
Natural Revegetation Alters Habitat Conditions, Bacterial Components, and Polycyclic Aromatic Hydrocarbon (PAH)-Degrading Communities in Aged PAH-Polluted Soils

Abstract
1. Introduction
2. Materials and Methods
2.1. Study Area and Sampling
2.2. Analysis of Physicochemical Properties and PAHs
2.3. DNA Extraction, Amplification, and Sequencing
2.4. Real-Time Quantitative PCR
2.5. Statistical Analysis
3. Results
3.1. Soil Properties and PAH Contents
3.2. Bacterial Community Structure and Diversity
3.3. Microbial Functional Profiling
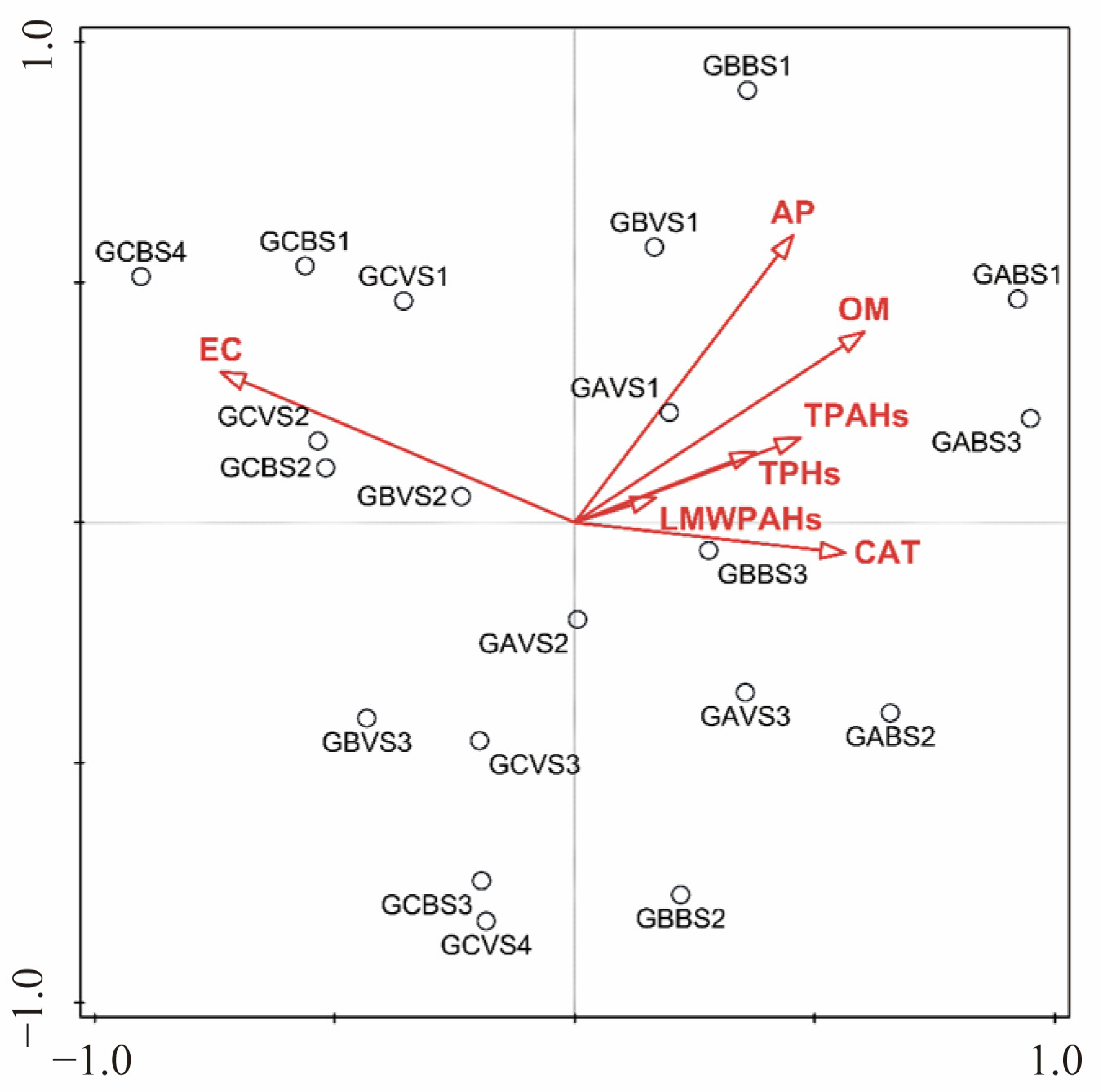
3.4. Correlations Between Environmental Factors and Bacterial Communities
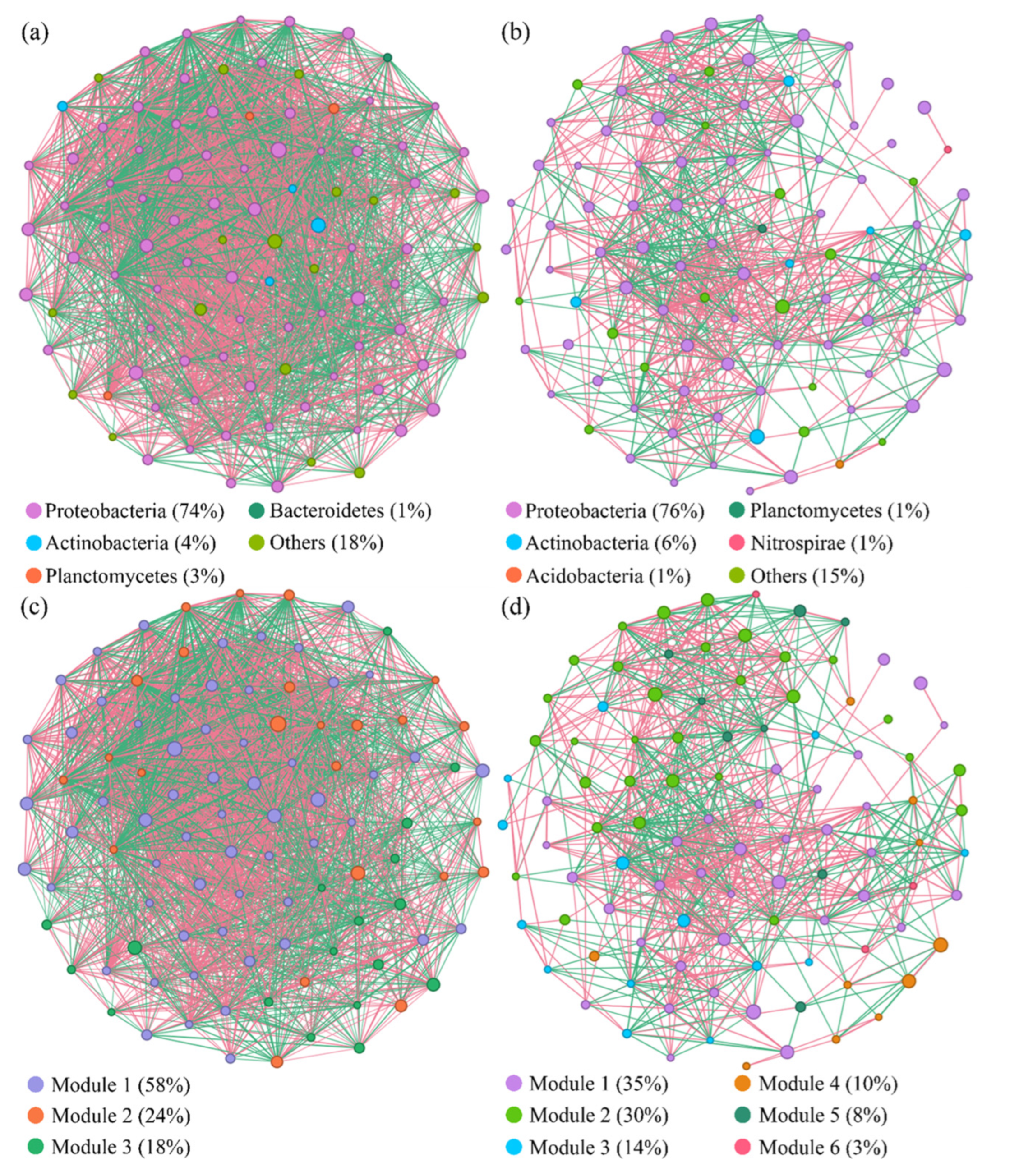
3.5. Bacterial Interaction Patterns
4. Discussion
4.1. Natural Revegetation Affected the Soil Properties and PAH Contents
4.2. Natural Revegetation Affected the Components and Diversity of Bacterial Communities
4.3. Natural Revegetation Stimulated the Biodegradation Potential of PAHs
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Famiyeh, L.; Chen, K.; Xu, J.S. A review on analysis methods, source identification, and cancer risk evaluation of atmospheric polycyclic aromatic hydrocarbons. Sci. Total Environ. 2021, 789, 147741. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Tang, J.; Jiang, X.; Wang, R.; Wang, L.; Bi, X. Occurrence, source apportionment and toxicity assessment of polycyclic aromatic hydrocarbons in surface sediments of the Chaohu Lake, China. Sci. Total Environ. 2019, 651, 2497–2506. [Google Scholar]
- Neff, J.M.; Stout, S.A.; Gunster, D.G. Ecological Risk Assessment of Polycyclic Aromatic Hydrocarbons in Sediments: Integrating Source Identification and Toxicity Evaluation. Environ. Assess. Manag. 2005, 24, 2449–2456. [Google Scholar]
- Francioli, D.; Schulz, E.; Buscot, F.; Wubet, T.; Heintz, B.A.; Reitz, T.; Kuramae, E.E. Dynamics of soil bacterial communities over a vegetation season relate to both soil nutrient status and plant growth phenology. Microb. Ecol. 2018, 75, 216–227. [Google Scholar] [CrossRef]
- Li, X.; Zheng, R.; Bu, Q.; Zhang, Y.; Yang, J.; Liu, J.; Wu, Z.; Liu, X. Comparison of PAH content, potential risk in vegetation, and bare soil near Daqing oil well and evaluating the effects of soil properties on PAHs. Environ. Sci. Pollut. Res. 2019, 26, 25071–25083. [Google Scholar] [CrossRef] [PubMed]
- Premnath, N.; Mohanrasu, K.; Rao, R.G.R.; Dinesh, G.H.; Prakash, G.S.; Pugazhendhi, A.; Jebasingh, S.E.J.; Govarthanan, M. A crucial review on polycyclic aromatic hydrocarbons—Environmental occurrence and strategies for microbial degradation. Chemosphere 2021, 280, 130793. [Google Scholar] [CrossRef]
- Sakshi; Haritash, A.K. A comprehensive review of metabolic and genomic aspects of PAH degradation. Arch. Microbiol. 2020, 202, 2033–2058. [Google Scholar] [CrossRef]
- Chen, T.; Dong, Y.; Huang, W.; Zhang, Y.; Liu, X.; Wang, J.; Li, H.; Guo, S. Dynamics of microbial community and functional genes during bioremediation of PAHs-contaminated soil by two biostimulants. Biochem. Eng. J. 2024, 208, 109356. [Google Scholar] [CrossRef]
- Muangchinda, C.; Chavanich, S.; Viyakarn, V.; Watanabe, K.; Imura, S.; Vangnai, A.S.; Pinyakong, O. Abundance and diversity of functional genes involved in the degradation of aromatic hydrocarbons in Antarctic soils and sediments around Syowa Station. Environ. Sci. Pollut. Res. 2015, 22, 4725–4735. [Google Scholar] [CrossRef]
- Liao, Q.; Liu, H.; Lu, C.; Zhang, Y.; Wang, L.; Li, X.; Chen, J.; Li, Z. Root exudates enhance the PAH degradation and degrading gene abundance in soils. Sci. Total Environ. 2021, 764, 144436. [Google Scholar] [CrossRef]
- Cristaldi, A.; Conti, G.O.; Jho, E.H.; Zuccarello, P.; Grasso, A.; Copat, C.; Ferrante, M. Phytoremediation of contaminated soils by heavy metals and PAHs. A brief review. Environ. Technol. Innov. 2017, 8, 309–326. [Google Scholar] [CrossRef]
- Li, J.; Luo, C.; Zhang, D.; Li, X.; Chen, Y.; Wang, H.; Ma, Y. Diversity of the active phenanthrene degraders in PAH-polluted soil is shaped by ryegrass rhizosphere and root exudates. Soil Biol. Biochem. 2019, 128, 100–110. [Google Scholar] [CrossRef]
- Goswami, M.; Deka, S. Plant growth-promoting rhizobacteria—Alleviators of abiotic stresses in soil: A review. Pedosphere 2020, 30, 40–61. [Google Scholar] [CrossRef]
- Oleńska, E.; Małek, W.; Wójcik, M.; Swiecicka, I.; Thijs, S.; Vangronsveld, J. Beneficial features of plant growth-promoting rhizobacteria for improving plant growth and health in challenging conditions: A methodical review. Sci. Total Environ. 2020, 743, 140682. [Google Scholar] [CrossRef] [PubMed]
- Urana, R.; Dahiya, A.; Sharma, P.; Sharma, S.; Singh, D.; Kumar, V. Effects of Plant Growth Promoting Rhizobacteria on Phytoremediation of Phenanthrene Contaminated Sodic Soil. Polycycl. Aromat. Compd. 2019, 40, 1–10. [Google Scholar] [CrossRef]
- Yu, Y.; Zhang, Y.; Zhao, N.; Li, C.; Sheng, Y.; Liu, H.; Wang, W.; Li, X.; Zou, H.; Zhang, X. Remediation of Crude Oil-Polluted Soil by the Bacterial Rhizosphere Community of Suaeda Salsa Revealed by 16S rRNA Genes. Int. J. Environ. Res. Public Health 2020, 17, 1471. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.-J.; Zhang, S.-W.; Wang, L.; Yan, H.; Xu, J.; Ma, L.-Y.; Guo, Q.-J.; Zhang, X.-H.; Li, X.-F. Concentration and Risk Evaluation of Polycyclic Aromatic Hydrocarbons in Urban Soil in the Typical Semi-Arid City of Xi’an in Northwest China. Int. J. Environ. Res. Public Health 2018, 15, 607. [Google Scholar] [CrossRef]
- Delgado-González, C.R.; Madariaga-Navarrete, A.; Rodríguez-Laguna, R.; Islas-Pelcastre, M.; Méndez-Bautista, J.I.; Fernández-Luqueño, F. Microorganism rhizosphere interactions and their impact on the bioremediation of saline soils: A review. Int. J. Environ. Sci. Technol. 2022, 19, 12775–12790. [Google Scholar] [CrossRef]
- HJ/T 166-2004; The Technical Specification for Soil Environmental Monitoring. China Environmental Publishing House: Beijing, China, 2004.
- Nakhli, S.A.A.; Panta, S.; Brown, J.D.; Sattar, M.A.; Sobeih, W.; Del Grosso, S.J.; Spokas, K.A. Quantifying biochar content in a field soil with varying organic matter content using a two-temperature loss on ignition method. Sci. Total Environ. 2019, 658, 1106–1116. [Google Scholar] [CrossRef]
- Wang, J.; Yang, C.-X.; Zhang, H.-O.; Li, Y.-F.; Liu, X.-M.; Zhao, Q.-S.; Zhang, L.-P. Improving Soil Properties by Sand Application in the Saline-Alkali Area of the Middle and Lower Reaches of the Yellow River, China. Sustainability 2023, 15, 9437. [Google Scholar] [CrossRef]
- Li, X.; Qu, C.; Bian, Y.; Zhang, Y.; Christie, P.; Hu, B.; Zhou, J.; Li, Z.; Liu, X.; Jiang, X. New insights into the responses of soil microorganisms to polycyclic aromatic hydrocarbon stress by combining enzyme activity and sequencing analysis with metabolomics. Environ. Pollut. 2019, 255, 113312. [Google Scholar] [CrossRef]
- Tanhan, P.; Saengwilai, P.; Chotigeat, W. Assessment of dynamic microbial community structure and rhizosphere interactions during bioaugmented phytoremediation of petroleum contaminated soil by a newly designed rhizobox system. Int. J. Phytoremediat. 2022, 24, 1505–1517. [Google Scholar]
- Li, Y.; Li, W.; Ji, L.; Zhang, Y.; Wang, X.; Li, J.; Li, G.; Liu, X.; Jiang, C. Effects of Salinity on the Biodegradation of Polycyclic Aromatic Hydrocarbons in Oilfield Soils Emphasizing Degradation Genes and Soil Enzymes. Front. Microbiol. 2022, 12, 798318. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Wang, Y.-H.; Tian, J.-M.; Li, X.-F.; Zhang, L.-L.; Liu, Y.-Q.; Chen, J.-H.; Zhou, Y.-Y. Characteristics of PAHs in soils under different land-use types and their associated health risks in the northern Taihu Basin, China. J. Soils Sediments 2022, 22, 134–145. [Google Scholar] [CrossRef]
- Shen, F.-T.; Lin, J.-L.; Huang, C.-C.; Ho, M.-J.; Arun, A.B.; Young, C.-C. Molecular detection and phylogenetic analysis of the catechol 1,2-dioxygenase gene from Gordonia spp. Syst. Appl. Microbiol. 2009, 32, 291–300. [Google Scholar] [CrossRef]
- Jia, W.; Cheng, L.; Tan, Q.; Li, Y.; Zhang, H.; Liu, X.; Wang, J.; Chen, Z.; Li, S. Response of the soil microbial community to petroleum hydrocarbon stress shows a threshold effect: Research on aged realistic contaminated fields. Front. Microbiol. 2023, 14, 1188229. [Google Scholar] [CrossRef]
- Barberán, A.; Bates, S.T.; Casamayor, E.O.; Fierer, N. Using network analysis to explore co-occurrence patterns in soil microbial communities. ISME J. 2012, 6, 343–351. [Google Scholar] [CrossRef]
- Zhao, Y.; Duan, F.-A.; Cui, Z.; Li, X.; Zhang, Y.; Wang, L.; Liu, H.; Chen, J.; Guo, S. Insights into the vertical distribution of the microbiota in steel plant soils with potentially toxic elements and PAHs contamination after 60 years operation: Abundance, structure, co-occurrence network and functionality. Sci. Total Environ. 2021, 786, 147338. [Google Scholar] [CrossRef]
- Ding, G.-C.; Heuer, H.; Zühlke, S.; Spiteller, M.; Pronk, G.J.; Heister, K.; Kögel-Knabner, I.; Smalla, K. Soil Type-Dependent Responses to Phenanthrene as Revealed by Determining the Diversity and Abundance of Polycyclic Aromatic Hydrocarbon Ring-Hydroxylating Dioxygenase Genes by Using a Novel PCR Detection System. Appl. Environ. Microbiol. 2010, 76, 4765–4771. [Google Scholar] [CrossRef]
- Maliszewska-Kordybach, B. Polycyclic aromatic hydrocarbons in agricultural soils in Poland: Preliminary proposals for criteria to evaluate the level of soil contamination. Appl. Geochem. 1996, 11, 121–127. [Google Scholar] [CrossRef]
- Hesham, A.; Mawad, A.M.M.; Mostafa, Y.M.; Shoreit, A. Biodegradation Ability and Catabolic Genes of Petroleum-Degrading Sphingomonas koreensis Strain ASU-06 Isolated from Egyptian Oily Soil. Biomed Res. Int. 2014, 2014, 127674. [Google Scholar] [CrossRef] [PubMed]
- Jiao, S.; Liu, Z.; Lin, Y.; Yang, J.; Chen, W.; Wei, G. Bacterial communities in oil contaminated soils: Biogeography and co-occurrence patterns. Soil Biol. Biochem. 2016, 98, 64–73. [Google Scholar] [CrossRef]
- Zhao, Q.Q.; Bai, J.H.; Gao, Y.C.; Zhang, G.L.; Wang, X.M.; Cui, B.-S. Shifts in the soil bacterial community along a salinity gradient in the Yellow River Delta. Land Degrad. Dev. 2020, 31, 2255–2267. [Google Scholar] [CrossRef]
- Zhang, L.; Yang, J.; Ge, A.-H.; Li, Y.; Wang, X.; Chen, Z.; Liu, H. Salinity drives niche differentiation of soil bacteria and archaea in Hetao Plain, China. J. Environ. Manag. 2024, 370, 122977. [Google Scholar] [CrossRef] [PubMed]
- Lau, J.A.; Lennon, J.T. Rapid responses of soil microorganisms improve plant fitness in novel environments. Proc. Natl. Acad. Sci. USA 2012, 109, 14058–14062. [Google Scholar] [CrossRef] [PubMed]
- Lareen, A.; Burton, F.; Schäfer, P. Plant root-microbe communication in shaping root microbiomes. Plant Mol. Biol. 2016, 90, 575–587. [Google Scholar] [CrossRef]
- Yin, Y.W.; Xia, N.; Wei, H.F. Study on the absorption of PAHs by Suaeda heteroptera under hydroponic conditions. Appl. Ecol. Environ. Res. 2024, 22, 1959–1970. [Google Scholar] [CrossRef]
- Wang, L.L.; Zhang, G.H.; Zhu, P.Z.; Li, X.F.; Liu, Y.Q.; Wang, J.H.; Chen, Y.M. Correlations between plant and soil for their C, N, P contents and stoichiometry on the steep gully slopes. Ecol. Indic. 2023, 154, 110823. [Google Scholar] [CrossRef]
- Zhang, X.M.; Wang, Y.D.; Zhao, Y.; Xu, X.W.; Li, S.Y.; Liu, J.H. Litter decomposition and nutrient dynamics of three woody halophytes in the Taklimakan Desert Highway Shelterbelt. Arid Land Res. Manag. 2017, 31, 335–351. [Google Scholar] [CrossRef]
- Yuan, H.; Li, T.; Ding, X.; Zhao, G.; Ye, S.; Han, J. Distribution, sources and potential toxicological significance of polycyclic aromatic hydrocarbons (PAHs) in surface soils of the Yellow River Delta, China. Mar. Pollut. Bull. 2014, 83, 258–264. [Google Scholar] [CrossRef]
- Yuan, Z.; Liu, G.; Da, C.; Ye, S.; Han, J.; Liu, H. Occurrence, sources, and potential toxicity of polycyclic aromatic hydrocarbons in surface soils from the Yellow River Delta Natural Reserve, China. Arch. Environ. Contam. Toxicol. 2015, 68, 330–341. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Guo, S.; Wang, J. Assessment of the human health risk of polycyclic aromatic hydrocarbons in soils from areas of crude oil exploitation. Environ. Res. 2021, 193, 110617. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Qian, F.; Bao, Y. Variations of microbiota and metabolites in rhizosphere soil of Carmona microphylla at the co-contaminated site with polycyclic aromatic hydrocarbons and heavy metals. Ecotoxicol. Environ. Saf. 2025, 290, 117734. [Google Scholar] [CrossRef] [PubMed]
- Alagic, S.C.; Maluckov, B.S.; Radojicic, V.B. How can plants manage polycyclic aromatic hydrocarbons? May these effects represent a useful tool for an effective soil remediation? A review. Clean Technol. Environ. Policy 2015, 17, 597–614. [Google Scholar] [CrossRef]
- Yuan, L.M.; Wu, Y.Q.; Fan, Q.H.; Zhang, X.H.; Li, S.Y.; Wang, J.H.; Chen, Y.M. Remediating petroleum hydrocarbons in highly saline-alkali soils using three native plant species. J. Environ. Manag. 2023, 339, 117567. [Google Scholar] [CrossRef]
- Gul, N.; Wani, I.A.; Mir, R.A.; Nowshehri, J.A.; Aslam, S.; Gupta, R.; Verma, S.; Aslam, S. Plant growth promoting microorganisms mediated abiotic stress tolerance in crop plants: A critical appraisal. Plant Growth Regul. 2023, 100, 7–24. [Google Scholar] [CrossRef]
- Gul, N.; Wani, I.A.; Mir, R.A.; Hakeem, K.R.; Qadri, R.A.; Nazir, R. The role of rhizodeposits in shaping rhizomicrobiome. Environ. Microbiol. Rep. 2020, 12, 160–172. [Google Scholar]
- Pan, Y.Q.; Kang, P.; Tan, M.; Li, Y.F.; Liu, X.M.; Wang, J.H.; Zhang, X.H.; Chen, Y.M. Root exudates and rhizosphere soil bacterial relationships of Nitraria tangutorum are linked to k-strategists bacterial community under salt stress. Front. Plant Sci. 2022, 13, 987578. [Google Scholar] [CrossRef]
- Parasar, B.J.; Sharma, I.; Agarwala, N. Root exudation drives abiotic stress tolerance in plants by recruiting beneficial microbes. Appl. Soil Ecol. 2024, 198, 105432. [Google Scholar] [CrossRef]
- Li, J.J.; Yang, L.; Mao, S.L.; Zhang, Y.H.; Wang, X.M.; Liu, Y.Q.; Chen, J.H.; Zhou, Y.Y. Assembly and enrichment of rhizosphere and bulk soil microbiomes in Robinia pseudoacacia plantations during long-term vegetation restoration. Appl. Soil Ecol. 2023, 187, 104846. [Google Scholar] [CrossRef]
- Shi, S.; Nuccio, E.E.; Shi, Z.J.; Zhou, J.; Yuan, C.; Firestone, M.K.; Pett-Ridge, J. The interconnected rhizosphere: High network complexity dominates rhizosphere assemblages. Ecol. Lett. 2016, 19, 926–936. [Google Scholar] [CrossRef] [PubMed]
- Graham, E.D.; Tully, B.J. Marine Dadabacteria exhibit genome streamlining and phototrophy-driven niche partitioning. ISME J. 2021, 15, 1248–1256. [Google Scholar] [CrossRef] [PubMed]
- Picariello, E.; Baldantoni, D.; De Nicola, F. Annual dynamics of indigenous microbial communities of forest soils after severe PAH contamination. Appl. Soil Ecol. 2023, 186, 104812. [Google Scholar] [CrossRef]
- Guo, Y.; Chen, X.; Wu, Y.; Zhang, L.; Li, J.; Liu, X.; Wang, J. Natural revegetation of a semiarid habitat alters taxonomic and functional diversity of soil microbial communities. Sci. Total Environ. 2018, 635, 598–606. [Google Scholar] [CrossRef]
- Gao, Y.; Yuan, L.; Du, J.; Li, X.; Zhang, Y.; Wang, L.; Liu, H.; Chen, J.; Guo, S. Bacterial community profile of the crude oil-contaminated saline soil in the Yellow River Delta Natural Reserve, China. Chemosphere 2022, 289, 133207. [Google Scholar] [CrossRef]
- Wang, L.M.; Wang, J.J.; Guo, D.F.; Li, X.F.; Zhang, Y.H.; Liu, Y.Q.; Chen, J.H. Catabolic Activity and Structural Diversity of Bacterial Community in Soil Covered by Halophytic Vegetation. Curr. Microbiol. 2020, 77, 1821–1828. [Google Scholar] [CrossRef]
- Qin, H.; Liu, Y.; Chen, C.; Chen, A.; Liang, Y.; Cornell, C.R. Differential contribution of microbial and plant-derived organic matter to soil organic carbon sequestration over two decades of natural revegetation and cropping. Sci. Total Environ. 2024, 949, 174960. [Google Scholar] [CrossRef]
- Shi, X.L.; Guo, P.; Chen, Y.X.; Li, J.; Zhang, Y.; Wang, L.; Liu, H. Integrated Analysis of Soil Metagenome and Soil Metabolome Reveals the Differential Responses of Sorghum and Peanut Rhizosphere Microbes to Salt Stress. J. Soil Sci. Plant Nutr. 2024, 24, 2959–2971. [Google Scholar] [CrossRef]
- Kawasaki, A.; Watson, E.R.; Kertesz, M.A. Indirect effects of polycyclic aromatic hydrocarbon contamination on microbial communities in legume and grass rhizospheres. Plant Soil 2012, 358, 162–175. [Google Scholar] [CrossRef]
- Hamdi, H.; Benzarti, S.; Aoyama, I.; Jedidi, N. Rehabilitation of degraded soils containing aged PAHs based on phytoremediation with alfalfa (Medicago sativa L.). Int. Biodeterior. Biodegrad. 2012, 67, 40–47. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Item | GAB | GAV | GBB | GBV | GCB | GCV |
|---|---|---|---|---|---|---|
| pH | 7.71 ± 0.03 a | 7.81 ± 0.08 a | 7.81 ± 0.16 a | 7.74 ± 0.09 a | 7.96 ± 0.25 a | 7.85 ± 0.22 a |
| EC (ms·cm−1) | 1.25 ± 0.70 b | 1.24 ± 1.05 b | 2.06 ± 0.37 b | 1.98 ± 1.17 b | 7.23 ± 2.32 a | 2.96 ± 1.84 b |
| OM (g·kg−1) | 132.65 ± 77.84 ab | 138.00 ± 24.66 ab | 169.32 ± 68.35 a | 102.27 ± 69.95 ab | 19.87 ± 26.10 b | 36.95 ± 11.01 b |
| CAT (μmol·h−1·g−1) | 4.07 ± 0.46 ab | 4.81 ± 1.16 a | 3.52 ± 0.17 b | 3.49 ± 1.09 b | 2.39 ± 0.61 b | 3.42 ± 0.36 b |
| AN (g·kg−1) | 50.37 ± 6.26 a | 49.21 ± 6.10 a | 47.48 ± 17.57 a | 42.71 ± 18.21 a | 41.25 ± 6.08 a | 39.51 ± 16.86 a |
| AP (g·kg−1) | 7.19 ± 1.96 c | 7.20 ± 1.17 c | 7.79 ± 4.45 c | 5.86 ± 2.88 c | 4.05 ± 0.94 a | 5.60 ± 2.40 b |
| TPHs (g·kg−1) | 5.69 ± 7.82 a | 2.43 ± 0.89 a | 5.88 ± 7.92 a | 2.41 ± 1.23 a | 1.74 ± 2.07 a | 0.94 ± 0.19 a |
| TPAHs (mg·kg−1) | 2.30 ± 0.64 a | 1.13 ± 0.21 b | 2.04 ± 0.56 ab | 0.90 ± 0.38 b | 1.77 ± 0.43 a | 0.79 ± 0.20 b |
| LMW PAHs (mg·kg−1) | 0.46 ± 0.05 ab | 0.38 ± 0.11 b | 0.63 ± 0.07 a | 0.27 ± 0.13 b | 0.62 ± 0.25 a | 0.21 ± 0.11 b |
| HMW PAHs (mg·kg−1) | 1.84 ± 0.63 a | 0.76 ± 0.10 a | 1.07 ± 0.04 a | 0.63 ± 0.26 a | 1.16 ± 0.19 a | 0.58 ± 0.10 a |
| TEQBaP (mg·kg−1) | 0.39 ± 0.13 a | 0.03 ± 0.002 b | 0.11 ± 0.06 b | 0.08 ± 0.06 b | 0.15 ± 0.09 b | 0.06 ± 0.05 b |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, J.; Liang, H.; Huang, L.; Li, Q.; Ji, L.; Xing, Y.; Zhou, C.; Wang, J.; Fu, X. Natural Revegetation Alters Habitat Conditions, Bacterial Components, and Polycyclic Aromatic Hydrocarbon (PAH)-Degrading Communities in Aged PAH-Polluted Soils. Microorganisms 2025, 13, 1098. https://doi.org/10.3390/microorganisms13051098
Huang J, Liang H, Huang L, Li Q, Ji L, Xing Y, Zhou C, Wang J, Fu X. Natural Revegetation Alters Habitat Conditions, Bacterial Components, and Polycyclic Aromatic Hydrocarbon (PAH)-Degrading Communities in Aged PAH-Polluted Soils. Microorganisms. 2025; 13(5):1098. https://doi.org/10.3390/microorganisms13051098
Chicago/Turabian StyleHuang, Jinrong, Heng Liang, Lilong Huang, Qi Li, Lei Ji, Yingna Xing, Chang Zhou, Jianing Wang, and Xiaowen Fu. 2025. "Natural Revegetation Alters Habitat Conditions, Bacterial Components, and Polycyclic Aromatic Hydrocarbon (PAH)-Degrading Communities in Aged PAH-Polluted Soils" Microorganisms 13, no. 5: 1098. https://doi.org/10.3390/microorganisms13051098
APA StyleHuang, J., Liang, H., Huang, L., Li, Q., Ji, L., Xing, Y., Zhou, C., Wang, J., & Fu, X. (2025). Natural Revegetation Alters Habitat Conditions, Bacterial Components, and Polycyclic Aromatic Hydrocarbon (PAH)-Degrading Communities in Aged PAH-Polluted Soils. Microorganisms, 13(5), 1098. https://doi.org/10.3390/microorganisms13051098