
Comparison of Two DNA Labeling Dyes Commonly Used to Detect Metabolically Active Bacteria

 , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Types and Sample Collection
2.2. BrdU and PMA Treatment of Natural Samples
2.2.1. BrdU Treatment
2.2.2. Water Sample and Produce Wash Filtration
2.2.3. PMA Treatment
2.3. BrdU and PMA Treatment of Manufactured Samples
2.4. Immunocapturing of BrdU Treated Samples
2.5. DNA Extraction, 16S rRNA Gene Amplification, and Sequencing
2.6. 16S rRNA Gene Sequencing Analysis
3. Results
3.1. Sequencing Dataset
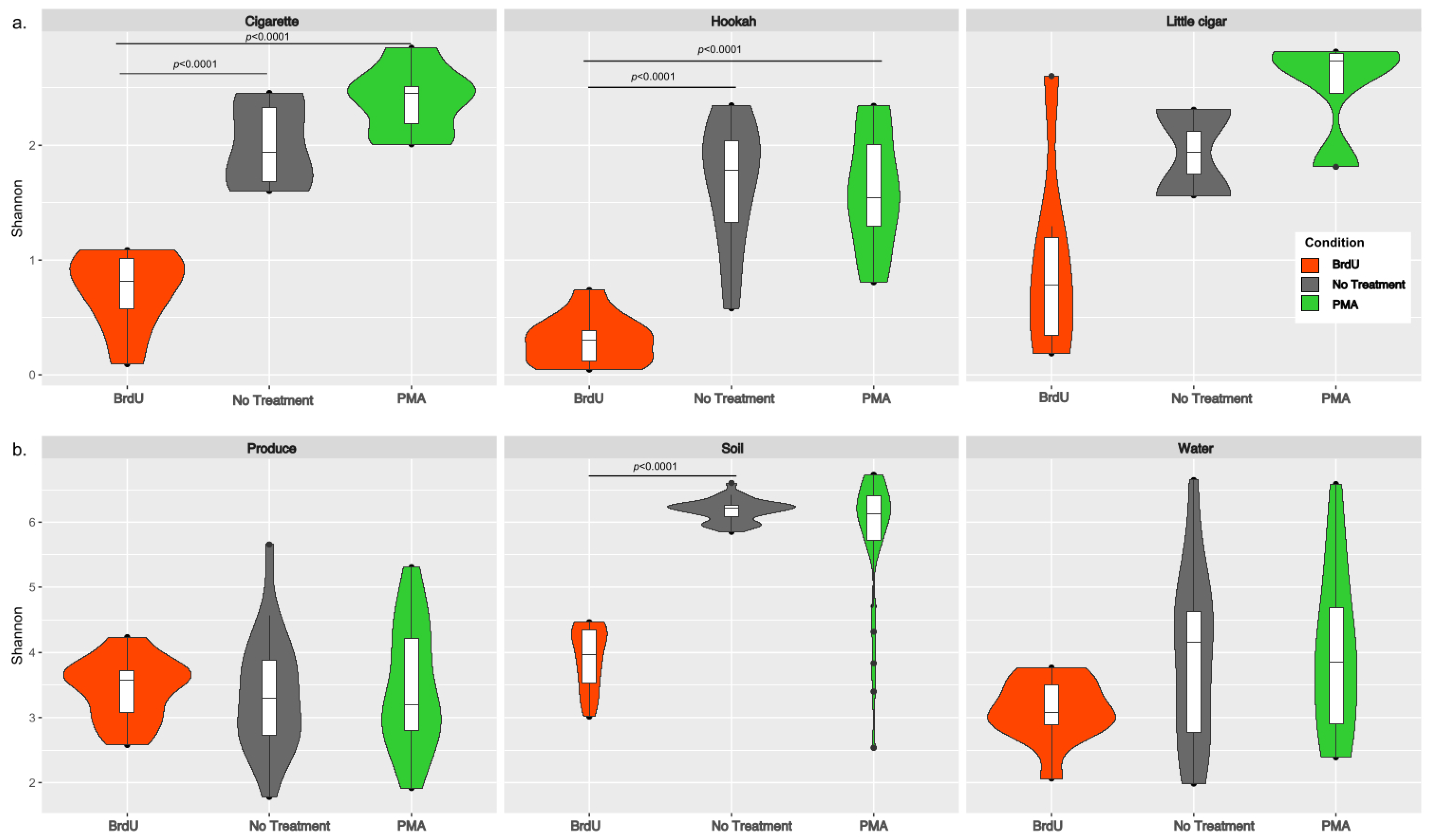
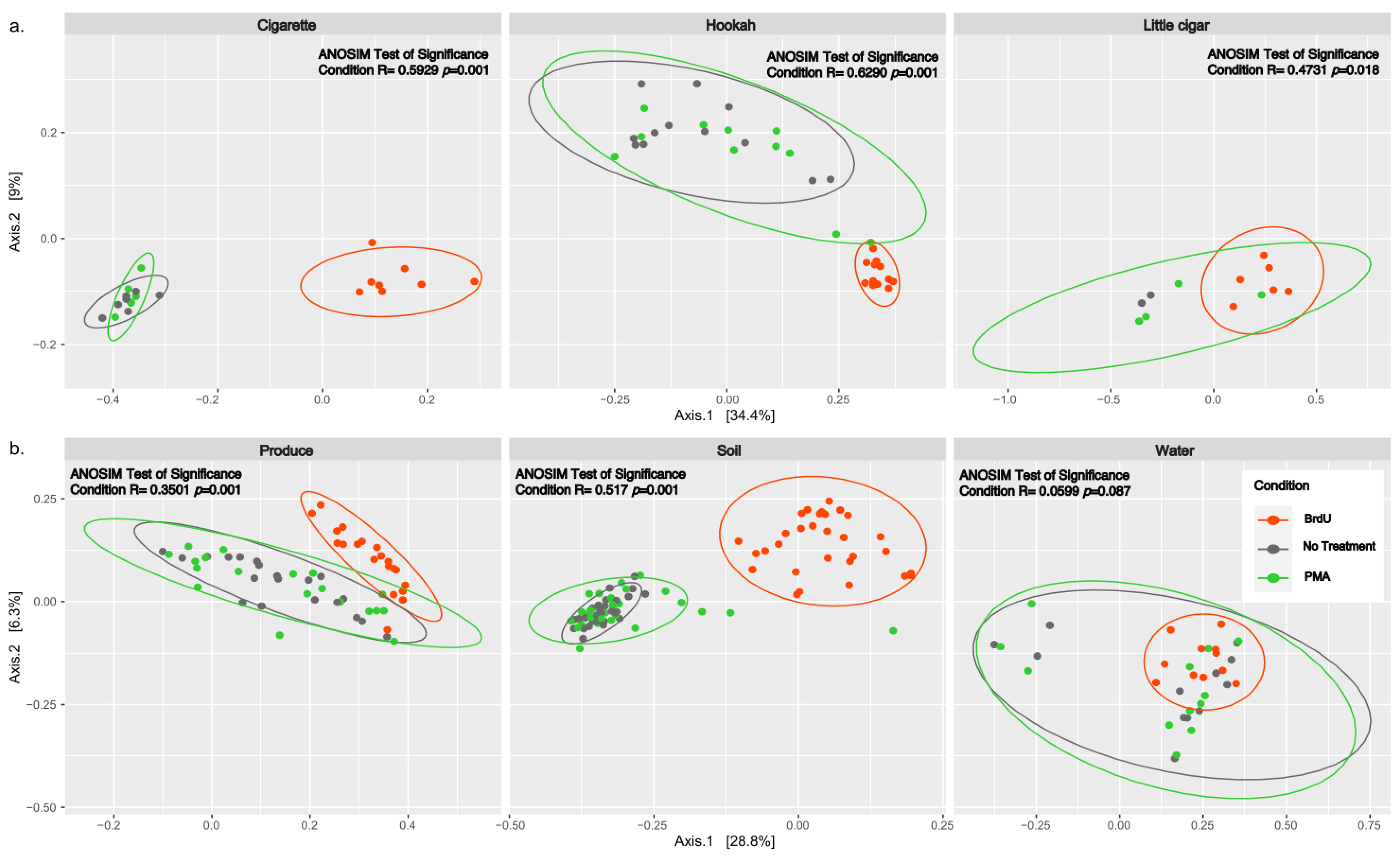
3.2. Diversity Measures Between Sample Types and Treatments
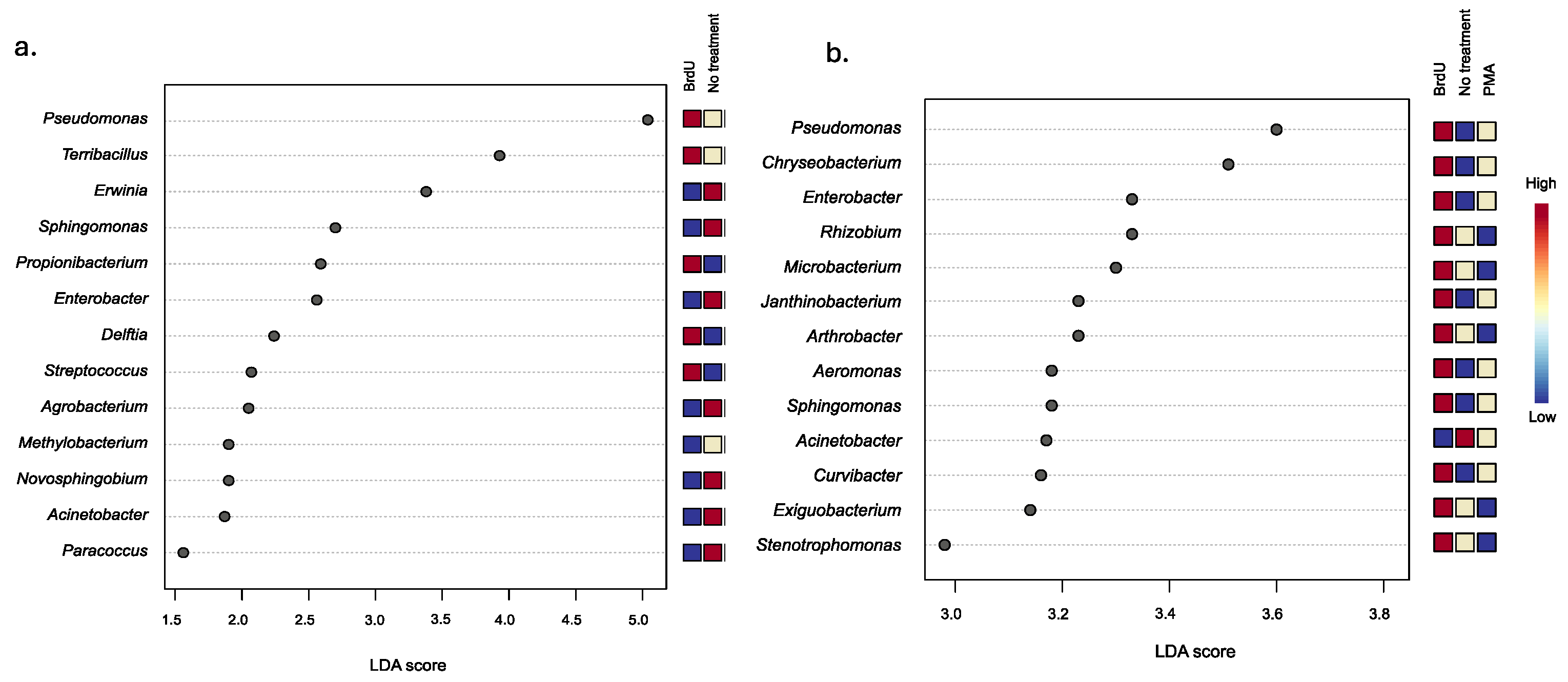
3.3. Bacterial Taxonomic Variations Between Treatments and Across Sample Types
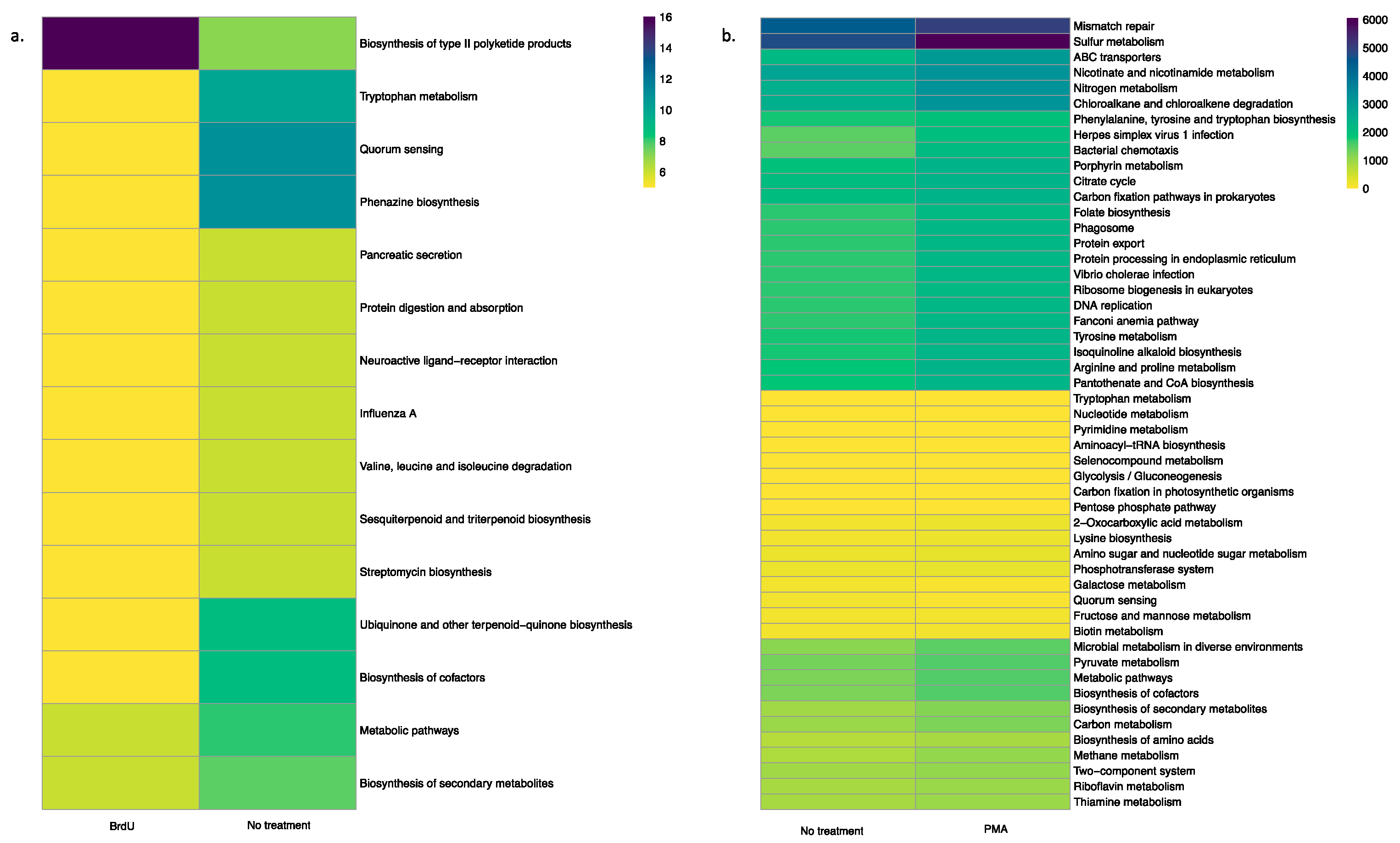
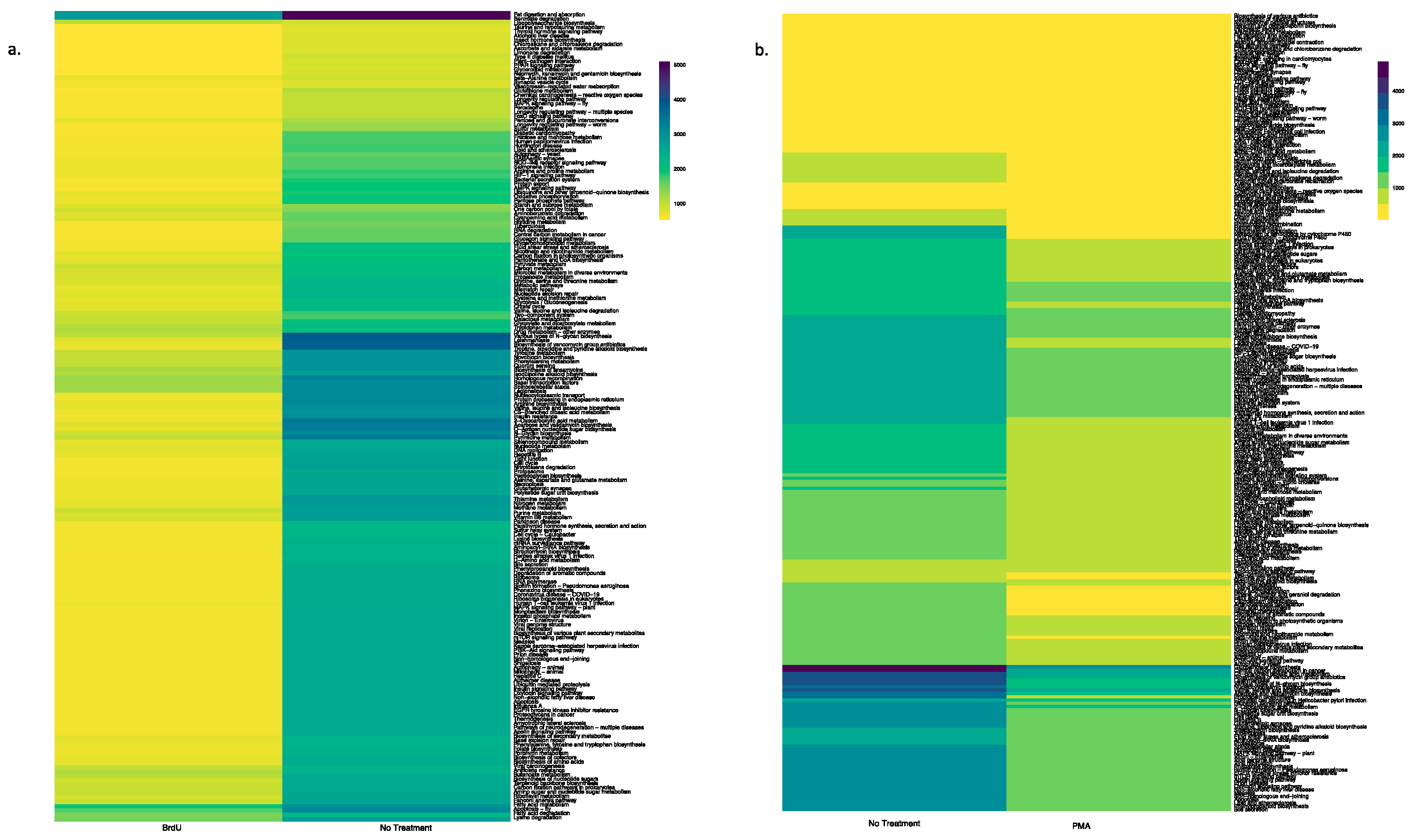
3.4. Predictive Functional Profile of Bacterial Communities Between Treatments
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rappé, M.S.; Giovannoni, S.J. The Uncultured Microbial Majority. Annu. Rev. Microbiol. 2003, 57, 369–394. [Google Scholar] [CrossRef] [PubMed]
- Franco-Duarte, R.; Černáková, L.; Kadam, S.; Kaushik, K.S.; Salehi, B.; Bevilacqua, A.; Corbo, M.R.; Antolak, H.; Dybka-Stępień, K.; Leszczewicz, M.; et al. Advances in Chemical and Biological Methods to Identify Microorganisms—From Past to Present. Microorganisms 2019, 7, 130. [Google Scholar] [CrossRef]
- Xu, Y.-G.; Liu, Z.-M.; Zhang, B.-Q.; Qu, M.; Mo, C.-S.; Luo, J.; Li, S.-L. Development of a Novel Target-Enriched Multiplex PCR (Tem-PCR) Assay for Simultaneous Detection of Five Foodborne Pathogens. Food Control 2016, 64, 54–59. [Google Scholar] [CrossRef]
- Schuster, S.C. Next-Generation Sequencing Transforms Today’s Biology. Nat. Methods 2008, 5, 16–18. [Google Scholar] [CrossRef] [PubMed]
- Shokralla, S.; Spall, J.L.; Gibson, J.F.; Hajibabaei, M. Next-Generation Sequencing Technologies for Environmental DNA Research. Mol. Ecol. 2012, 21, 1794–1805. [Google Scholar] [CrossRef]
- Tan, B.; Ng, C.M.; Nshimyimana, J.P.; Loh, L.-L.; Gin, K.Y.-H.; Thompson, J.R. Next-Generation Sequencing (NGS) for Assessment of Microbial Water Quality: Current Progress, Challenges, and Future Opportunities. Front. Microbiol. 2015, 6. [Google Scholar] [CrossRef]
- Li, R.; Tun, H.M.; Jahan, M.; Zhang, Z.; Kumar, A.; Dilantha Fernando, W.G.; Farenhorst, A.; Khafipour, E. Comparison of DNA-, PMA-, and RNA-Based 16S rRNA Illumina Sequencing for Detection of Live Bacteria in Water. Sci. Rep. 2017, 7, 5752. [Google Scholar] [CrossRef]
- Carini, P.; Marsden, P.J.; Leff, J.W.; Morgan, E.E.; Strickland, M.S.; Fierer, N. Relic DNA Is Abundant in Soil and Obscures Estimates of Soil Microbial Diversity. Nat. Microbiol. 2016, 2, 16242. [Google Scholar] [CrossRef]
- Krohn, C.; Jansriphibul, K.; Dias, D.A.; Rees, C.A.; Akker, B.V.D.; Boer, J.C.; Plebanski, M.; Surapaneni, A.; O’Carroll, D.; Richard, S.; et al. Dead in the Water—Role of Relic DNA and Primer Choice for Targeted Sequencing Surveys of Anaerobic Sewage Sludge Intended for Biological Monitoring. Water Res. 2024, 253, 121354. [Google Scholar] [CrossRef]
- Chattopadhyay, S.; Ramachandran, P.; Malayil, L.; Mongodin, E.F.; Sapkota, A.R. Conventional Tobacco Products Harbor Unique And Heterogenous Microbiomes. Environ. Res. 2022, 220, 115205. [Google Scholar] [CrossRef]
- Lennon, J.T.; Muscarella, M.E.; Placella, S.A.; Lehmkuhl, B.K. How, When, and Where Relic DNA Affects Microbial Diversity. mBio 2018, 9, e00637-18. [Google Scholar] [CrossRef] [PubMed]
- Hice, S.A.; Santoscoy, M.C.; Soupir, M.L.; Cademartiri, R. Distinguishing between Metabolically Active and Dormant Bacteria on Paper. Appl. Microbiol. Biotechnol. 2018, 102, 367–375. [Google Scholar] [CrossRef] [PubMed]
- Steele, M.; Odumeru, J. Irrigation Water as Source of Foodborne Pathogens on Fruit and Vegetables. J. Food Prot. 2004, 67, 2839–2849. [Google Scholar] [CrossRef] [PubMed]
- Malayil, L.; Chattopadhyay, S.; Bui, A.; Panse, M.; Cagle, R.; Mongodin, E.F.; Sapkota, A.R. Viable Bacteria Abundant in Cigarettes Are Aerosolized in Mainstream Smoke. Environ. Res. 2022, 212, 113462. [Google Scholar] [CrossRef]
- Urbach, E.; Vergin, K.L.; Giovannoni, S.J. Immunochemical Detection and Isolation of DNA from Metabolically Active Bacteria. Appl. Environ. Microbiol. 1999, 65, 1207–1213. [Google Scholar] [CrossRef]
- Nocker, A.; Cheung, C.-Y.; Camper, A.K. Comparison of Propidium Monoazide with Ethidium Monoazide for Differentiation of Live vs. Dead Bacteria by Selective Removal of DNA from Dead Cells. J. Microbiol. Methods 2006, 67, 310–320. [Google Scholar] [CrossRef]
- Galand, P.; Alonso-Saez, L.; Bertilsson, S.; Lovejoy, C.; Casamayor, E. Contrasting Activity Patterns Determined by BrdU Incorporation in Bacterial Ribotypes from the Arctic Ocean in Winter. Front. Microbiol. 2013, 4, 118. [Google Scholar] [CrossRef]
- Heise, J.; Nega, M.; Alawi, M.; Wagner, D. Propidium Monoazide Treatment to Distinguish between Live and Dead Methanogens in Pure Cultures and Environmental Samples. J. Microbiol. Methods 2016, 121, 11–23. [Google Scholar] [CrossRef]
- Brauge, T.; Midelet-Bourdin, G.; Soumet, C. Viability Detection of Foodborne Bacterial Pathogens in Food Environment by PMA-qPCR and by Microscopic Observation. Methods Mol. Biol. 2019, 1918, 117–128. [Google Scholar] [CrossRef]
- Morgado, M.E.; Hudson, C.L.; Chattopadhyay, S.; Ta, K.; East, C.; Purser, N.; Allard, S.; Ferrier, M.D.; Sapkota, A.R.; Sharma, M.; et al. The Effect of a First Flush Rainwater Harvesting and Subsurface Irrigation System on E. Coli and Pathogen Concentrations in Irrigation Water, Soil, and Produce. Sci. Total Environ. 2022, 843, 156976. [Google Scholar] [CrossRef]
- Malayil, L.; Chattopadhyay, S.; Kulkarni, P.; Hittle, L.; Clark, P.I.; Mongodin, E.F.; Sapkota, A.R. Mentholation Triggers Brand-Specific Shifts in the Bacterial Microbiota of Commercial Cigarette Products. Appl. Microbiol. Biotechnol. 2020, 104, 6287–6297. [Google Scholar] [CrossRef]
- Fadrosh, D.W.; Ma, B.; Gajer, P.; Sengamalay, N.; Ott, S.; Brotman, R.M.; Ravel, J. An Improved Dual-Indexing Approach for Multiplexed 16S rRNA Gene Sequencing on the Illumina MiSeq Platform. Microbiome 2014, 2, 6. [Google Scholar] [CrossRef]
- Masella, A.P.; Bartram, A.K.; Truszkowski, J.M.; Brown, D.G.; Neufeld, J.D. PANDAseq: Paired-End Assembler for Illumina Sequences. BMC Bioinform. 2012, 13, 31. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME Allows Analysis of High-Throughput Community Sequencing Data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A Versatile Open Source Tool for Metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef]
- Paulson, J.N.; Stine, O.C.; Bravo, H.C.; Pop, M. Differential Abundance Analysis for Microbial Marker-Gene Surveys. Nat. Methods 2013, 10, 1200–1202. [Google Scholar] [CrossRef]
- Shannon, C.E. A Mathematical Theory of Communication. Bell Syst. Tech. J. 1948, 27, 379–423. [Google Scholar] [CrossRef]
- KO (KEGG ORTHOLOGY) Database. Available online: https://www.kegg.jp/kegg/ko.html (accessed on 30 June 2023).
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic Biomarker Discovery and Explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef]
- Malayil, L.; Ramachandran, P.; Chattopadhyay, S.; Cagle, R.; Hittle, L.; Ottesen, A.; Mongodin, E.F.; Sapkota, A.R. Metabolically-Active Bacteria in Reclaimed Water and Ponds Revealed Using Bromodeoxyuridine DNA Labeling Coupled with 16S rRNA and Shotgun Sequencing. Water Res. 2020, 184, 116185. [Google Scholar] [CrossRef]
- Nocker, A.; Camper, A.K. Selective Removal of DNA from Dead Cells of Mixed Bacterial Communities by Use of Ethidium Monoazide. Appl. Environ. Microbiol. 2006, 72, 1997–2004. [Google Scholar] [CrossRef]
- Hellman, M.; Berg, J.; Brandt, K.K.; Hallin, S. Survey of Bromodeoxyuridine Uptake among Environmental Bacteria and Variation in Uptake Rates in a Taxonomically Diverse Set of Bacterial Isolates. J. Microbiol. Methods 2011, 86, 376–378. [Google Scholar] [CrossRef] [PubMed]
- Dygert, H.P. Snuff—A Source of Pathogenic Bacteria in Chronic Bronchitis. N. Engl. J. Med. 1957, 257, 311–313. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S.; Pham, K.; Moe, L.; McNees, R. Exploring the Microbial Diversity and Composition of Three Cigar Product Categories. Microb. Ecol. 2024, 87, 107. [Google Scholar] [CrossRef]
- Taylor, M.J.; Bentham, R.H.; Ross, K.E. Limitations of Using Propidium Monoazide with qPCR to Discriminate between Live and Dead Legionella in Biofilm Samples. Microbiol. Insights 2014, 7, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yan, Y.; Thompson, K.N.; Bae, S.; Accorsi, E.K.; Zhang, Y.; Shen, J.; Vlamakis, H.; Hartmann, E.M.; Huttenhower, C. Whole Microbial Community Viability Is Not Quantitatively Reflected by Propidium Monoazide Sequencing Approach. Microbiome 2021, 9, 17. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Natural Samples | Manufactured Samples | |
|---|---|---|
| Initial number of samples | 186 (n = 36 water, n = 90 soil, and n = 60 produce) | 78 (n = 24 cigarette tobacco, n = 36 hookah, and n = 18 little cigar) |
| Total number of raw seq. | 5,975,496 | 1,007,780 |
| Initial number of OTUs | 15,071 | 1216 |
| Min number of reads | 445 | 61 |
| Max number of reads | 62,676 | 261,593 |
| Number of sequences per sample (average ± SD) | 32,126.32 (±11,403.18 SD) | 13,996.94 (±31,018.68 SD) |
| Good’s coverage cutoff | 0.90 | 0.85 |
| Final number of OTUs | 8791 | 609 |
| Final number of sequences | 5,582,816 | 638,852 |
| Final number of samples | 184 (n = 36 water, n = 89 soil, and n = 59 produce) | 68 (n = 21 cigarette tobacco, n = 35 hookah, and n = 12 little cigar) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Malayil, L.; Chattopadhyay, S.; Sripathi, N.; Mongodin, E.F.; Sapkota, A.R. Comparison of Two DNA Labeling Dyes Commonly Used to Detect Metabolically Active Bacteria. Microorganisms 2025, 13, 1015. https://doi.org/10.3390/microorganisms13051015
Malayil L, Chattopadhyay S, Sripathi N, Mongodin EF, Sapkota AR. Comparison of Two DNA Labeling Dyes Commonly Used to Detect Metabolically Active Bacteria. Microorganisms. 2025; 13(5):1015. https://doi.org/10.3390/microorganisms13051015
Chicago/Turabian StyleMalayil, Leena, Suhana Chattopadhyay, Neha Sripathi, Emmanuel F. Mongodin, and Amy R. Sapkota. 2025. "Comparison of Two DNA Labeling Dyes Commonly Used to Detect Metabolically Active Bacteria" Microorganisms 13, no. 5: 1015. https://doi.org/10.3390/microorganisms13051015
APA StyleMalayil, L., Chattopadhyay, S., Sripathi, N., Mongodin, E. F., & Sapkota, A. R. (2025). Comparison of Two DNA Labeling Dyes Commonly Used to Detect Metabolically Active Bacteria. Microorganisms, 13(5), 1015. https://doi.org/10.3390/microorganisms13051015