
Revealing the Characteristics and Correlations Among Microbial Communities, Functional Genes, and Vital Metabolites Through Metagenomics in Henan Mung Bean Sour


Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Sample Collection
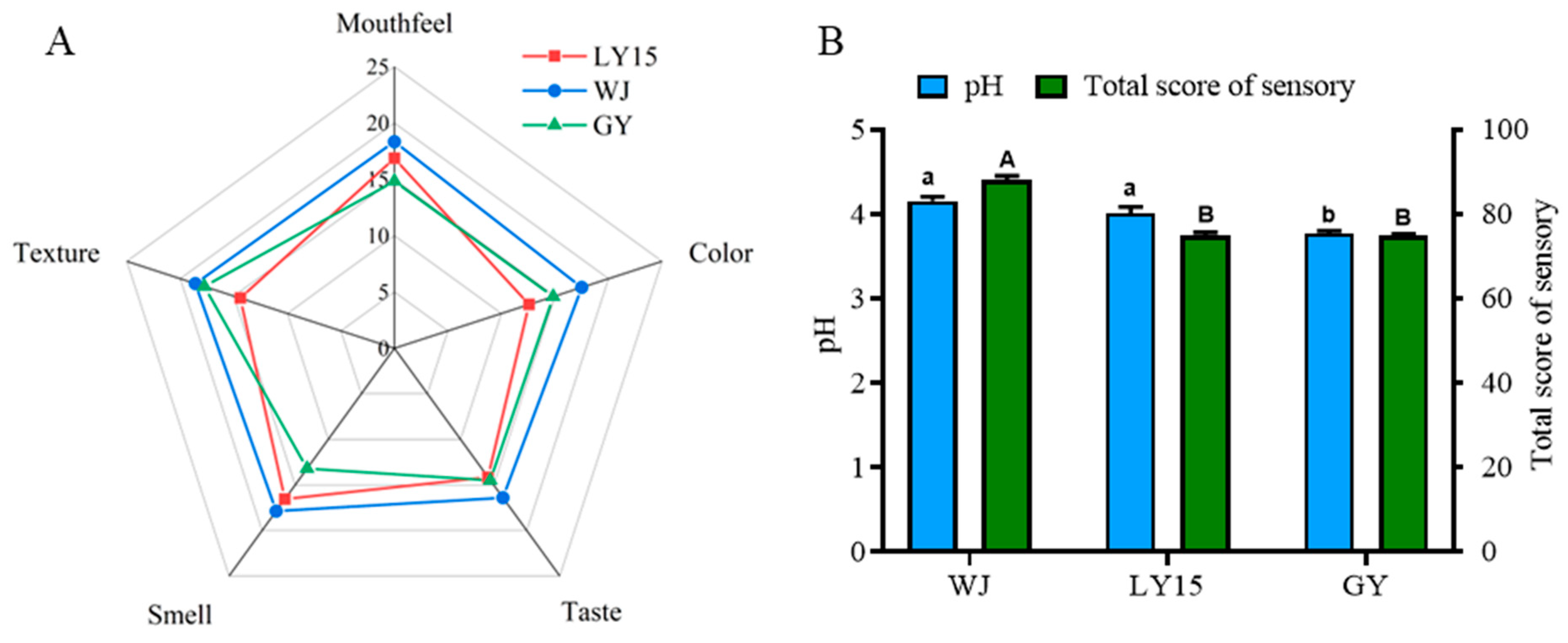
2.3. Sensory Evaluation of Henan Mung Bean Sour
2.4. Total Titrable Acid and pH Measurement
2.5. Determination of Free Amino Acids and Vital Metabolites
2.6. Microbial Community and Functional Annotation by Metagenomics
2.6.1. DNA Extraction and Metagenomic Sequencing
2.6.2. Bioinformatics Analysis
2.7. Statistical Analysis
3. Results and Discussion
3.1. Sensory Evaluation and pH Changes of Different HMBS
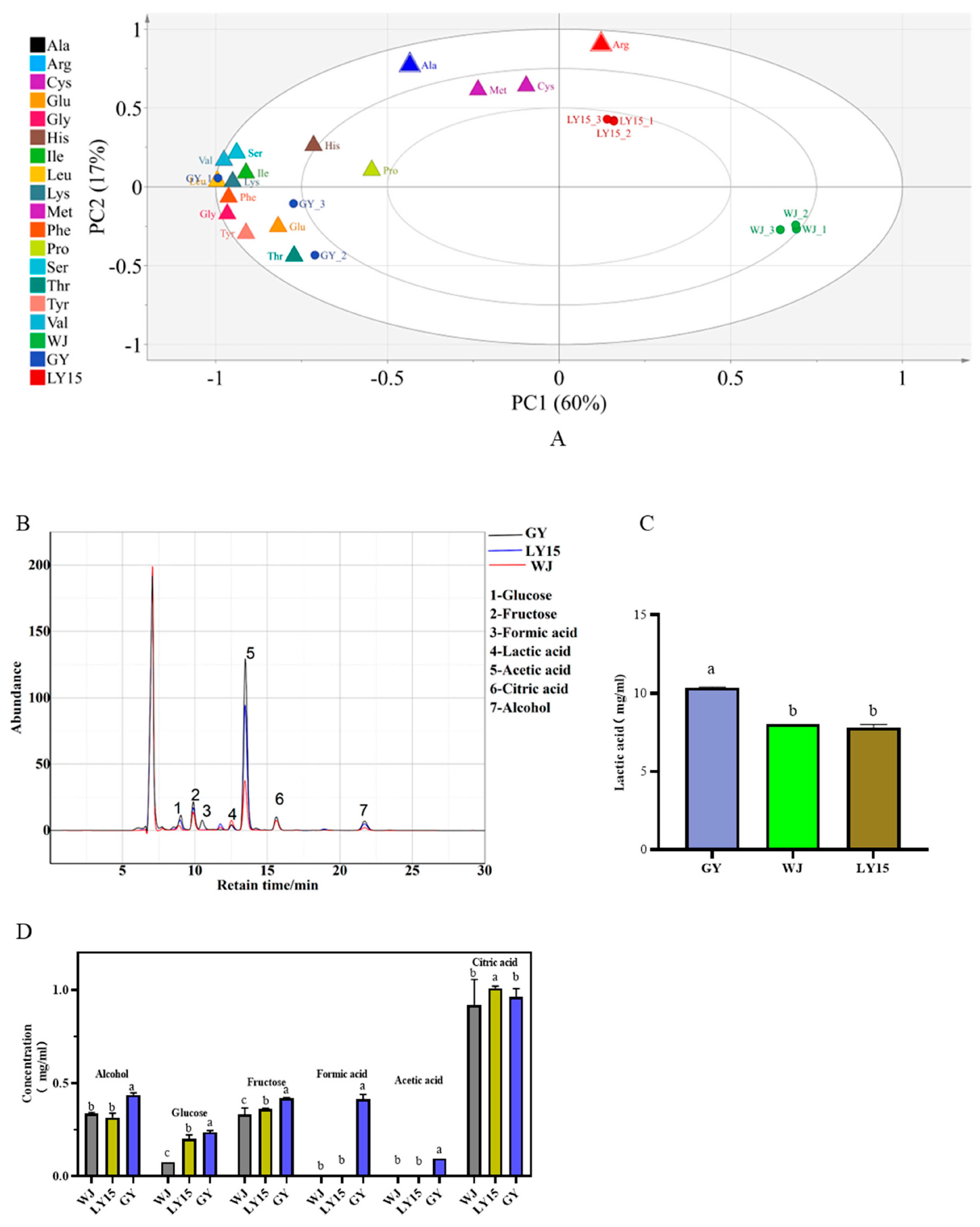
3.2. Role of Characteristic Free Amino Acids
3.3. Comparison of the Content of Organic Acids, Sugar, and Alcohol
3.4. Microbial Community Composition Analysis
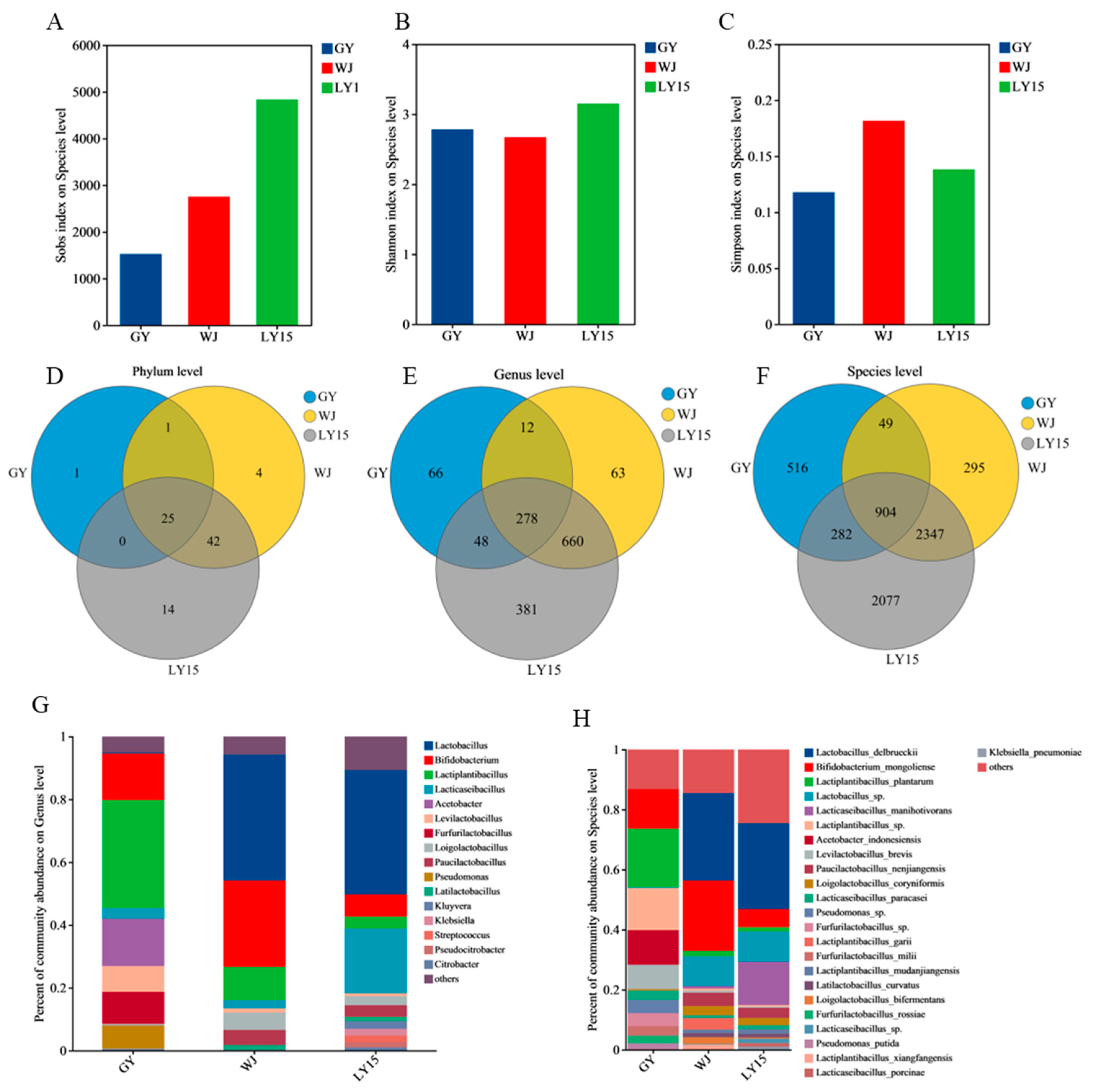
3.4.1. Change in Microbial Diversity During Fermentation
3.4.2. The Changes of Microbial Composition in HMBS
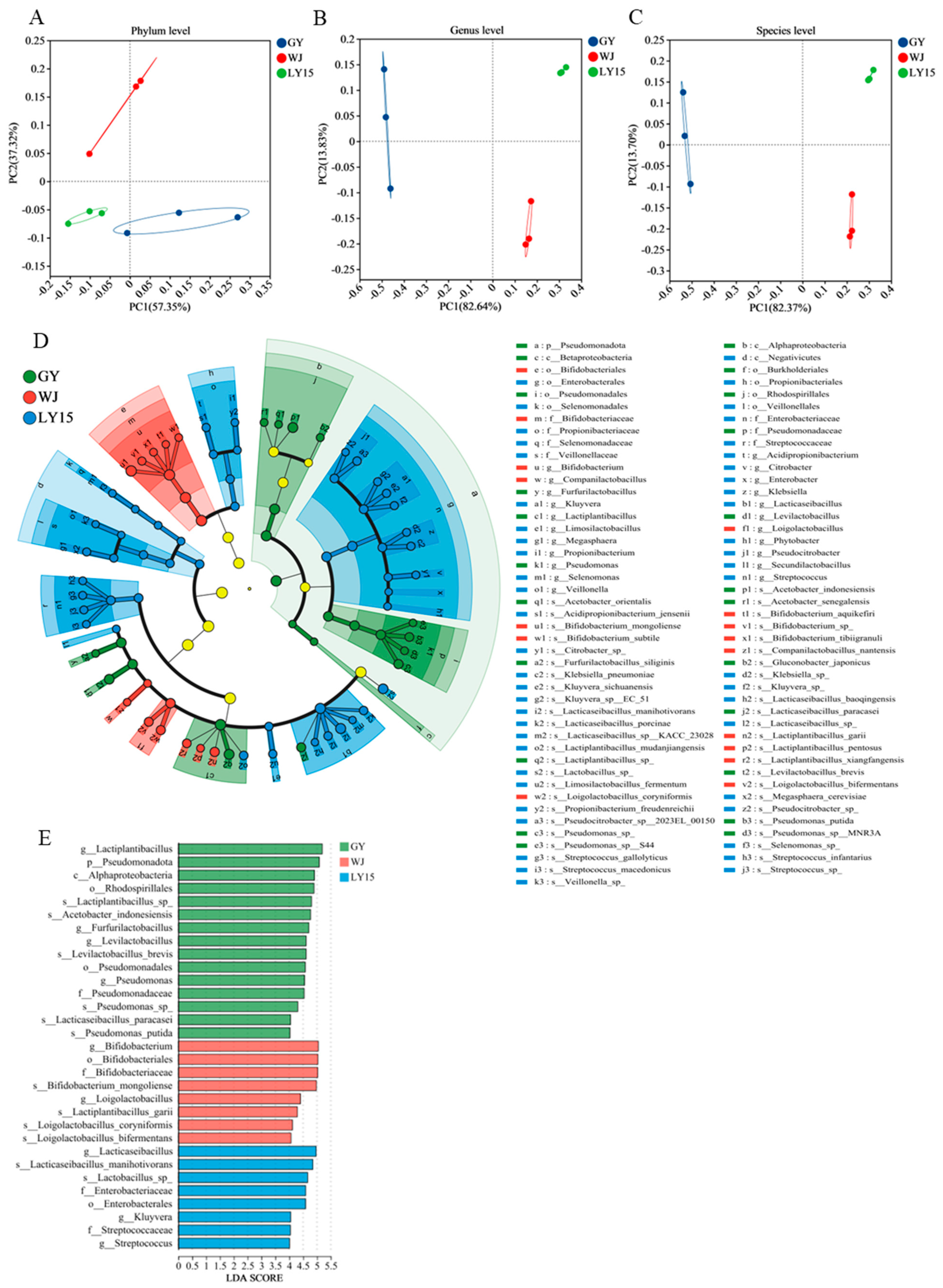
3.4.3. Screening out the Characteristic Members at Phylum, Genus, and Species Level in HMBS
3.5. Functional Gene Category by Blasting to the EggNOG, KEGG, and CAZy Databases
3.6. Analysis of the Differences in Carbohydrate and Amino Acid Metabolic Pathways
3.7. Correlation Analysis Between Dominant Genera and Vital Metabolites
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Abdeldaiem, A.M.; Ali, A.H.; Shah, N.; Ayyash, M.; Mousa, A.H. Physicochemical analysis, rheological properties, and sensory evaluation of yogurt drink supplemented with roasted barley powder. LWT 2023, 173, 114319. [Google Scholar] [CrossRef]
- An, T.; Chen, M.; Zu, Z.; Chen, Q.; Lu, H.; Yue, P.; Gao, X. Untargeted and targeted metabolomics reveal changes in the chemical constituents of instant dark tea during liquid-state fermentation by Eurotium cristatum. Food Res. Int. 2021, 148, 110623. [Google Scholar] [CrossRef] [PubMed]
- Bao, T.; Deng, S.; Yu, K.; Li, W.; Dong, A. Metagenomic insights into seasonal variations in the soil microbial community and function in a Larix gmelinii forest of Mohe, China. J. For. Res. 2020, 32, 371–383. [Google Scholar] [CrossRef]
- Behera, B.C.; Mishra, R.; Mohapatra, S. Microbial citric acid: Production, properties, application, and future perspectives. Food Front. 2021, 2, 62–76. [Google Scholar] [CrossRef]
- Chen, J.-B.; Li, G.; Chen, X.; Liao, L.-H.; He, Y.-Q.; Ye, F.; Chen, G.-X. Nutritional value and antioxidant activity of Artemisia princeps, an edible plant frequently used in folk food in the Xiangxi region. Food Med. Homol. 2025, 2, 9420051. [Google Scholar] [CrossRef]
- Chen, Y.-W.; Yu, Y.-H. Differential effects of Bacillus subtilis– and Bacillus licheniformis–fermented products on growth performance, intestinal morphology, intestinal antioxidant and barrier function gene expression, cecal microbiota community, and microbial carbohydrate-active enzyme composition in broilers. Poult. Sci. 2023, 102, 102670. [Google Scholar]
- Chen, Z.; Liang, N.; Zhang, H.; Li, H.; Guo, J.; Zhang, Y.; Chen, Y.; Wang, Y.; Shi, N. Resistant starch and the gut microbiome: Exploring beneficial interactions and dietary impacts. Food Chem. X 2024, 21, 101118. [Google Scholar] [CrossRef]
- Cui, J.; Xia, P.; Zhang, L.; Hu, Y.; Xie, Q.; Xiang, H. A novel fermented soybean, inoculated with selected Bacillus, Lactobacillus and Hansenula strains, showed strong antioxidant and anti-fatigue potential activity. Food Chem. 2020, 333, 127527. [Google Scholar] [CrossRef]
- De Vuyst, L.; Leroy, F. Functional role of yeasts, lactic acid bacteria and acetic acid bacteria in cocoa fermentation processes. FEMS Microbiol. Rev. 2020, 44, 432–453. [Google Scholar] [CrossRef]
- Derrien, M.; Turroni, F.; Ventura, M.; van Sinderen, D. Insights into endogenous Bifidobacterium species in the human gut microbiota during adulthood. Trends Microbiol. 2022, 30, 940–947. [Google Scholar] [CrossRef]
- Du, X.; Xu, Y.; Jiang, Z.; Zhu, Y.; Li, Z.; Ni, H.; Chen, F. Removal of the fishy malodor from Bangia fusco-purpurea via fermentation of Saccharomyces cerevisiae, Acetobacter pasteurianus, and Lactobacillus plantarum. J. Food Biochem. 2021, 45, 13728. [Google Scholar]
- Echegaray, N.; Yilmaz, B.; Sharma, H.; Kumar, M.; Pateiro, M.; Ozogul, F.; Lorenzo, J.M. A novel approach to Lactiplantibacillus plantarum: From probiotic properties to the omics insights. Microbiol. Res. 2023, 268, 127289. [Google Scholar]
- Gao, P.; Zhang, Z.; Jiang, Q.; Hu, X.; Zhang, X.; Yu, P.; Yang, F.; Liu, S.; Xia, W. Metabolomics ravels flavor compound formation and metabolite transformation in rapid fermentation of salt-free fish sauce from catfish frames induced by mixed microbial cultures. Food Chem. 2025, 463, 141246. [Google Scholar] [PubMed]
- Gao, R.; Peng, P.; Yu, L.; Wan, B.; Liang, X.; Liu, P.; Liao, W.; Miao, L. Metagenomic analysis reveals the correlations between microbial communities and flavor compounds during the brewing of traditional Fangxian huangjiu. Food Biosci. 2024, 58, 103816. [Google Scholar]
- Han, D.; Yang, Y.; Guo, Z.; Dai, S.; Jiang, M.; Zhu, Y.; Wang, Y.; Yu, Z.; Wang, K.; Rong, C.; et al. A Review on the Interaction of Acetic Acid Bacteria and Microbes in Food Fermentation: A Microbial Ecology Perspective. Foods 2024, 13, 2534. [Google Scholar] [CrossRef]
- Hao, H.; Yan, R.; Miao, Z.; Wang, B.; Sun, J.; Sun, B. Volatile organic compounds mediated endogenous microbial interactions in Chinese baijiu fermentation. Int. J. Food Microbiol. 2022, 383, 109955. [Google Scholar] [CrossRef]
- Hong, J.H.; Jin, Y.H.; Pawluk, A.M.; Mah, J.-H. Metagenomic and culturomic analyses of bacterial species contributing to tyramine formation in Cheonggukjang. LWT 2024, 201, 116265. [Google Scholar]
- Hu, Y.; Zhang, L.; Wen, R.; Chen, Q.; Kong, B. Role of lactic acid bacteria in flavor development in traditional Chinese fermented foods: A review. Crit. Rev. Food Sci. Nutr. 2020, 62, 2741–2755. [Google Scholar]
- Jung, S.; Lee, J.-H. Characterization of transcriptional response of Lactobacillus plantarum under acidic conditions provides insight into bacterial adaptation in fermentative environments. Sci. Rep. 2020, 10, 19203. [Google Scholar]
- Kim, S.Y.; Hyeonbin, O.; Lee, P.; Kim, Y.-S. The quality characteristics, antioxidant activity, and sensory evaluation of reduced-fat yogurt and nonfat yogurt supplemented with basil seed gum as a fat substitute. J. Dairy Sci. 2020, 103, 1324–1336. [Google Scholar]
- Kircher, B.; Woltemate, S.; Gutzki, F.; Schlüter, D.; Geffers, R.; Bähre, H.; Vital, M. Predicting butyrate- and propionate-forming bacteria of gut microbiota from sequencing data. Gut Microbes. 2022, 14, 2149019. [Google Scholar] [CrossRef] [PubMed]
- GB5009.124-2016; Determination of Amino Acids in Food Safety National Standards. National Standards of the People’s Republic of China: Beijing, China, 2016.
- Kuang, X.; Su, H.; Li, W.; Lin, L.; Lin, W.; Luo, L. Effects of microbial community structure and its co-occurrence on the dynamic changes of physicochemical properties and free amino acids in the Cantonese soy sauce fermentation process. Food Res. Int. 2022, 156, 111347. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.S.; Song, M.W.; Kim, K.-T.; Hong, W.-S.; Paik, H.-D. Antioxidant Effect and Sensory Evaluation of Yogurt Supplemented with Hydroponic Ginseng Root Extract. Foods 2021, 10, 639. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Oh, J.; Jeong, Y.-S. Lactobacillus plantarum-mediated conversion of flavonoid glycosides into flavonols, quercetin, and kaempferol in Cudrania tricuspidata leaves. Food Sci. Biotechnol. 2015, 24, 1817–1821. [Google Scholar] [CrossRef]
- Li, H.; Li, B.; Gao, L.; Ge, R.; Cui, X.; Zhou, J.; Li, Z. Gamma-aminobutyric acid (GABA) promotes characteristics of Levilactobacillus sp. LB-2. LWT 2023, 184, 115014. [Google Scholar] [CrossRef]
- Li, S.; Feng, X.; Hao, X.; Zhu, Y.; Zou, L.; Chen, X.; Yao, Y. A comprehensive review of mung bean proteins: Extraction, characterization, biological potential, techno-functional properties, modifications, and applications. Compr. Rev. Food Sci. Food Saf. 2023, 22, 3292–3327. [Google Scholar] [CrossRef]
- Li, W.; Shen, X.; Wang, J.; Sun, X.; Yuan, Q. Engineering microorganisms for the biosynthesis of dicarboxylic acids. Biotechnol. Adv. 2021, 48, 107710. [Google Scholar] [CrossRef]
- Li, X.; Wu, Y.; Shu, L.; Zhao, L.; Cao, L.; Li, X.; Tie, S.; Tian, P.; Gu, S. Unravelling the correlations among the microbial community, physicochemical properties, and volatile compounds of traditional mung bean sour liquid. LWT 2024, 198, 115971. [Google Scholar] [CrossRef]
- Liu, M.; Deng, N.; Hou, X.; Zhang, B.; Li, H.; Wang, J. Characterisation of flavour profiles and microbial communities of fermented peppers with different fermentation years by combining flavouromics and metagenomics. Food Chem. 2024, 443, 138550. [Google Scholar] [CrossRef]
- Nyanzi, R.; Jooste, P.J.; Buys, E.M. Invited review: Probiotic yogurt quality criteria, regulatory framework, clinical evidence, and analytical aspects. J. Dairy Sci. 2021, 104, 1–19. [Google Scholar] [CrossRef]
- Peng, X.; Liao, Y.; Ren, K.; Liu, Y.; Wang, M.; Yu, A.; Tian, T.; Liao, P.; Huang, Z.; Wang, H.; et al. Fermentation performance, nutrient composition, and flavor volatiles in soy milk after mixed culture fermentation. Process Biochem. 2022, 121, 286–297. [Google Scholar] [CrossRef]
- Que, Z.; Jin, Y.; Huang, J.; Zhou, R.; Wu, C. Flavor compounds of traditional fermented bean condiments: Classes, synthesis, and factors involved in flavor formation. Trends Food Sci. Technol. 2023, 133, 160–175. [Google Scholar]
- Ricke, S.C.; Dittoe, D.K.; Richardson, K.E. Formic Acid as an Antimicrobial for Poultry Production: A Review. Front. Vet. Sci. 2020, 7, 563. [Google Scholar]
- Shangpliang, H.N.J.; Tamang, J.P. Metagenome-assembled genomes for biomarkers of bio-functionalities in Laal dahi, an Indian ethnic fermented milk product. Int. J. Food Microbiol. 2023, 402, 110300. [Google Scholar]
- Tarahi, M.; Abdolalizadeh, L.; Hedayati, S. Mung bean protein isolate: Extraction, structure, physicochemical properties, modifications, and food applications. Food Chem. 2024, 444, 138626. [Google Scholar]
- Tokatli DemİRok, N.; Alpaslan, M.; YikmiŞ, S. Some lactobacillus, leuconostoc and acetobacter strains in traditional turkish yoghurt, cheese, kefir samples as a probiotic candidate. Int. J. Agric. Environ. Food Sci. 2023, 7, 326–334. [Google Scholar]
- Wang, X.; Wang, X.; Lou, H.; Li, Y.; khashaba, R.; Zhao, R. Understanding the correlation between formation of flavor compounds and dominant bacteria during Luoyang mung bean sour fermentation. Food Biosci. 2024, 60, 104374. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, Y.; Qiu, S.; Wang, B.; Zeng, H. Metagenomic and flavoromic profiling reveals the correlation between the microorganisms and volatile flavor compounds in Monascus-fermented cheese. Food Res. Int. 2024, 188, 114483. [Google Scholar]
- Xia, A.; Yang, Y.; Guo, M.; Lee, Y.-K.; Yang, B.; Liu, X.; Zhao, J.; Zhang, H.; Chen, W. Unveiling of the key pathway in flavor formation in fermented milk of Lactococcus lactis subsp. lactis via genomics and metabolomics. Food Biosci. 2023, 56, 103159. [Google Scholar]
- Xia, F.; Hu, S.; Zheng, X.; Wang, M.W.; Zhang, C.C.; Wu, Z.N.; Sun, Y.J. New insights into metabolomics profile generation in fermented tea: The relevance of bacteria and metabolites in Fuzhuan brick tea. J. Sci. Food Agric. 2022, 102, 350–359. [Google Scholar] [CrossRef]
- Xu, H.; Chen, Y.; Ding, S.; Qin, Y.; Jiang, L.; Zhou, H.; Deng, F.; Wang, R. Changes in texture qualities and pectin characteristics of fermented minced pepper during natural and inoculated fermentation process. Int. J. Food Sci. Technol. 2021, 56, 6073–6085. [Google Scholar]
- Xu, J.; Peng, S.; Xiong, Y.; Zheng, Z.; Liu, M.; Xu, J.; Chen, W.; Liu, M.; Kong, J.; Wang, C.; et al. A review on fermented vegetables: Microbial community and potential upgrading strategy via inoculated fermentation. Compr. Rev. Food Sci. Food Saf. 2024, 23, 13362. [Google Scholar]
- Xu, Y.; Yu, Y.; Tian, Y.; Su, Y.; Li, X.; Zhang, Z.; Zhu, H.; Han, J.; Zhang, H.; Liu, L.; et al. Analysis of Beijing Douzhir Microbiota by High-Throughput Sequencing and Isolation of Acidogenic, Starch-Flocculating Strains. Front. Microbiol. 2018, 9, 1142. [Google Scholar]
- Yu, M.; Li, Z.; Rong, T.; Tian, Z.; Deng, D.; Lu, H.; Zhang, R.; Ma, X. Integrated metagenomics-metabolomics analysis reveals the cecal microbial composition, function, and metabolites of pigs fed diets with different starch sources. Food Res. Int. 2022, 154, 110951. [Google Scholar]
- Yu, Y.; Xu, Y.; Li, L.; Chen, S.; An, K.; Yu, Y.; Xu, Z.-L. Isolation of lactic acid bacteria from Chinese pickle and evaluation of fermentation characteristics. LWT 2023, 180, 114627. [Google Scholar]
- Zhao, Y.-L.; Tang, R.; Liu, S.; Han, S.-T.; Feng, J.; Chi, K.-X.; Yang, G.; Hou, X.-Y.; Fang, Y.-W. Effects of urolithin A-producing Streptococcus thermophilus FUA329 fermentation on the composition and antioxidant bioactivities of black tea. Food Med. Homol. 2025, 2, 9420041. [Google Scholar]
- Glen, D.; Shraddha, S.; Daniel, P.; Ghada, Y.; Silvio, W.; Christian, K. Ecology and evolution of metabolic cross-feeding interactions in bacteria. Nat. Prod. Rep. 2018, 35, 455–488. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Amino Acid | GY | WJ | LY15 | GY | WJ | LY15 | ||
|---|---|---|---|---|---|---|---|---|
| TVA | VIP | |||||||
| Umamiiii | T | |||||||
| Asp | 10 | 66 ± 16.46 a | 3.66 ± 0.57 b | 10.3333 ± 0.57 b | 6.6 | 0.366 | 1.033 | 0.91 |
| Glu | 3 | 109 ± 25.15 a | 56.66 ± 2.08 b | 70 ± 1.01 b | 36.33 | 18.88 | 23.33 | 0.92 |
| TUAA | 175 | 59 | 80 | |||||
| Sweet | ||||||||
| Ser | 15 | 18.33 ± 1.15 c | 8.33 ± 1.15 b | 14.33 ± 0.57 a | 1.22 | 0.55 | 0.96 | 1.02 |
| Gly | 13 | 29 ± 4 c | 5.66 ± 0.57 b | 12.66 ± 2.08 a | 2.23 | 0.43 | 0.92 | 1.03 |
| Thr | 26 | 21 ± 3.46 a | 4 ± 1.10 b | 5.66 ± 0.57 b | 0.80 | 0.15 | 0.21 | 1.01 |
| Met | 6 | 11.33 ± 3.21 | 9.66 ± 0.57 | 12 ± 1.12 | 3.66 | 3.22 | 4 | 0.90 |
| Ala | 30 | 31 ± 1.03 b | 28 ± 2.64 b | 43.66 ± 0.57 a | 5.16 | 4.66 | 7.16 | 1.18 |
| Pro | 50 | 7.66 ± 1.15 a | 5.66 ± 0.57 b | 7.33 ± 0.57 a | 0.25 | 0.18 | 0.24 | 0.59 |
| TSAA | 117 | 59 | 93 | |||||
| Bitter | ||||||||
| His | 90 | 12.33 ± 0.57 a | 10.66 ± 1.15 b | 12.66 ± 0.57 a | 6.16 | 5.33 | 6.33 | 0.83 |
| Arg | 5 | 0.5 ± 0.04 b | 0.51 ± 0.02 b | 14.33 ± 2.08 a | 0.1 | 0.102 | 2.86 | 1.28 |
| Lys | 5 | 20.66 ± 1.52 c | 8.33 ± 0.05 b | 13 ± 1.11 a | 4.132 | 1.66 | 2.6 | 0.99 |
| Val | 4 | 30 ± 3.60 a | 9 ± 7.81 b | 21 ± 1.00 a | 7.56 | 2.25 | 5.25 | 1.04 |
| Trp | 9 | 1.33 ± 0.15 | 0.66 ± 0.57 | 0.66 ± 0.05 | 0.14 | 0.07 | 0.07 | |
| Phe | 9 | 22 ± 1.02 a | 13 ± 1 c | 16 ± 1.13 b | 2.44 | 1.44 | 1.77 | 1.00 |
| Ile | 9 | 13 ± 2.64 a | 7.67 ± 0.57 b | 9.33 ± 0.57 b | 1.44 | 0.85 | 1.03 | 0.96 |
| Leu | 19 | 34 ± 2.64 a | 16.66 ± 0.57 c | 22.33 ± 2.08 b | 1.78 | 0.87 | 1.17 | 1.04 |
| TBAA | 132 | 63 | 107 | |||||
| Astringent | ||||||||
| Tyr | 9.1 | 13.66 ± 2.51 a | 1.33 ± 0.57 b | 0.66 ± 0.04 b | 1.50 | 0.14 | 0.07 | 1.03 |
| TAAA | 13 | 1 | 0.66 | |||||
| Genus | GY (%) | LY15 (%) | WJ (%) |
|---|---|---|---|
| Lactobacillus | 0.24 ± 0.03b | 41.23 ± 0.56a | 40.48 ± 2.32a |
| Bifidobacterium | 16.38 ± 5.78b | 7.75 ± 2.11c | 28.98 ± 9.72a |
| Lactiplantibacillus | 34.40 ± 9.79a | 3.78 ± 0.48c | 10.24 ± 0.45b |
| Lacticaseibacillus | 3.25 ± 0.41b | 20.58 ± 2.49a | 2.27 ± 0.09b |
| Acetobacter | 15.27 ± 4.37a | Nd | Nd |
| Furfurilactobacillus | 10.29 ± 2.43a | Nd | Nd |
| Levilactobacillus | 8.06 ± 1.41a | 0.64 ± 0.03c | 1.00 ± 0.29b |
| Loigolactobacillus | 0.49 ± 0.07c | 3.03 ± 0.21b | 5.50 ± 1.50a |
| Paucilactobacillus | Nd | 3.80 ± 0.82 | 4.77 ± 3.52 |
| Pseudomonas | 7.40 ± 2.74 | Nd | Nd |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, X.; Li, Y.; Zuo, L.; Li, P.; Lou, H.; Zhao, R. Revealing the Characteristics and Correlations Among Microbial Communities, Functional Genes, and Vital Metabolites Through Metagenomics in Henan Mung Bean Sour. Microorganisms 2025, 13, 845. https://doi.org/10.3390/microorganisms13040845
Wang X, Li Y, Zuo L, Li P, Lou H, Zhao R. Revealing the Characteristics and Correlations Among Microbial Communities, Functional Genes, and Vital Metabolites Through Metagenomics in Henan Mung Bean Sour. Microorganisms. 2025; 13(4):845. https://doi.org/10.3390/microorganisms13040845
Chicago/Turabian StyleWang, Xunda, Yue Li, Lei Zuo, Pengna Li, Haiwei Lou, and Renyong Zhao. 2025. "Revealing the Characteristics and Correlations Among Microbial Communities, Functional Genes, and Vital Metabolites Through Metagenomics in Henan Mung Bean Sour" Microorganisms 13, no. 4: 845. https://doi.org/10.3390/microorganisms13040845
APA StyleWang, X., Li, Y., Zuo, L., Li, P., Lou, H., & Zhao, R. (2025). Revealing the Characteristics and Correlations Among Microbial Communities, Functional Genes, and Vital Metabolites Through Metagenomics in Henan Mung Bean Sour. Microorganisms, 13(4), 845. https://doi.org/10.3390/microorganisms13040845