


Host-Directed Therapies Based on Protease Inhibitors to Control Mycobacterium tuberculosis and HIV Coinfection




Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
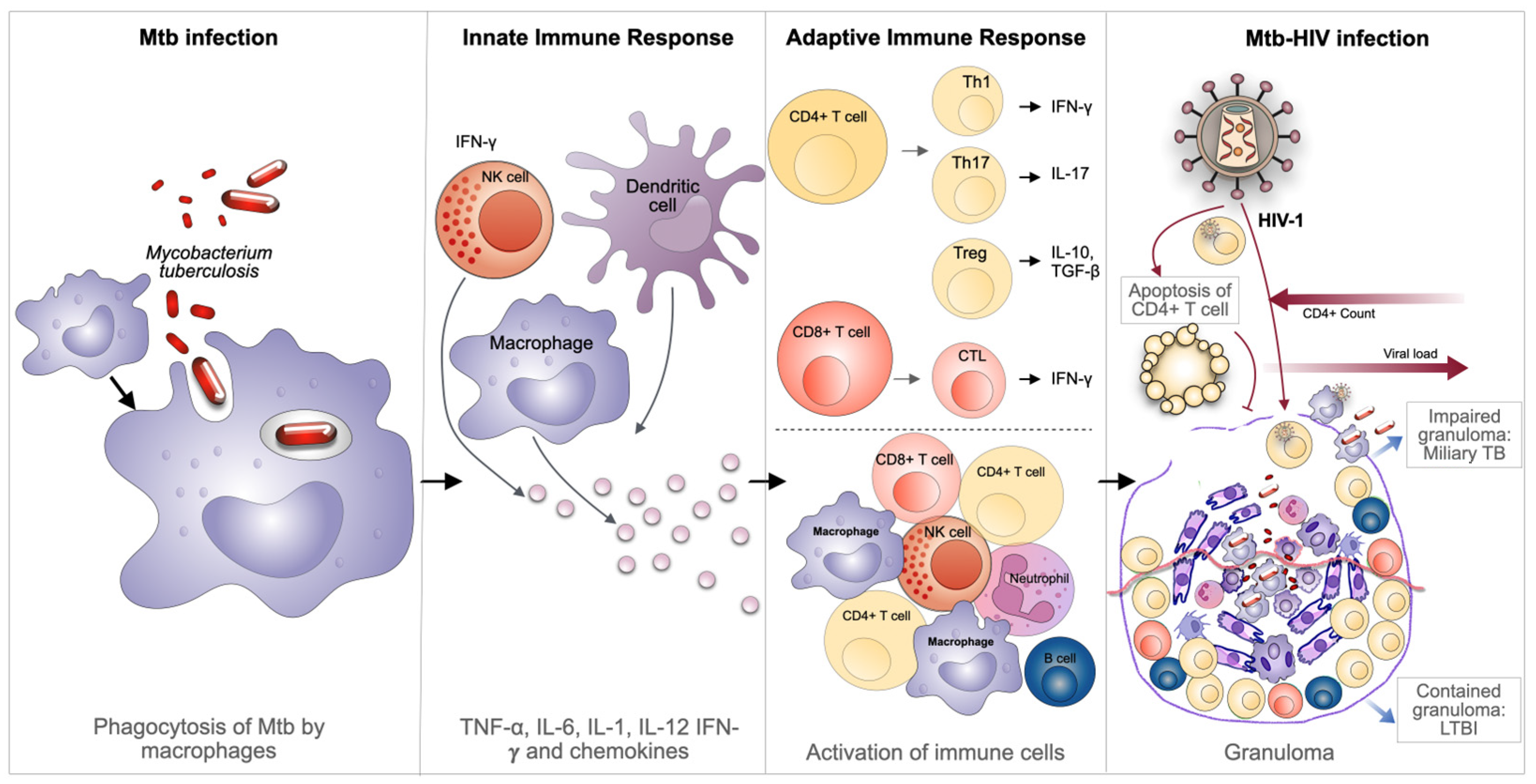
2. Immuno-Pathogenesis of Mtb-HIV Coinfection
3. Current Therapeutic Approaches in TB and During HIV Coinfection
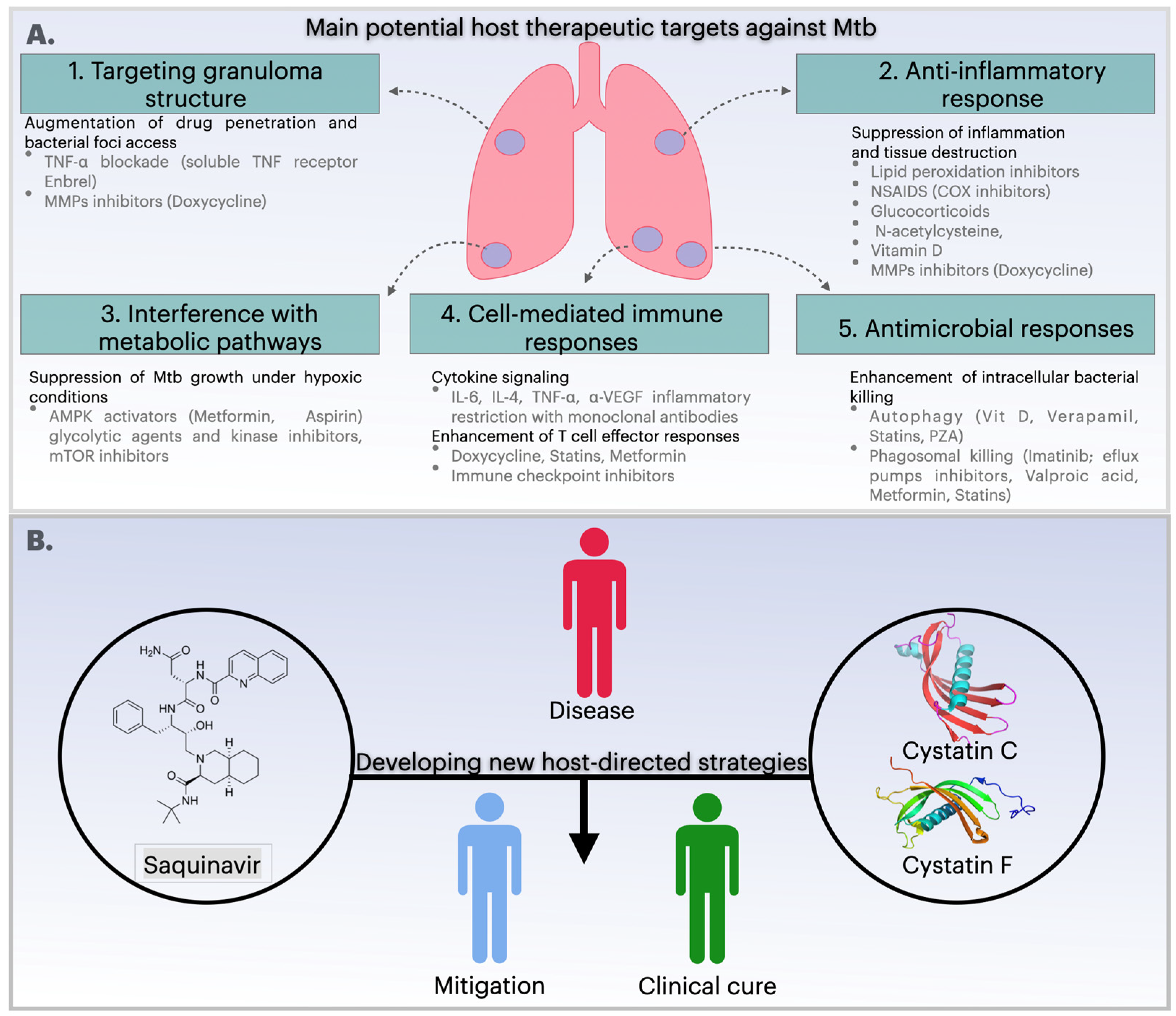
4. Host-Directed Therapies: Current Status and Recent Progress for the Treatment of Infections
5. Protease Inhibitors as a Strategy to Control Infectious Diseases
6. New Host-Directed Strategies Based on Protease Inhibitors for Mtb and Coinfection with HIV
6.1. Repurposing Saquinavir as a Host-Directed Strategy to Control Mycobacterium tuberculosis Infection
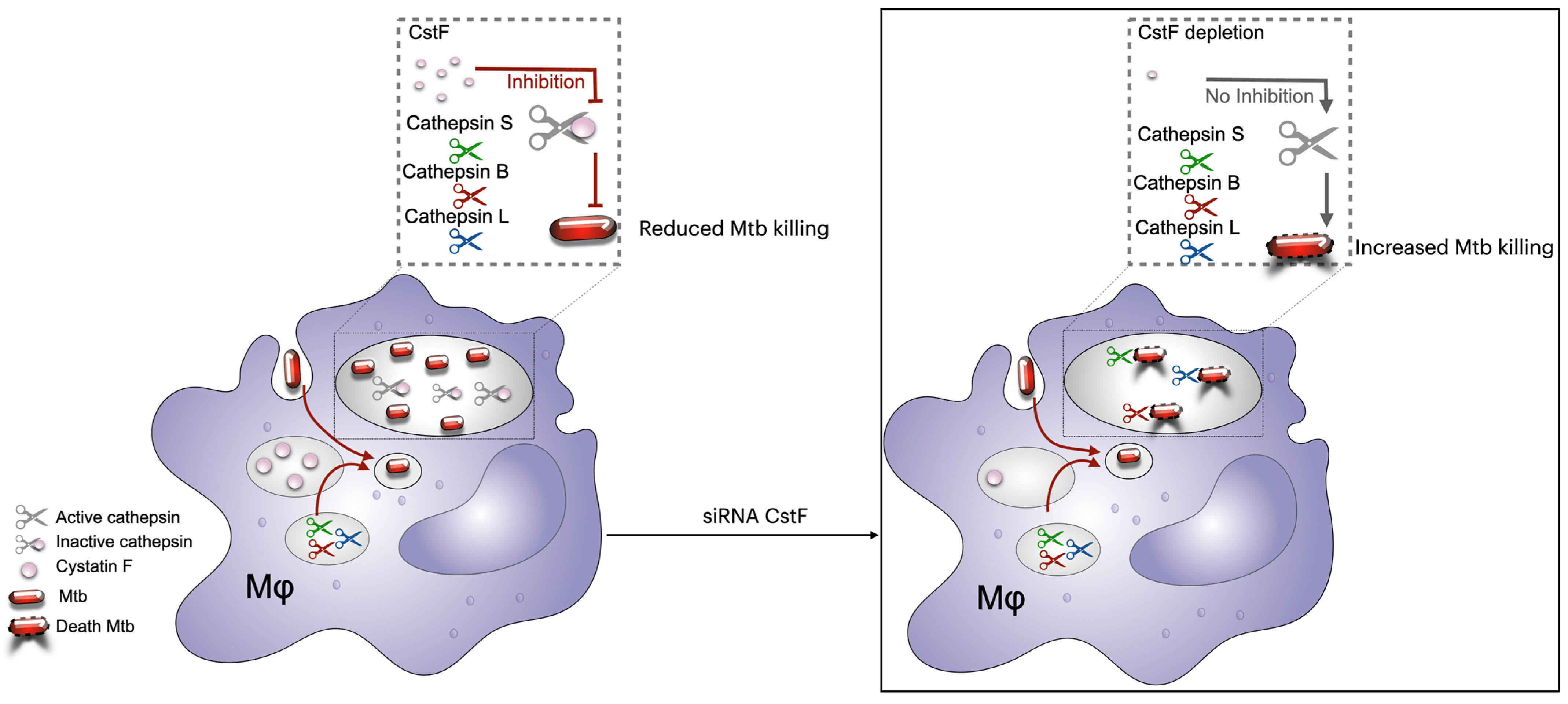
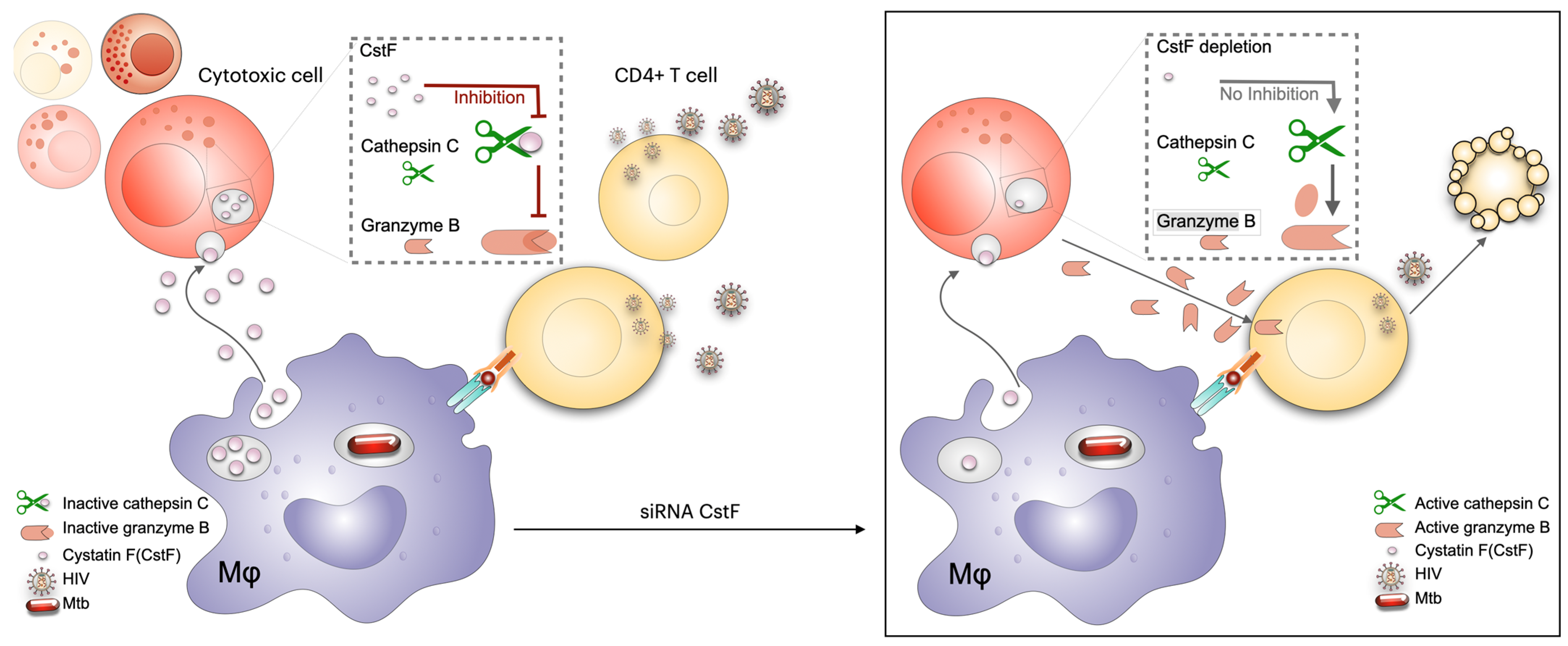
6.2. Modulation of Cystatins C and F as a Host-Directed Strategy to Control Mtb Mono-Infection or Coinfection with HIV
6.3. Matrix Metalloproteinase Inhibitors to Treat TB Immunopathology
7. Conclusions and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- World Health Organization. 2024 Global Tuberculosis Report. 2024. Available online: https://www.who.int/teams/global-tuberculosis-programme/tb-reports/global-tuberculosis-report-2024 (accessed on 14 January 2025).
- Houben, R.M.G.J.; Dodd, P.J. The Global Burden of Latent Tuberculosis Infection: A Re-Estimation Using Mathematical Modelling. PLoS Med. 2016, 13, e1002152. [Google Scholar] [CrossRef]
- Kiazyk, S.; Ball, T.B. Latent Tuberculosis Infection: An Overview. Can. Commun. Dis. Rep. 2017, 43, 62–66. [Google Scholar] [CrossRef] [PubMed]
- UNAIDS. 2024 Report. The Urgency of Now: Aids at a Crossroads; Joint United Nations Programme on HIV/AIDS: Geneva, Switzerland, 2024; Available online: https://www.unaids.org/sites/default/files/media_asset/2024-unaids-global-aids-update_en.pdf (accessed on 14 March 2025).
- Kitenge, M.K.; Fatti, G.; Eshun-Wilson, I.; Aluko, O.; Nyasulu, P. Prevalence and Trends of Advanced HIV Disease among Antiretroviral Therapy-Naïve and Antiretroviral Therapy-Experienced Patients in South Africa between 2010-2021: A Systematic Review and Meta-Analysis. BMC Infect. Dis. 2023, 23, 549. [Google Scholar] [CrossRef]
- Abdulrahman, S.A.; Ganasegeran, K.; Rampal, L.; Martins, O.F. HIV Treatment Adherence—A Shared Burden for Patients, Health-Care Providers, and Other Stakeholders. AIDS Rev. 2019, 21, 28–39. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. HIV Drug Resistance: Brief Report 2024. 2024. Available online: https://iris.who.int/bitstream/handle/10665/376039/9789240086319-eng.pdf?sequence=1 (accessed on 14 March 2025).
- Mandell, L.; Woodhead, M.; Ewig, S.; Torres, A. Respiratory Infections; CRC Press: Boca Raton, FL, USA, 2006; ISBN 9780429073366. [Google Scholar]
- Russell, D.G. Mycobacterium tuberculosis: Here Today, and Here Tomorrow. Nat. Rev. Mol. Cell Biol. 2001, 2, 569–577. [Google Scholar] [CrossRef] [PubMed]
- Hosseinian, K.; Gerami, A.; Bral, M.; Venketaraman, V. Mycobacterium tuberculosis-Human Immunodeficiency Virus Infection and the Role of T Cells in Protection. Vaccines 2024, 12, 730. [Google Scholar] [CrossRef]
- Stamm, C.E.; Collins, A.C.; Shiloh, M.U. Sensing of Mycobacterium tuberculosis and Consequences to Both Host and Bacillus. Immunol. Rev. 2015, 264, 204–219. [Google Scholar] [CrossRef]
- Woo, M.; Wood, C.; Kwon, D.; Park, K.-H.P.; Fejer, G.; Delorme, V. Mycobacterium tuberculosis Infection and Innate Responses in a New Model of Lung Alveolar Macrophages. Front. Immunol. 2018, 9, 438. [Google Scholar] [CrossRef] [PubMed]
- Domingo-Gonzalez, R.; Prince, O.; Cooper, A.; Khader, S.A. Cytokines and Chemokines in Mycobacterium tuberculosis Infection. Microbiol. Spectr. 2016, 4, 4–5. [Google Scholar] [CrossRef]
- Nisa, A.; Kipper, F.C.; Panigrahy, D.; Tiwari, S.; Kupz, A.; Subbian, S. Different Modalities of Host Cell Death and Their Impact on Mycobacterium tuberculosis Infection. Am. J. Physiol. Cell Physiol. 2022, 323, C1444–C1474. [Google Scholar] [CrossRef]
- Navasardyan, I.; Miwalian, R.; Petrosyan, A.; Yeganyan, S.; Venketaraman, V. HIV-TB Coinfection: Current Therapeutic Approaches and Drug Interactions. Viruses 2024, 16, 321. [Google Scholar] [CrossRef] [PubMed]
- de Martino, M.; Lodi, L.; Galli, L.; Chiappini, E. Immune Response to Mycobacterium tuberculosis: A Narrative Review. Front. Pediatr. 2019, 7, 350. [Google Scholar] [CrossRef] [PubMed]
- Cronan, M.R. In the Thick of It: Formation of the Tuberculous Granuloma and Its Effects on Host and Therapeutic Responses. Front. Immunol. 2022, 13, 820134. [Google Scholar] [CrossRef]
- Ramakrishnan, L. Revisiting the Role of the Granuloma in Tuberculosis. Nat. Rev. Immunol. 2012, 12, 352–366. [Google Scholar] [CrossRef] [PubMed]
- Azevedo-Pereira, J.M.; Pires, D.; Calado, M.; Mandal, M.; Santos-Costa, Q.; Anes, E. HIV/Mtb Co-Infection: From the Amplification of Disease Pathogenesis to an “Emerging Syndemic”. Microorganisms 2023, 11, 853. [Google Scholar] [CrossRef]
- Bares, S.H.; Swindells, S. Latent Tuberculosis and HIV Infection. Curr. Infect. Dis. Rep. 2020, 22, 17. [Google Scholar] [CrossRef]
- Diedrich, C.R.; O’Hern, J.; Wilkinson, R.J. HIV-1 and the Mycobacterium tuberculosis Granuloma: A Systematic Review and Meta-Analysis. Tuberculosis 2016, 98, 62–76. [Google Scholar] [CrossRef]
- Jones, R.M.; Adams, K.N.; Eldesouky, H.E.; Sherman, D.R. The Evolving Biology of Mycobacterium tuberculosis Drug Resistance. Front. Cell. Infect. Microbiol. 2022, 12, 1027394. [Google Scholar] [CrossRef]
- Sun, W.; Gui, X.; Wu, Z.; Zhang, Y.; Yan, L. Prediction of Drug Resistance Profile of Multidrug-Resistant Mycobacterium tuberculosis (MDR-MTB) Isolates from Newly Diagnosed Case by Whole Genome Sequencing (WGS): A Study from a High Tuberculosis Burden Country. BMC Infect. Dis. 2022, 22, 499. [Google Scholar] [CrossRef]
- World Health Organization. HIV Drug Resistance Report 2021; World Health Organization: Geneva, Switzerland, 2021; Available online: https://www.who.int/publications/i/item/9789240038608 (accessed on 14 January 2025).
- Olivença, F.; Nunes, A.; Macedo, R.; Pires, D.; Silveiro, C.; Anes, E.; Miragaia, M.; Gomes, J.P.; Catalão, M.J. Uncovering Beta-Lactam Susceptibility Patterns in Clinical Isolates of Mycobacterium tuberculosis through Whole-Genome Sequencing. Microbiol. Spectr. 2022, 10, e0067422. [Google Scholar] [CrossRef]
- Pires, D.; Valente, E.; Simões, M.F.; Carmo, N.; Testa, B.; Constantino, L.; Anes, E. Esters of Pyrazinoic Acid Are Active against Pyrazinamide-Resistant Strains of Mycobacterium tuberculosis and Other Naturally Resistant Mycobacteria In Vitro and Ex Vivo within Macrophages. Antimicrob. Agents Chemother. 2015, 59, 7693–7699. [Google Scholar] [CrossRef]
- Pais, J.P.; Magalhães, M.; Antoniuk, O.; Barbosa, I.; Freire, R.; Pires, D.; Valente, E.; Testa, B.; Anes, E.; Constantino, L. Benzoic Acid Derivatives as Prodrugs for the Treatment of Tuberculosis. Pharmaceuticals 2022, 15, 1118. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention. Treatment for TB Disease. Available online: https://www.cdc.gov/tb/treatment/index.html (accessed on 30 October 2023).
- Seung, K.J.; Keshavjee, S.; Rich, M.L. Multidrug-Resistant Tuberculosis and Extensively Drug-Resistant Tuberculosis. Cold Spring Harb. Perspect. Med. 2015, 5, a017863. [Google Scholar] [CrossRef]
- Beloor Suresh, A.; Rosani, A.; Patel, P.; Wadhwa, R. Rifampin. 2025. Available online: https://www.ncbi.nlm.nih.gov/books/NBK557488/ (accessed on 14 March 2025).
- Koch, A.; Mizrahi, V.; Warner, D.F. The Impact of Drug Resistance on Mycobacterium tuberculosis Physiology: What Can We Learn from Rifampicin? Emerg. Microbes Infect. 2014, 3, e17. [Google Scholar] [CrossRef] [PubMed]
- Timmins, G.S.; Deretic, V. Mechanisms of Action of Isoniazid. Mol. Microbiol. 2006, 62, 1220–1227. [Google Scholar] [CrossRef]
- Marrakchi, H.; Lanéelle, M.-A.; Daffé, M. Mycolic Acids: Structures, Biosynthesis, and Beyond. Chem. Biol. 2014, 21, 67–85. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Shi, W.; Zhang, W.; Mitchison, D. Mechanisms of Pyrazinamide Action and Resistance. Microbiol. Spectr. 2014, 2. [Google Scholar] [CrossRef]
- Palomino, J.C.; Martin, A. Drug Resistance Mechanisms in Mycobacterium tuberculosis. Antibiotics 2014, 3, 317–340. [Google Scholar] [CrossRef]
- Olivença, F.; Pires, D.; Silveiro, C.; Gama, B.; Holtreman, F.; Anes, E.; Catalão, M.J. Ethambutol and Meropenem/Clavulanate Synergy Promotes Enhanced Extracellular and Intracellular Killing of Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2024, 68, e0158623. [Google Scholar] [CrossRef]
- Ennassiri, W.; Jaouhari, S.; Sabouni, R.; Cherki, W.; Charof, R.; Filali-Maltouf, A.; Lahlou, O. Analysis of Isoniazid and Rifampicin Resistance in Mycobacterium tuberculosis Isolates in Morocco Using GenoType® MTBDRplus Assay. J. Glob. Antimicrob. Resist. 2018, 12, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Lai, R.P.J.; Meintjes, G.; Wilkinson, R.J. HIV-1 Tuberculosis-Associated Immune Reconstitution Inflammatory Syndrome. Semin. Immunopathol. 2016, 38, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Sueki, H.; Mizukawa, Y.; Aoyama, Y. Immune Reconstitution Inflammatory Syndrome in Non-HIV Immunosuppressed Patients. J. Dermatol. 2018, 45, 3–9. [Google Scholar] [CrossRef]
- Manzardo, C.; Guardo, A.C.; Letang, E.; Plana, M.; Gatell, J.M.; Miro, J.M. Opportunistic Infections and Immune Reconstitution Inflammatory Syndrome in HIV-1-Infected Adults in the Combined Antiretroviral Therapy Era: A Comprehensive Review. Expert. Rev. Anti Infect. Ther. 2015, 13, 751–767. [Google Scholar] [CrossRef]
- Bruchfeld, J.; Correia-Neves, M.; Kallenius, G. Tuberculosis and HIV Coinfection. Cold Spring Harb. Perspect. Med. 2015, 5, a017871. [Google Scholar] [CrossRef]
- Panel on Antiretroviral Guidelines for Adults and Adolescents. In Guidelines for the Use of Antiretroviral Agents in Adults and Adolescents with HIV; Department of Health and Human Services: Washington, DC, USA, 2024. Available online: https://clinicalinfo.hiv.gov/en/guidelines/adult-and-adolescent-arv (accessed on 14 January 2025).
- Niemi, M.; Backman, J.T.; Fromm, M.F.; Neuvonen, P.J.; Kivistö, K.T. Pharmacokinetic Interactions with Rifampicin: Clinical Relevance. Clin. Pharmacokinet. 2003, 42, 819–850. [Google Scholar] [CrossRef] [PubMed]
- Schutz, C.; Meintjes, G.; Almajid, F.; Wilkinson, R.J.; Pozniak, A. Clinical Management of Tuberculosis and HIV-1 Co-Infection. Eur. Respir. J. 2010, 36, 1460–1481. [Google Scholar] [CrossRef]
- Horne, D.J.; Spitters, C.; Narita, M. Experience with Rifabutin Replacing Rifampin in the Treatment of Tuberculosis. Int. J. Tuberc. Lung Dis. 2011, 15, 1485–1489. [Google Scholar] [CrossRef]
- Ignatius, E.H.; Swindells, S. Are We There Yet? Short-Course Regimens in TB and HIV: From Prevention to Treatment of Latent to XDR TB. Curr. HIV/AIDS Rep. 2020, 17, 589–600. [Google Scholar] [CrossRef]
- Swindells, S.; Ramchandani, R.; Gupta, A.; Benson, C.A.; Leon-Cruz, J.; Mwelase, N.; Jean Juste, M.A.; Lama, J.R.; Valencia, J.; Omoz-Oarhe, A.; et al. One Month of Rifapentine plus Isoniazid to Prevent HIV-Related Tuberculosis. N. Engl. J. Med. 2019, 380, 1001–1011. [Google Scholar] [CrossRef]
- World Health Organization. WHO Consolidated Guidelines on Tuberculosis Tuberculosis Preventive Treatment; World Health Organization: Geneva, Switzerland, 2020; ISBN 9789240001503. [Google Scholar]
- Pais, J.P.; Antoniuk, O.; Pires, D.; Delgado, T.; Fortuna, A.; Costa, P.J.; Anes, E.; Constantino, L. Synthesis, Activity, Toxicity, and In Silico Studies of New Antimycobacterial N-Alkyl Nitrobenzamides. Pharmaceuticals 2024, 17, 608. [Google Scholar] [CrossRef] [PubMed]
- Carr, W.; Kurbatova, E.; Starks, A.; Goswami, N.; Allen, L.; Winston, C. Interim Guidance: 4-Month Rifapentine-Moxifloxacin Regimen for the Treatment of Drug-Susceptible Pulmonary Tuberculosis—United States, 2022. MMWR Morb. Mortal. Wkly. Rep. 2022, 71, 285–289. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention. TB Treatment for Persons with HIV. Available online: https://www.cdc.gov/tb/hcp/clinical-care/hiv.html?CDC_AAref_Val=https://www.cdc.gov/tb/topic/treatment/tbhiv.htm (accessed on 14 January 2025).
- Alexandrova, L.; Zicari, S.; Matyugina, E.; Khandazhinskaya, A.; Smirnova, T.; Andreevskaya, S.; Chernousova, L.; Vanpouille, C.; Kochetkov, S.; Margolis, L. Dual-Targeted Anti-TB/Anti-HIV Heterodimers. Antivir. Res. 2017, 145, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Mukherjee, T.; Radhakrishnan, R.; Paidipally, P.; Ansari, D.; John, S.; Vankayalapati, R.; Tripathi, D.; Yi, G. HIV-Differentiated Metabolite N-Acetyl-L-Alanine Dysregulates Human Natural Killer Cell Responses to Mycobacterium tuberculosis Infection. Int. J. Mol. Sci. 2023, 24, 7267. [Google Scholar] [CrossRef]
- Migliori, G.B.; Tiberi, S. WHO Drug-Resistant TB Guidelines 2022: What Is New? Int. J. Tuberc. Lung Dis. 2022, 26, 590–591. [Google Scholar] [CrossRef]
- Conradie, F.; Diacon, A.H.; Ngubane, N.; Howell, P.; Everitt, D.; Crook, A.M.; Mendel, C.M.; Egizi, E.; Moreira, J.; Timm, J.; et al. Treatment of Highly Drug-Resistant Pulmonary Tuberculosis. N. Engl. J. Med. 2020, 382, 893–902. [Google Scholar] [CrossRef]
- Schaberg, T.; Rebhan, K.; Lode, H. Risk Factors for Side-Effects of Isoniazid, Rifampin and Pyrazinamide in Patients Hospitalized for Pulmonary Tuberculosis. Eur. Respir. J. 1996, 9, 2026–2030. [Google Scholar] [CrossRef] [PubMed]
- Awofeso, N. Anti-Tuberculosis Medication Side-Effects Constitute Major Factor for Poor Adherence to Tuberculosis Treatment. Bull. World Health Organ. 2008, 86, 240. [Google Scholar] [CrossRef] [PubMed]
- Nortey, A.N.; Adjoda, A.; Alhassan, A.; Scott, G.Y. Adherence Patterns, Risk Factors and Complications among Patients with Tuberculosis: A Cross-Sectional Study at Nsawam Government Hospital. BMJ Public Health 2024, 2, e000618. [Google Scholar] [CrossRef]
- Sant´Anna, F.M.; Araújo-Pereira, M.; Schmaltz, C.A.S.; Arriaga, M.B.; Andrade, B.B.; Rolla, V.C. Impact of Adverse Drug Reactions on the Outcomes of Tuberculosis Treatment. PLoS ONE 2023, 18, e0269765. [Google Scholar] [CrossRef]
- Yang, T.W.; Park, H.O.; Jang, H.N.; Yang, J.H.; Kim, S.H.; Moon, S.H.; Byun, J.H.; Lee, C.E.; Kim, J.W.; Kang, D.H. Side Effects Associated with the Treatment of Multidrug-Resistant Tuberculosis at a Tuberculosis Referral Hospital in South Korea. Medicine 2017, 96, e7482. [Google Scholar] [CrossRef]
- Walker, T.M.; Watson, J.A.; Moore, D.A.J.; Frick, M.; Jamrozik, E. Tuberculosis Preventive Therapy: Scientific and Ethical Considerations for Trials of Ultra-Short Regimens. Lancet Infect. Dis. 2025. [Google Scholar] [CrossRef] [PubMed]
- Derendinger, B.; Dippenaar, A.; de Vos, M.; Huo, S.; Alberts, R.; Tadokera, R.; Limberis, J.; Sirgel, F.; Dolby, T.; Spies, C.; et al. Bedaquiline Resistance in Patients with Drug-Resistant Tuberculosis in Cape Town, South Africa: A Retrospective Longitudinal Cohort Study. Lancet Microbe 2023, 4, e972–e982. [Google Scholar] [CrossRef]
- World Health Organization. Global Tuberculosis Report 2023; World Health Organization: Geneva, Switzerland, 2023; ISBN 9789240083851. [Google Scholar]
- Zumla, A.; Rao, M.; Wallis, R.S.; Kaufmann, S.H.E.; Rustomjee, R.; Mwaba, P.; Vilaplana, C.; Yeboah-Manu, D.; Chakaya, J.; Ippolito, G.; et al. Host-Directed Therapies for Infectious Diseases: Current Status, Recent Progress, and Future Prospects. Lancet Infect. Dis. 2016, 16, e47–e63. [Google Scholar] [CrossRef]
- Casadevall, A.; Pirofski, L.A. Host-Pathogen Interactions: Basic Concepts of Microbial Commensalism, Colonization, Infection, and Disease. Infect. Immun. 2000, 68, 6511–6518. [Google Scholar] [CrossRef] [PubMed]
- GBD 2013 Mortality and Causes of Death Collaborators. Global, Regional, and National Age-Sex Specific All-Cause and Cause-Specific Mortality for 240 Causes of Death, 1990–2013: A Systematic Analysis for the Global Burden of Disease Study 2013. Lancet 2015, 385, 117–171. [Google Scholar] [CrossRef]
- Wallis, R.S.; O’Garra, A.; Sher, A.; Wack, A. Host-Directed Immunotherapy of Viral and Bacterial Infections: Past, Present and Future. Nat. Rev. Immunol. 2023, 23, 121–133. [Google Scholar] [CrossRef] [PubMed]
- Shapira, T.; Christofferson, M.; Av-Gay, Y. The Antimicrobial Activity of Innate Host-Directed Therapies: A Systematic Review. Int. J. Antimicrob. Agents 2024, 63, 107138. [Google Scholar] [CrossRef]
- Goletti, D.; Ong, C.W.M.; Friedland, J.S. Host-Directed Therapies: Old and New Approaches for the Treatment of Infections. Int. J. Infect. Dis. 2024, 146, 107130. [Google Scholar] [CrossRef]
- Liu, B.; Zhang, W.; Xia, B.; Jing, S.; Du, Y.; Zou, F.; Li, R.; Lu, L.; Chen, S.; Li, Y.; et al. Broadly Neutralizing Antibody-Derived CAR T Cells Reduce Viral Reservoir in Individuals Infected with HIV-1. J. Clin. Investig. 2021, 131, e150211. [Google Scholar] [CrossRef]
- Kamyshnyi, A.; Koval, H.; Kobevko, O.; Buchynskyi, M.; Oksenych, V.; Kainov, D.; Lyubomirskaya, K.; Kamyshna, I.; Potters, G.; Moshynets, O. Therapeutic Effectiveness of Interferon-A2b against COVID-19 with Community-Acquired Pneumonia: The Ukrainian Experience. Int. J. Mol. Sci. 2023, 24, 6887. [Google Scholar] [CrossRef]
- Muir, A.J.; Arora, S.; Everson, G.; Flisiak, R.; George, J.; Ghalib, R.; Gordon, S.C.; Gray, T.; Greenbloom, S.; Hassanein, T.; et al. A Randomized Phase 2b Study of Peginterferon Lambda-1a for the Treatment of Chronic HCV Infection. J. Hepatol. 2014, 61, 1238–1246. [Google Scholar] [CrossRef] [PubMed]
- Tam, A.R.; Zhang, R.R.; Lung, K.-C.; Liu, R.; Leung, K.-Y.; Liu, D.; Fan, Y.; Lu, L.; Lam, A.H.-Y.; Chung, T.W.-H.; et al. Early Treatment of High-Risk Hospitalized Coronavirus Disease 2019 (COVID-19) Patients with a Combination of Interferon Beta-1b and Remdesivir: A Phase 2 Open-Label Randomized Controlled Trial. Clin. Infect. Dis. 2023, 76, e216–e226. [Google Scholar] [CrossRef]
- Mapamba, D.A.; Sauli, E.; Mrema, L.; Lalashowi, J.; Magombola, D.; Buza, J.; Olomi, W.; Wallis, R.S.; Ntinginya, N.E. Impact of N-Acetyl Cysteine (NAC) on Tuberculosis (TB) Patients-A Systematic Review. Antioxidants 2022, 11, 2298. [Google Scholar] [CrossRef]
- Vita, S.; Rosati, S.; Ascoli Bartoli, T.; Beccacece, A.; D’Abramo, A.; Mariano, A.; Scorzolini, L.; Goletti, D.; Nicastri, E. Monoclonal Antibodies for Pre- and Postexposure Prophylaxis of COVID-19: Review of the Literature. Pathogens 2022, 11, 882. [Google Scholar] [CrossRef]
- Raghuveer, T.S.; Zackula, R. Nirsevimab for Prevention of RSV Hospitalizations in Infants. N. Engl. J. Med. 2024, 390, 1152. [Google Scholar] [CrossRef]
- Ferraccioli, G.; Gremese, E.; Goletti, D.; Petrone, L.; Cantini, F.; Ugel, S.; Canè, S.; Bronte, V. Immune-Guided Therapy of COVID-19. Cancer Immunol. Res. 2022, 10, 384–402. [Google Scholar] [CrossRef] [PubMed]
- Goulenok, T.; Gaudemer, A.; Rouzaud, D.; Chauveheid, M.-P.; Alexandra, J.-F.; Sacre, K.; Papo, T. Infliximab to Treat Severe Paradoxical Reaction in HIV-Negative Tuberculous Meningoencephalitis. Neurology 2022, 98, 118–119. [Google Scholar] [CrossRef]
- Barnard, C. Tuberculous Meningitis; Cortisone Treatment as an Adjunct to the Antibiotics; the Effect on the Clinical Features and the Cerebrospinal Fluid. S. Afr. Med. J. 1953, 27, 219–220. [Google Scholar] [PubMed]
- Chandra, H.; Rahman, A.; Yadav, P.; Maurya, G.; Kumar Shukla, S. Effect of Adjunct Vitamin D Treatment in Vitamin D Deficient Pulmonary Tuberculosis Patients: A Randomized, Double Blind, Active Controlled Clinical Trial. Indian. J. Tuberc. 2024, 71, 170–178. [Google Scholar] [CrossRef]
- Dutta, N.K.; Bruiners, N.; Pinn, M.L.; Zimmerman, M.D.; Prideaux, B.; Dartois, V.; Gennaro, M.L.; Karakousis, P.C. Statin Adjunctive Therapy Shortens the Duration of TB Treatment in Mice. J. Antimicrob. Chemother. 2016, 71, 1570–1577. [Google Scholar] [CrossRef]
- Mai, N.T.H.; Dobbs, N.; Phu, N.H.; Colas, R.A.; Thao, L.T.P.; Thuong, N.T.T.; Nghia, H.D.T.; Hanh, N.H.H.; Hang, N.T.; Heemskerk, A.D.; et al. A Randomised Double Blind Placebo Controlled Phase 2 Trial of Adjunctive Aspirin for Tuberculous Meningitis in HIV-Uninfected Adults. eLife 2018, 7, e33478. [Google Scholar] [CrossRef] [PubMed]
- Bourigault, M.-L.; Vacher, R.; Rose, S.; Olleros, M.L.; Janssens, J.-P.; Quesniaux, V.F.; Garcia, I. Tumor Necrosis Factor Neutralization Combined with Chemotherapy Enhances Mycobacterium tuberculosis Clearance and Reduces Lung Pathology. Am. J. Clin. Exp. Immunol. 2013, 2, 124–134. [Google Scholar]
- Wallis, R.S.; Kyambadde, P.; Johnson, J.L.; Horter, L.; Kittle, R.; Pohle, M.; Ducar, C.; Millard, M.; Mayanja-Kizza, H.; Whalen, C.; et al. A Study of the Safety, Immunology, Virology, and Microbiology of Adjunctive Etanercept in HIV-1-Associated Tuberculosis. AIDS 2004, 18, 257–264. [Google Scholar] [CrossRef]
- Wu, S.; Sun, J. Vitamin D, Vitamin D Receptor, and Macroautophagy in Inflammation and Infection. Discov. Med. 2011, 11, 325–335. [Google Scholar]
- Mily, A.; Rekha, R.S.; Kamal, S.M.M.; Akhtar, E.; Sarker, P.; Rahim, Z.; Gudmundsson, G.H.; Agerberth, B.; Raqib, R. Oral Intake of Phenylbutyrate with or without Vitamin D3 Upregulates the Cathelicidin LL-37 in Human Macrophages: A Dose Finding Study for Treatment of Tuberculosis. BMC Pulm. Med. 2013, 13, 23. [Google Scholar] [CrossRef] [PubMed]
- Mily, A.; Rekha, R.S.; Kamal, S.M.M.; Arifuzzaman, A.S.M.; Rahim, Z.; Khan, L.; Haq, M.A.; Zaman, K.; Bergman, P.; Brighenti, S.; et al. Significant Effects of Oral Phenylbutyrate and Vitamin D3 Adjunctive Therapy in Pulmonary Tuberculosis: A Randomized Controlled Trial. PLoS ONE 2015, 10, e0138340. [Google Scholar] [CrossRef] [PubMed]
- Wejse, C.; Gomes, V.F.; Rabna, P.; Gustafson, P.; Aaby, P.; Lisse, I.M.; Andersen, P.L.; Glerup, H.; Sodemann, M. Vitamin D as Supplementary Treatment for Tuberculosis: A Double-Blind, Randomized, Placebo-Controlled Trial. Am. J. Respir. Crit. Care Med. 2009, 179, 843–850. [Google Scholar] [CrossRef]
- Martineau, A.R.; Timms, P.M.; Bothamley, G.H.; Hanifa, Y.; Islam, K.; Claxton, A.P.; Packe, G.E.; Moore-Gillon, J.C.; Darmalingam, M.; Davidson, R.N.; et al. High-Dose Vitamin D(3) during Intensive-Phase Antimicrobial Treatment of Pulmonary Tuberculosis: A Double-Blind Randomised Controlled Trial. Lancet 2011, 377, 242–250. [Google Scholar] [CrossRef]
- Cubillos-Angulo, J.M.; Nogueira, B.M.F.; Arriaga, M.B.; Barreto-Duarte, B.; Araújo-Pereira, M.; Fernandes, C.D.; Vinhaes, C.L.; Villalva-Serra, K.; Nunes, V.M.; Miguez-Pinto, J.P.; et al. Host-Directed Therapies in Pulmonary Tuberculosis: Updates on Anti-Inflammatory Drugs. Front. Med. 2022, 9, 970408. [Google Scholar] [CrossRef]
- Marzo, E.; Vilaplana, C.; Tapia, G.; Diaz, J.; Garcia, V.; Cardona, P.-J. Damaging Role of Neutrophilic Infiltration in a Mouse Model of Progressive Tuberculosis. Tuberculosis 2014, 94, 55–64. [Google Scholar] [CrossRef]
- Byrne, S.T.; Denkin, S.M.; Zhang, Y. Aspirin and Ibuprofen Enhance Pyrazinamide Treatment of Murine Tuberculosis. J. Antimicrob. Chemother. 2007, 59, 313–316. [Google Scholar] [CrossRef] [PubMed]
- Vilaplana, C.; Marzo, E.; Tapia, G.; Diaz, J.; Garcia, V.; Cardona, P.-J. Ibuprofen Therapy Resulted in Significantly Decreased Tissue Bacillary Loads and Increased Survival in a New Murine Experimental Model of Active Tuberculosis. J. Infect. Dis. 2013, 208, 199–202. [Google Scholar] [CrossRef] [PubMed]
- Shim, D.; Kim, H.; Shin, S.J. Mycobacterium tuberculosis Infection-Driven Foamy Macrophages and Their Implications in Tuberculosis Control as Targets for Host-Directed Therapy. Front. Immunol. 2020, 11, 910. [Google Scholar] [CrossRef]
- Parihar, S.P.; Guler, R.; Khutlang, R.; Lang, D.M.; Hurdayal, R.; Mhlanga, M.M.; Suzuki, H.; Marais, A.D.; Brombacher, F. Statin Therapy Reduces the Mycobacterium tuberculosis Burden in Human Macrophages and in Mice by Enhancing Autophagy and Phagosome Maturation. J. Infect. Dis. 2014, 209, 754–763. [Google Scholar] [CrossRef]
- Skerry, C.; Pinn, M.L.; Bruiners, N.; Pine, R.; Gennaro, M.L.; Karakousis, P.C. Simvastatin Increases the in Vivo Activity of the First-Line Tuberculosis Regimen. J. Antimicrob. Chemother. 2014, 69, 2453–2457. [Google Scholar] [CrossRef]
- Wallis, R.S.; Hafner, R. Advancing Host-Directed Therapy for Tuberculosis. Nat. Rev. Immunol. 2015, 15, 255–263. [Google Scholar] [CrossRef]
- Kang, Y.A.; Choi, N.-K.; Seong, J.-M.; Heo, E.Y.; Koo, B.K.; Hwang, S.-S.; Park, B.-J.; Yim, J.-J.; Lee, C.-H. The Effects of Statin Use on the Development of Tuberculosis among Patients with Diabetes Mellitus. Int. J. Tuberc. Lung Dis. 2014, 18, 717–724. [Google Scholar] [CrossRef]
- Pandey, A.K.; Sassetti, C.M. Mycobacterial Persistence Requires the Utilization of Host Cholesterol. Proc. Natl. Acad. Sci. USA 2008, 105, 4376–4380. [Google Scholar] [CrossRef]
- Guerra-De-Blas, P.D.C.; Bobadilla-Del-Valle, M.; Sada-Ovalle, I.; Estrada-García, I.; Torres-González, P.; López-Saavedra, A.; Guzmán-Beltrán, S.; Ponce-de-León, A.; Sifuentes-Osornio, J. Simvastatin Enhances the Immune Response Against Mycobacterium tuberculosis. Front. Microbiol. 2019, 10, 2097. [Google Scholar] [CrossRef]
- Rodrigues Díez, R.; Rodrigues-Díez, R.; Lavoz, C.; Rayego-Mateos, S.; Civantos, E.; Rodríguez-Vita, J.; Mezzano, S.; Ortiz, A.; Egido, J.; Ruiz-Ortega, M. Statins Inhibit Angiotensin II/Smad Pathway and Related Vascular Fibrosis, by a TGF-β-Independent Process. PLoS ONE 2010, 5, e14145. [Google Scholar] [CrossRef]
- Ma, Y.-X.; Li, W.-H.; Xie, Q. Rosuvastatin Inhibits TGF-Beta1 Expression and Alleviates Myocardial Fibrosis in Diabetic Rats. Pharmazie 2013, 68, 355–358. [Google Scholar]
- Sojka, D.; Šnebergerová, P.; Robbertse, L. Protease Inhibition-An Established Strategy to Combat Infectious Diseases. Int. J. Mol. Sci. 2021, 22, 5762. [Google Scholar] [CrossRef]
- Drag, M.; Salvesen, G.S. Emerging Principles in Protease-Based Drug Discovery. Nat. Rev. Drug Discov. 2010, 9, 690–701. [Google Scholar] [CrossRef]
- Bond, J.S. Proteases: History, Discovery, and Roles in Health and Disease. J. Biol. Chem. 2019, 294, 1643–1651. [Google Scholar] [CrossRef] [PubMed]
- Rawlings, N.D.; Bateman, A. How to Use the MEROPS Database and Website to Help Understand Peptidase Specificity. Protein Sci. 2021, 30, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Barrett, A.J.; Rawling, N.D.; Woessner, J.F. Handbook of Proteolytic Enzymes. Volume 1 Aspartic and Metallo Peptidases Editor Biographies XXI, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2004. [Google Scholar]
- Puente, X.S.; Sánchez, L.M.; Overall, C.M.; López-Otín, C. Human and Mouse Proteases: A Comparative Genomic Approach. Nat. Rev. Genet. 2003, 4, 544–558. [Google Scholar] [CrossRef]
- Wang, L.; Main, K.; Wang, H.; Julien, O.; Dufour, A. Biochemical Tools for Tracking Proteolysis. J. Proteome Res. 2021, 20, 5264–5279. [Google Scholar] [CrossRef]
- Fortelny, N.; Cox, J.H.; Kappelhoff, R.; Starr, A.E.; Lange, P.F.; Pavlidis, P.; Overall, C.M. Network Analyses Reveal Pervasive Functional Regulation Between Proteases in the Human Protease Web. PLoS Biol. 2014, 12, e1001869. [Google Scholar] [CrossRef]
- Kappelhoff, R.; Puente, X.S.; Wilson, C.H.; Seth, A.; López-Otín, C.; Overall, C.M. Overview of Transcriptomic Analysis of All Human Proteases, Non-Proteolytic Homologs and Inhibitors: Organ, Tissue and Ovarian Cancer Cell Line Expression Profiling of the Human Protease Degradome by the CLIP-CHIPTM DNA Microarray. Biochim. Biophys. Acta BBA—Mol. Cell Res. 2017, 1864, 2210–2219. [Google Scholar] [CrossRef]
- Renslo, A.R.; McKerrow, J.H. Drug Discovery and Development for Neglected Parasitic Diseases. Nat. Chem. Biol. 2006, 2, 701–710. [Google Scholar] [CrossRef]
- McKerrow, J.H.; Caffrey, C.; Kelly, B.; Loke, P.; Sajid, M. Proteases in Parasitic Diseases. Annu. Rev. Pathol. 2006, 1, 497–536. [Google Scholar] [CrossRef] [PubMed]
- Kitchen, V.S.; Skinner, C.; Ariyoshi, K.; Lane, E.A.; Duncan, I.B.; Burckhardt, J.; Burger, H.U.; Bragman, K.; Pinching, A.J.; Weber, J.N. Safety and Activity of Saquinavir in HIV Infection. Lancet 1995, 345, 952–955. [Google Scholar] [CrossRef] [PubMed]
- Kosalaraksa, P.; Ananworanich, J.; Puthanakit, T.; Pinyakorn, S.; Lumbiganon, P.; Chuanjaroen, T.; Chobkarjing, U.; Phanuphak, P.; Pancharoen, C.; Bunupuradah, T.; et al. Long-Term Lopinavir/Ritonavir Monotherapy in HIV-Infected Children. Pediatr. Infect. Dis. J. 2013, 32, 350–353. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Osswald, H.L.; Prato, G. Recent Progress in the Development of HIV-1 Protease Inhibitors for the Treatment of HIV/AIDS. J. Med. Chem. 2016, 59, 5172–5208. [Google Scholar] [CrossRef] [PubMed]
- McKerrow, J.H. Update on Drug Development Targeting Parasite Cysteine Proteases. PLoS Negl. Trop. Dis. 2018, 12, e0005850. [Google Scholar] [CrossRef]
- Richardson, P.G. Update on Proteasome Inhibitors in Multiple Myeloma. Clin. Adv. Hematol. Oncol. 2014, 12, 179–181. [Google Scholar]
- Bibo-Verdugo, B.; Jiang, Z.; Caffrey, C.R.; O’Donoghue, A.J. Targeting Proteasomes in Infectious Organisms to Combat Disease. FEBS J. 2017, 284, 1503–1517. [Google Scholar] [CrossRef]
- Hilgenfeld, R. From SARS to MERS: Crystallographic Studies on Coronaviral Proteases Enable Antiviral Drug Design. FEBS J. 2014, 281, 4085–4096. [Google Scholar] [CrossRef]
- Rut, W.; Lv, Z.; Zmudzinski, M.; Patchett, S.; Nayak, D.; Snipas, S.J.; El Oualid, F.; Huang, T.T.; Bekes, M.; Drag, M.; et al. Activity Profiling and Crystal Structures of Inhibitor-Bound SARS-CoV-2 Papain-like Protease: A Framework for Anti-COVID-19 Drug Design. Sci. Adv. 2020, 6, eabd4596. [Google Scholar] [CrossRef]
- Freitas, B.T.; Durie, I.A.; Murray, J.; Longo, J.E.; Miller, H.C.; Crich, D.; Hogan, R.J.; Tripp, R.A.; Pegan, S.D. Characterization and Noncovalent Inhibition of the Deubiquitinase and DeISGylase Activity of SARS-CoV-2 Papain-Like Protease. ACS Infect. Dis. 2020, 6, 2099–2109. [Google Scholar] [CrossRef]
- Shin, D.; Mukherjee, R.; Grewe, D.; Bojkova, D.; Baek, K.; Bhattacharya, A.; Schulz, L.; Widera, M.; Mehdipour, A.R.; Tascher, G.; et al. Papain-like Protease Regulates SARS-CoV-2 Viral Spread and Innate Immunity. Nature 2020, 587, 657–662. [Google Scholar] [CrossRef] [PubMed]
- Almeleebia, T.M.; Shahrani, M.A.; Alshahrani, M.Y.; Ahmad, I.; Alkahtani, A.M.; Alam, M.J.; Kausar, M.A.; Saeed, A.; Saeed, M.; Iram, S. Identification of New Mycobacterium tuberculosis Proteasome Inhibitors Using a Knowledge-Based Computational Screening Approach. Molecules 2021, 26, 2326. [Google Scholar] [CrossRef]
- Tyagi, R.; Srivastava, M.; Jain, P.; Pandey, R.P.; Asthana, S.; Kumar, D.; Raj, V.S. Development of Potential Proteasome Inhibitors against Mycobacterium tuberculosis. J. Biomol. Struct. Dyn. 2022, 40, 2189–2203. [Google Scholar] [CrossRef] [PubMed]
- Mehra, R.; Chib, R.; Munagala, G.; Yempalla, K.R.; Khan, I.A.; Singh, P.P.; Khan, F.G.; Nargotra, A. Discovery of New Mycobacterium tuberculosis Proteasome Inhibitors Using a Knowledge-Based Computational Screening Approach. Mol. Divers. 2015, 19, 1003–1019. [Google Scholar] [CrossRef]
- Zhou, B.; Gao, Y.; Zhao, H.; Liu, B.; Zhang, H.; Fang, C.; Yuan, H.; Wang, J.; Li, Z.; Zhao, Y.; et al. Structural Insights into Bortezomib-Induced Activation of the Caseinolytic Chaperone-Protease System in Mycobacterium tuberculosis. Nat. Commun. 2025, 16, 3466. [Google Scholar] [CrossRef]
- Benaroudj, N.; Raynal, B.; Miot, M.; Ortiz-Lombardia, M. Assembly and Proteolytic Processing of Mycobacterial ClpP1 and ClpP2. BMC Biochem. 2011, 12, 61. [Google Scholar] [CrossRef]
- Raju, R.M.; Unnikrishnan, M.; Rubin, D.H.F.; Krishnamoorthy, V.; Kandror, O.; Akopian, T.N.; Goldberg, A.L.; Rubin, E.J. Mycobacterium tuberculosis ClpP1 and ClpP2 Function Together in Protein Degradation and Are Required for Viability in Vitro and During Infection. PLoS Pathog. 2012, 8, e1002511. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhao, N.; Xu, X.; Zhou, Y.; Luo, B.; Zhang, J.; Sui, J.; Huang, J.; Qiu, Z.; Zhang, X.; et al. Discovery and Mechanistic Study of Novel Mycobacterium tuberculosis ClpP1P2 Inhibitors. J. Med. Chem. 2023, 66, 16597–16614. [Google Scholar] [CrossRef]
- Moreira, W.; Santhanakrishnan, S.; Ngan, G.J.Y.; Low, C.B.; Sangthongpitag, K.; Poulsen, A.; Dymock, B.W.; Dick, T. Towards Selective Mycobacterial ClpP1P2 Inhibitors with Reduced Activity against the Human Proteasome. Antimicrob. Agents Chemother. 2017, 61, e02307-16. [Google Scholar] [CrossRef]
- Moreira, W.; Ngan, G.J.Y.; Low, J.L.; Poulsen, A.; Chia, B.C.S.; Ang, M.J.Y.; Yap, A.; Fulwood, J.; Lakshmanan, U.; Lim, J.; et al. Target Mechanism-Based Whole-Cell Screening Identifies Bortezomib as an Inhibitor of Caseinolytic Protease in Mycobacteria. mBio 2015, 6. [Google Scholar] [CrossRef]
- Moreira, W.; Santhanakrishnan, S.; Dymock, B.W.; Dick, T. Bortezomib Warhead-Switch Confers Dual Activity against Mycobacterial Caseinolytic Protease and Proteasome and Selectivity against Human Proteasome. Front. Microbiol. 2017, 8, 746. [Google Scholar] [CrossRef] [PubMed]
- Lupoli, T.J.; Vaubourgeix, J.; Burns-Huang, K.; Gold, B. Targeting the Proteostasis Network for Mycobacterial Drug Discovery. ACS Infect. Dis. 2018, 4, 478–498. [Google Scholar] [CrossRef]
- Pires, D.; Marques, J.; Pombo, J.P.; Carmo, N.; Bettencourt, P.; Neyrolles, O.; Lugo-Villarino, G.; Anes, E. Role of Cathepsins in Mycobacterium tuberculosis Survival in Human Macrophages. Sci. Rep. 2016, 6, 32247. [Google Scholar] [CrossRef]
- Pires, D.; Bernard, E.M.; Pombo, J.P.; Carmo, N.; Fialho, C.; Gutierrez, M.G.; Bettencourt, P.; Anes, E. Mycobacterium tuberculosis Modulates MiR-106b-5p to Control Cathepsin S Expression Resulting in Higher Pathogen Survival and Poor T-Cell Activation. Front. Immunol. 2017, 8, 1819. [Google Scholar] [CrossRef]
- Nsanzabana, C.; Rosenthal, P.J. In Vitro Activity of Antiretroviral Drugs against Plasmodium Falciparum. Antimicrob. Agents Chemother. 2011, 55, 5073–5077. [Google Scholar] [CrossRef]
- Castilho, V.V.S.; Gonçalves, K.C.S.; Rebello, K.M.; Baptista, L.P.R.; Sangenito, L.S.; Santos, H.L.C.; Branquinha, M.H.; Santos, A.L.S.; Menna-Barreto, R.F.S.; Guimarães, A.C.; et al. Docking Simulation between HIV Peptidase Inhibitors and Trypanosoma Cruzi Aspartyl Peptidase. BMC Res. Notes 2018, 11, 825. [Google Scholar] [CrossRef] [PubMed]
- Savarino, A. Expanding the Frontiers of Existing Antiviral Drugs: Possible Effects of HIV-1 Protease Inhibitors against SARS and Avian Influenza. J. Clin. Virol. 2005, 34, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.; Valente, S.; Calado, M.; Mandal, M.; Azevedo-Pereira, J.M.; Anes, E. Repurposing Saquinavir for Host-Directed Therapy to Control Mycobacterium tuberculosis Infection. Front. Immunol. 2021, 12, 647728. [Google Scholar] [CrossRef]
- Conus, S.; Simon, H.-U. Cathepsins and Their Involvement in Immune Responses. Swiss Med. Wkly. 2010, 140, w13042. [Google Scholar] [CrossRef]
- Soualhine, H.; Deghmane, A.-E.; Sun, J.; Mak, K.; Talal, A.; Av-Gay, Y.; Hmama, Z. Mycobacterium Bovis Bacillus Calmette-Guérin Secreting Active Cathepsin S Stimulates Expression of Mature MHC Class II Molecules and Antigen Presentation in Human Macrophages. J. Immunol. 2007, 179, 5137–5145. [Google Scholar] [CrossRef]
- Beers, C.; Burich, A.; Kleijmeer, M.J.; Griffith, J.M.; Wong, P.; Rudensky, A.Y. Cathepsin S Controls MHC Class II-Mediated Antigen Presentation by Epithelial Cells in Vivo. J. Immunol. 2005, 174, 1205–1212. [Google Scholar] [CrossRef]
- Chen, K.-L.; Chang, W.-S.W.; Cheung, C.H.A.; Lin, C.-C.; Huang, C.-C.; Yang, Y.-N.; Kuo, C.-P.; Kuo, C.-C.; Chang, Y.-H.; Liu, K.-J.; et al. Targeting Cathepsin S Induces Tumor Cell Autophagy via the EGFR-ERK Signaling Pathway. Cancer Lett. 2012, 317, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Espert, L.; Beaumelle, B.; Vergne, I. Autophagy in Mycobacterium tuberculosis and HIV Infections. Front. Cell Infect. Microbiol. 2015, 5, 49. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.; Park, H.; Heisler, J.; Maculins, T.; Roose-Girma, M.; Xu, M.; Mckenzie, B.; van Lookeren Campagne, M.; Newton, K.; Murthy, A. Autophagy Regulates Inflammatory Programmed Cell Death via Turnover of RHIM-Domain Proteins. eLife 2019, 8, e44452. [Google Scholar] [CrossRef]
- Pires, D.; Mandal, M.; Pinho, J.; Catalão, M.J.; Almeida, A.J.; Azevedo-Pereira, J.M.; Gaspar, M.M.; Anes, E. Liposomal Delivery of Saquinavir to Macrophages Overcomes Cathepsin Blockade by Mycobacterium tuberculosis and Helps Control the Phagosomal Replicative Niches. Int. J. Mol. Sci. 2023, 24, 1142. [Google Scholar] [CrossRef]
- Cooper, C.L.; van Heeswijk, R.P.G.; Gallicano, K.; Cameron, D.W. A Review of Low-Dose Ritonavir in Protease Inhibitor Combination Therapy. Clin. Infect. Dis. 2003, 36, 1585–1592. [Google Scholar] [CrossRef]
- Ribera, E.; Azuaje, C.; Lopez, R.M.; Domingo, P.; Curran, A.; Feijoo, M.; Pou, L.; Sánchez, P.; Sambeat, M.A.; Colomer, J.; et al. Pharmacokinetic Interaction between Rifampicin and the Once-Daily Combination of Saquinavir and Low-Dose Ritonavir in HIV-Infected Patients with Tuberculosis. J. Antimicrob. Chemother. 2007, 59, 690–697. [Google Scholar] [CrossRef]
- Turk, V.; Stoka, V.; Vasiljeva, O.; Renko, M.; Sun, T.; Turk, B.; Turk, D. Cysteine Cathepsins: From Structure, Function and Regulation to New Frontiers. Biochim. Biophys. Acta 2012, 1824, 68–88. [Google Scholar] [CrossRef]
- Russell, D.G.; Vanderven, B.C.; Glennie, S.; Mwandumba, H.; Heyderman, R.S. The Macrophage Marches on Its Phagosome: Dynamic Assays of Phagosome Function. Nat. Rev. Immunol. 2009, 9, 594–600. [Google Scholar] [CrossRef]
- Russell, D.G. New Ways to Arrest Phagosome Maturation. Nat. Cell Biol. 2007, 9, 357–359. [Google Scholar] [CrossRef]
- Welin, A.; Raffetseder, J.; Eklund, D.; Stendahl, O.; Lerm, M. Importance of Phagosomal Functionality for Growth Restriction of Mycobacterium tuberculosis in Primary Human Macrophages. J. Innate Immun. 2011, 3, 508–518. [Google Scholar] [CrossRef] [PubMed]
- Magister, S.; Kos, J. Cystatins in Immune System. J. Cancer 2013, 4, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Ochieng, J.; Chaudhuri, G. Cystatin Superfamily. J. Health Care Poor Underserved 2010, 21, 51–70. [Google Scholar] [CrossRef]
- Lautwein, A.; Burster, T.; Lennon-Duménil, A.-M.; Overkleeft, H.S.; Weber, E.; Kalbacher, H.; Driessen, C. Inflammatory Stimuli Recruit Cathepsin Activity to Late Endosomal Compartments in Human Dendritic Cells. Eur. J. Immunol. 2002, 32, 3348–3357. [Google Scholar] [CrossRef]
- Colbert, J.D.; Matthews, S.P.; Kos, J.; Watts, C. Internalization of Exogenous Cystatin F Supresses Cysteine Proteases and Induces the Accumulation of Single-Chain Cathepsin L by Multiple Mechanisms. J. Biol. Chem. 2011, 286, 42082–42090. [Google Scholar] [CrossRef]
- Ogrinc, T.; Dolenc, I.; Ritonja, A.; Turk, V. Purification of the Complex of Cathepsin L and the MHC Class II-Associated Invariant Chain Fragment from Human Kidney. FEBS Lett. 1993, 336, 555–559. [Google Scholar] [CrossRef]
- Anes, E.; Azevedo-Pereira, J.M.; Pires, D. Cathepsins and Their Endogenous Inhibitors in Host Defense During Mycobacterium tuberculosis and HIV Infection. Front. Immunol. 2021, 12, 726984. [Google Scholar] [CrossRef]
- Pires, D.; Calado, M.; Velez, T.; Mandal, M.; Catalão, M.J.; Neyrolles, O.; Lugo-Villarino, G.; Vérollet, C.; Azevedo-Pereira, J.M.; Anes, E. Modulation of Cystatin C in Human Macrophages Improves Anti-Mycobacterial Immune Responses to Mycobacterium tuberculosis Infection and Coinfection with HIV. Front. Immunol. 2021, 12, 742822. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.; Mandal, M.; Matos, A.I.; Peres, C.; Catalão, M.J.; Azevedo-Pereira, J.M.; Satchi-Fainaro, R.; Florindo, H.F.; Anes, E. Development of Chitosan Particles Loaded with SiRNA for Cystatin C to Control Intracellular Drug-Resistant Mycobacterium tuberculosis. Antibiotics 2023, 12, 729. [Google Scholar] [CrossRef]
- Mandal, M.; Pires, D.; Catalão, M.J.; Azevedo-Pereira, J.M.; Anes, E. Modulation of Cystatin F in Human Macrophages Impacts Cathepsin-Driven Killing of Multidrug-Resistant Mycobacterium tuberculosis. Microorganisms 2023, 11, 1861. [Google Scholar] [CrossRef]
- Ni, J.; Fernandez, M.A.; Danielsson, L.; Chillakuru, R.A.; Zhang, J.; Grubb, A.; Su, J.; Gentz, R.; Abrahamson, M. Cystatin F Is a Glycosylated Human Low Molecular Weight Cysteine Proteinase Inhibitor. J. Biol. Chem. 1998, 273, 24797–24804. [Google Scholar] [CrossRef]
- Langerholc, T.; Zavasnik-Bergant, V.; Turk, B.; Turk, V.; Abrahamson, M.; Kos, J. Inhibitory Properties of Cystatin F and Its Localization in U937 Promonocyte Cells. FEBS J. 2005, 272, 1535–1545. [Google Scholar] [CrossRef] [PubMed]
- Cohen, L.A.; Light, R.W. Tuberculous Pleural Effusion. Turk. Thorac. J. 2015, 16, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Werle, B.; Sauckel, K.; Nathanson, C.-M.; Bjarnadottir, M.; Spiess, E.; Ebert, W.; Abrahamson, M. Cystatins C, E/M and F in Human Pleural Fluids of Patients with Neoplastic and Inflammatory Lung Disorders. Biol. Chem. 2003, 384, 281–287. [Google Scholar] [CrossRef]
- Mandal, M.; Pires, D.; Calado, M.; Azevedo-Pereira, J.M.; Anes, E. Cystatin F Depletion in Mycobacterium tuberculosis-Infected Macrophages Improves Cathepsin C/Granzyme B-Driven Cytotoxic Effects on HIV-Infected Cells during Coinfection. Int. J. Mol. Sci. 2024, 25, 8141. [Google Scholar] [CrossRef]
- Rohlwink, U.K.; Walker, N.F.; Ordonez, A.A.; Li, Y.J.; Tucker, E.W.; Elkington, P.T.; Wilkinson, R.J.; Wilkinson, K.A. Matrix Metalloproteinases in Pulmonary and Central Nervous System Tuberculosis—A Review. Int. J. Mol. Sci. 2019, 20, 1350. [Google Scholar] [CrossRef]
- Tian, N.; Chu, H.; Li, Q.; Sun, H.; Zhang, J.; Chu, N.; Sun, Z. Host-Directed Therapy for Tuberculosis. Eur. J. Med. Res. 2025, 30, 267. [Google Scholar] [CrossRef]
- Elkington, P.; Shiomi, T.; Breen, R.; Nuttall, R.K.; Ugarte-Gil, C.A.; Walker, N.F.; Saraiva, L.; Pedersen, B.; Mauri, F.; Lipman, M.; et al. MMP-1 Drives Immunopathology in Human Tuberculosis and Transgenic Mice. J. Clin. Investig. 2011, 121, 1827–1833. [Google Scholar] [CrossRef]
- Walker, N.F.; Clark, S.O.; Oni, T.; Andreu, N.; Tezera, L.; Singh, S.; Saraiva, L.; Pedersen, B.; Kelly, D.L.; Tree, J.A.; et al. Doxycycline and HIV Infection Suppress Tuberculosis-Induced Matrix Metalloproteinases. Am. J. Respir. Crit. Care Med. 2012, 185, 989–997. [Google Scholar] [CrossRef]
- Walker, N.F.; Wilkinson, K.A.; Meintjes, G.; Tezera, L.B.; Goliath, R.; Peyper, J.M.; Tadokera, R.; Opondo, C.; Coussens, A.K.; Wilkinson, R.J.; et al. Matrix Degradation in Human Immunodeficiency Virus Type 1–Associated Tuberculosis and Tuberculosis Immune Reconstitution Inflammatory Syndrome: A Prospective Observational Study. Clin. Infect. Dis. 2017, 65, 121–132. [Google Scholar] [CrossRef]
- Miow, Q.H.; Vallejo, A.F.; Wang, Y.; Hong, J.M.; Bai, C.; Teo, F.S.W.; Wang, A.D.Y.; Loh, H.R.; Tan, T.Z.; Ding, Y.; et al. Doxycycline Host-Directed Therapy in Human Pulmonary Tuberculosis. J. Clin. Investig. 2021, 131, e141895. [Google Scholar] [CrossRef]
- Aaron, L.; Saadoun, D.; Calatroni, I.; Launay, O.; Mémain, N.; Vincent, V.; Marchal, G.; Dupont, B.; Bouchaud, O.; Valeyre, D.; et al. Tuberculosis in HIV-Infected Patients: A Comprehensive Review. Clin. Microbiol. Infect. 2004, 10, 388–398. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Wang, L.; Zimmerman, M.D.; Chen, K.-Y.; Huang, L.; Fu, D.-J.; Kaya, F.; Rakhilin, N.; Nazarova, E.V.; Bu, P.; et al. Matrix Metalloproteinase Inhibitors Enhance the Efficacy of Frontline Drugs against Mycobacterium tuberculosis. PLoS Pathog. 2018, 14, e1006974. [Google Scholar] [CrossRef]
- Ordonez, A.A.; Pokkali, S.; Sanchez-Bautista, J.; Klunk, M.H.; Urbanowski, M.E.; Kübler, A.; Bishai, W.R.; Elkington, P.T.; Jain, S.K. Matrix Metalloproteinase Inhibition in a Murine Model of Cavitary Tuberculosis Paradoxically Worsens Pathology. J. Infect. Dis. 2019, 219, 633–636. [Google Scholar] [CrossRef] [PubMed]
- Urbanowski, M.E.; Ihms, E.A.; Bigelow, K.; Kübler, A.; Elkington, P.T.; Bishai, W.R. Repetitive Aerosol Exposure Promotes Cavitary Tuberculosis and Enables Screening for Targeted Inhibitors of Extensive Lung Destruction. J. Infect. Dis. 2018, 218, 53–63. [Google Scholar] [CrossRef]
- Ordonez, A.A.; Pokkali, S.; Kim, S.; Carr, B.; Klunk, M.H.; Tong, L.; Saini, V.; Chang, Y.S.; McKevitt, M.; Smith, V.; et al. Adjunct Antibody Administration with Standard Treatment Reduces Relapse Rates in a Murine Tuberculosis Model of Necrotic Granulomas. PLoS ONE 2018, 13, e0197474. [Google Scholar] [CrossRef]
- Majeed, S.; Radotra, B.D.; Sharma, S. Adjunctive Role of MMP-9 Inhibition along with Conventional Anti-tubercular Drugs against Experimental Tuberculous Meningitis. Int. J. Exp. Pathol. 2016, 97, 230–237. [Google Scholar] [CrossRef]
- Di Caprio, R.; Lembo, S.; Di Costanzo, L.; Balato, A.; Monfrecola, G. Anti-Inflammatory Properties of Low and High Doxycycline Doses: An In Vitro Study. Mediat. Inflamm. 2015, 2015, 329418. [Google Scholar] [CrossRef]
- Navarro-Triviño, F.; Pérez-López, I.; Ruíz-Villaverde, R. Doxycycline, an Antibiotic or an Anti-Inflammatory Agent? The Most Common Uses in Dermatology. Actas Dermo-Sifiliográficas 2020, 111, 561–566. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mandal, M.; Pires, D.; Azevedo-Pereira, J.M.; Anes, E. Host-Directed Therapies Based on Protease Inhibitors to Control Mycobacterium tuberculosis and HIV Coinfection. Microorganisms 2025, 13, 1040. https://doi.org/10.3390/microorganisms13051040
Mandal M, Pires D, Azevedo-Pereira JM, Anes E. Host-Directed Therapies Based on Protease Inhibitors to Control Mycobacterium tuberculosis and HIV Coinfection. Microorganisms. 2025; 13(5):1040. https://doi.org/10.3390/microorganisms13051040
Chicago/Turabian StyleMandal, Manoj, David Pires, José Miguel Azevedo-Pereira, and Elsa Anes. 2025. "Host-Directed Therapies Based on Protease Inhibitors to Control Mycobacterium tuberculosis and HIV Coinfection" Microorganisms 13, no. 5: 1040. https://doi.org/10.3390/microorganisms13051040
APA StyleMandal, M., Pires, D., Azevedo-Pereira, J. M., & Anes, E. (2025). Host-Directed Therapies Based on Protease Inhibitors to Control Mycobacterium tuberculosis and HIV Coinfection. Microorganisms, 13(5), 1040. https://doi.org/10.3390/microorganisms13051040