.; Nicloux, M.; Hachem, T.; Magny, J.F.; Lapillonne, A.; Frange, P.; Nassif, X.; Bille, E. Recent actuality about Bacillus cereus and human milk bank: A new sensitive method for microbiological analysis of pasteurized milk. Eur. J. Clin. Microbiol. Infect. Dis. 2018, 37, 1297–1303. [Google Scholar] [CrossRef]
- Cormontagne, D.; Rigourd, V.; Vidic, J.; Rizzotto, F.; Bille, E.; Ramarao, N. Bacillus cereus Induces Severe Infections in Preterm Neonates: Implication at the Hospital and Human Milk Bank Level. Toxins 2021, 13, 123. [Google Scholar] [CrossRef]
- Ansari, J.A.; Ismail, M.; Farid, M. Investigation of the use of ultrasonication followed by heat for spore inactivation. Food Bioprod. Process. 2017, 104, 32–39. [Google Scholar] [CrossRef]
- Raso, J.; Palop, A.; Pagán, R.; Condón, S. Inactivation of Bacillus subtilis spores by combining ultrasonic waves under pressure and mild heat treatment. J. Appl. Microbiol. 1998, 85, 849–854. [Google Scholar] [CrossRef]
- Moro, G.E.; Billeaud, C.; Rachel, B.; Calvo, J.; Cavallarin, L.; Christen, L.; Escuder-Vieco, D.; Gaya, A.; Lembo, D.; Wesolowska, A.; et al. Processing of Donor Human Milk: Update and Recommendations From the European Milk Bank Association (EMBA). Front. Pediatr. 2019, 7, 49. [Google Scholar] [CrossRef] [PubMed]
- Kullmann, K.C.; Adams, A.C.; Feldman-Winter, L. Human Milk Sharing in the United States: A Scoping Review. Breastfeed. Med. 2022, 17, 723–735. [Google Scholar] [CrossRef]
- Unger, S.L.; O’Connor, D.L. Review of current best practices for human milk banking. Matern. Child Nutr. 2024, 20 (Suppl. 4), e13657. [Google Scholar] [CrossRef]
- Dalwadi, P.; Nathani, N.; Chauhan, K.; Mansuri, J.; Koringa, P.; Bhatt, V.; Kunjadiya, A.P. Whole-genome sequencing of bacteria accountable for lactational mastitis in humans combined with an examination of their antibiotic resistance profiles. Braz. J. Microbiol. 2024, 55, 3827–3838. [Google Scholar] [CrossRef] [PubMed]
- Fei, P.; Yuan, X.; Zhao, S.; Yang, T.; Xiang, J.; Chen, X.; Zhou, L.; Ji, M. Prevalence and Genetic Diversity of Bacillus cereus Isolated from Raw Milk and Cattle Farm Environments. Curr. Microbiol. 2019, 76, 1355–1360. [Google Scholar] [CrossRef] [PubMed]
- Arredondo-Alonso, S.; Willems, R.J.; van Schaik, W.; Schürch, A.C. On the (im)possibility of reconstructing plasmids from whole-genome short-read sequencing data. Microb. Genom. 2017, 3, e000128. [Google Scholar] [CrossRef]
- Shuster, B.; Khemmani, M.; Abe, K.; Huang, X.; Nakaya, Y.; Maryn, N.; Buttar, S.; Gonzalez, A.N.; Driks, A.; Sato, T.; et al. Contributions of crust proteins to spore surface properties in Bacillus subtilis. Mol. Microbiol. 2019, 111, 825–843. [Google Scholar] [CrossRef]
- Sowell, M.O.; Buchanan, C.E. Changes in penicillin-binding proteins during sporulation of Bacillus subtilis. J. Bacteriol. 1983, 153, 1331–1337. [Google Scholar] [CrossRef]
- Wang, K.H.; Isidro, A.L.; Domingues, L.; Eskandarian, H.A.; McKenney, P.T.; Drew, K.; Grabowski, P.; Chua, M.H.; Barry, S.N.; Guan, M.; et al. The coat morphogenetic protein SpoVID is necessary for spore encasement in Bacillus subtilis. Mol. Microbiol. 2009, 74, 634–649. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gene, M.; Guthrie, J.L.; Li, K.; Teatero, S.; Paterson, A.; Li, A.; Doyen, A.; Yamamura, D.; Khan, S.; Srigley, J.A.; et al. High Genetic Diversity Among Bacillus cereus Isolates Contaminating Donated Milk at a Canadian Human Milk Bank. Microorganisms 2025, 13, 1136. https://doi.org/10.3390/microorganisms13051136
Gene M, Guthrie JL, Li K, Teatero S, Paterson A, Li A, Doyen A, Yamamura D, Khan S, Srigley JA, et al. High Genetic Diversity Among Bacillus cereus Isolates Contaminating Donated Milk at a Canadian Human Milk Bank. Microorganisms. 2025; 13(5):1136. https://doi.org/10.3390/microorganisms13051136
Chicago/Turabian StyleGene, Mathew, Jennifer L. Guthrie, Kevin Li, Sarah Teatero, Aimee Paterson, Angel Li, Alain Doyen, Deborah Yamamura, Sarah Khan, Jocelyn A. Srigley, and et al. 2025. "High Genetic Diversity Among Bacillus cereus Isolates Contaminating Donated Milk at a Canadian Human Milk Bank" Microorganisms 13, no. 5: 1136. https://doi.org/10.3390/microorganisms13051136
APA StyleGene, M., Guthrie, J. L., Li, K., Teatero, S., Paterson, A., Li, A., Doyen, A., Yamamura, D., Khan, S., Srigley, J. A., Stone, D., O’Connor, D. L., Poutanen, S., Unger, S., McGeer, A., & Fittipaldi, N. (2025). High Genetic Diversity Among Bacillus cereus Isolates Contaminating Donated Milk at a Canadian Human Milk Bank. Microorganisms, 13(5), 1136. https://doi.org/10.3390/microorganisms13051136