
Advances in the Molecular Modification of Microbial ω-Transaminases for Asymmetric Synthesis of Bulky Chiral Amines


Abstract
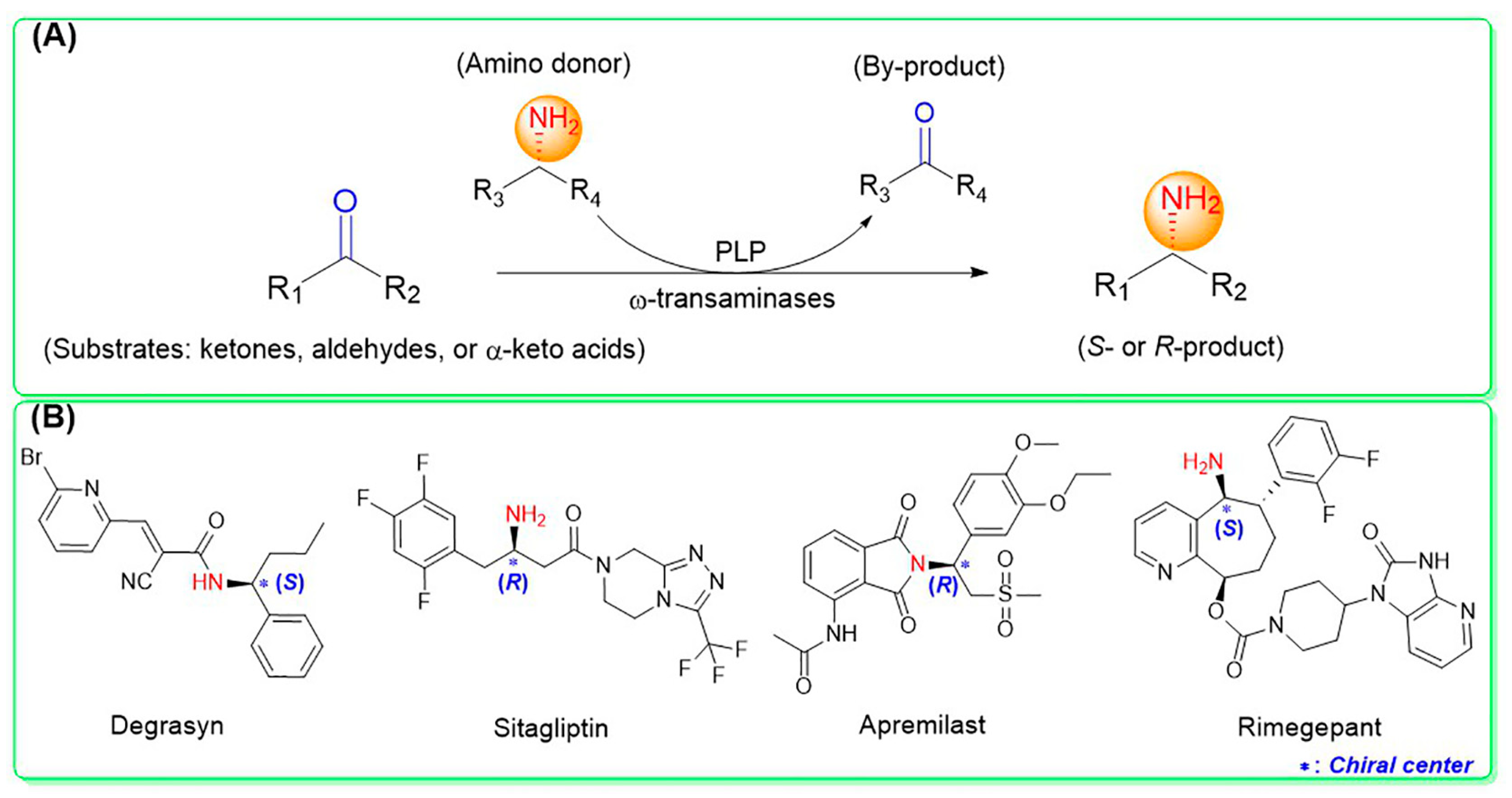
1. Introduction
2. Structural Characteristics, Catalytic Mechanism, and Substrate Recognition Mechanism
2.1. Structural Classification and Substrate Binding Features
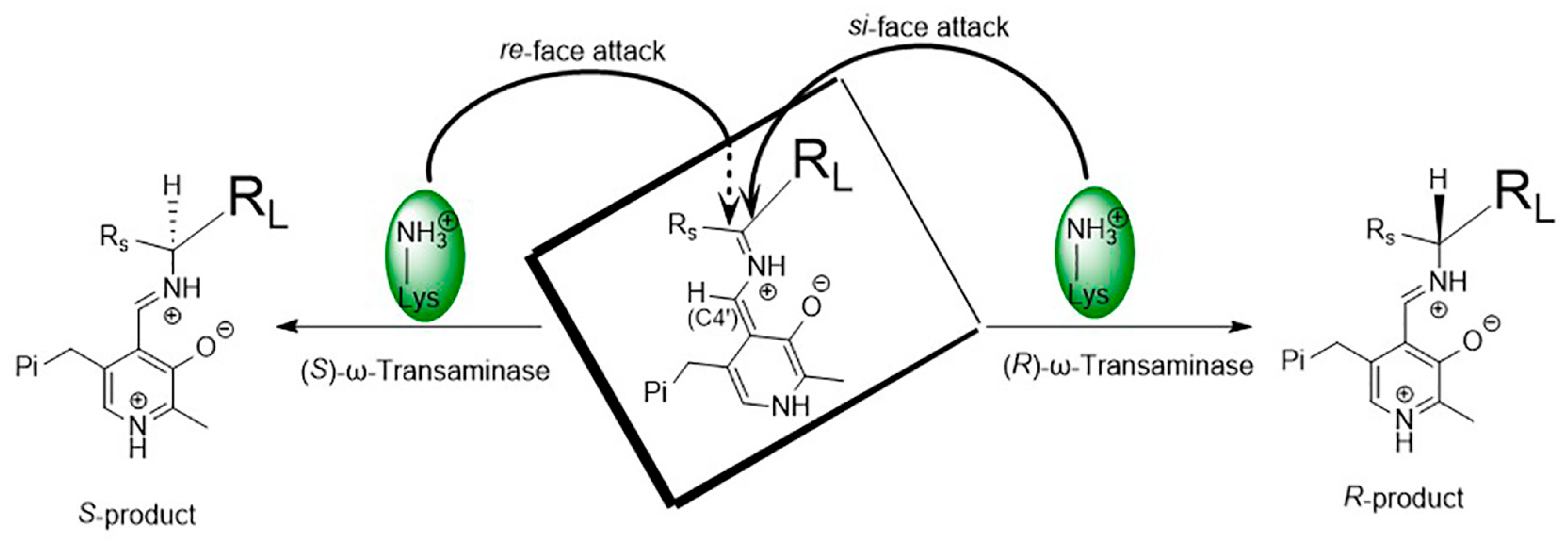
2.2. Catalytic Mechanisms
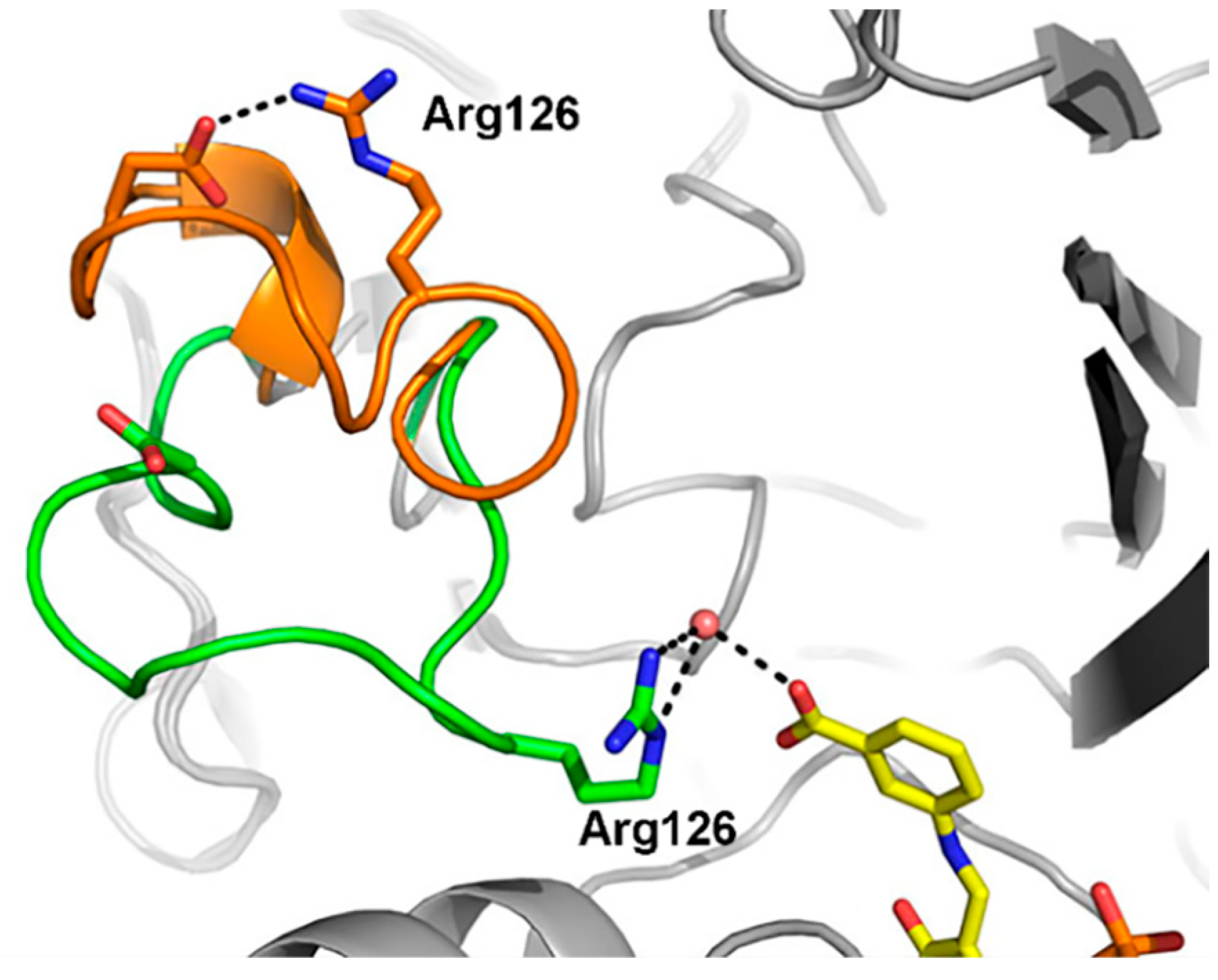
2.3. Substrate Recognition Mechanisms
3. Molecular Modification of ω-Transaminases to Expand the Substrate Scope
3.1. Substrate Specificity of ω-Transaminases
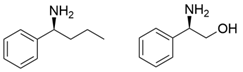
3.2. Methyl Ketones and Relevant Amines Bearing One Bulky Substituent

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzymes | Substrates or Products | Key Mutations | References |
|---|---|---|---|
| VfTA | 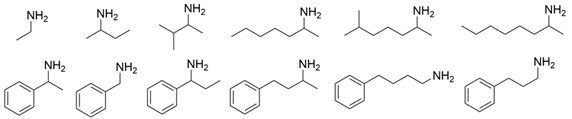 | P233L/V297A | [30] |
 | W57G and W147G | [31] | |
 | W57G/R415A | [32] | |
 | W57F/R415L/L417V | [36] | |
| HeTA |  | W56G, Y149F, F84A, and I258A | [33] |
| ExTA | 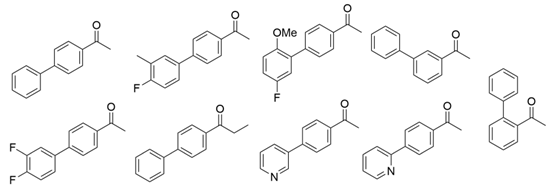 | T273S and K110R/L191F/N249F/E300K/K317E | [27] |
| MvTA |  | G68Y/F129A | [34] |
| OaTA |  | W58A and W58L | [35] |
| AtTA |  | H55A/G126F/S215P | [37] |
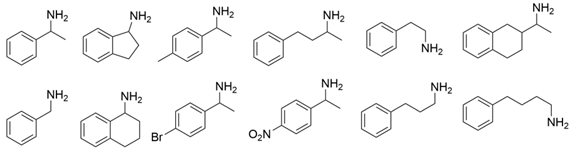
3.3. Ketones and Relevant Amines Bearing Two Bulky Substituents
| Enzymes | Substrates or Products | Key Mutations | References |
|---|---|---|---|
| VfTA | 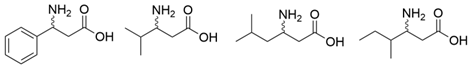 | Not mentioned | [38] |
 | F19W/W57F/F85A/R88K/V153A/K163F/I259V/R415F | [39] | |
 | F85L/V153A Y150M/V153A | [40] | |
 | L56V/W57C/F85V/V153A | [41] | |
 | ATA217+Y17M/F19H/V31M/V42F/N53C/L57A/K66P/F85V/S86N/R146H/Y165W/R203H/C260T/S284A/S286G/F291Y/P293A/A313L/I314R/E316W/G320A/G394P/C414V/P416A/V422A/C424A | [42] | |
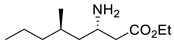
| CvTA | 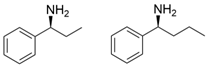 | F88L/C418G F88L/C418L | [43] |
 | L59A/F88A | [44] | |
 | L59A | [45] | |
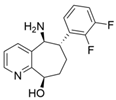 | L59A/F88A/V234A/L380A/Y89D/N86H/Y85M/T91I/P83S/K90G/S417I/S424A/F301S/G164S/T452S/M180V/F449L/F320H/Y322T | [46] | |
| ArTA | 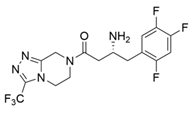 | S8P/Y60F/L61Y/H62T/V65A/V69T/D81G/M94I/I96L/F122M/S124/S126T/G136F/Y150S/V152C/A169L/V199I/A209L/G215C/G217N/S223P/L269P/L273/T282S/A284G/P297S/V306I/S321P | [7] |
| CcTA | 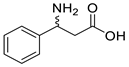 | V227G and N285A | [48] |
| BvTA | 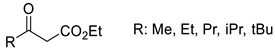 | L57A/W58F/F86M/A154S/I260V | [49] |
| PdTA | 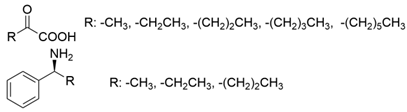 | V153A | [50] |
| GzTA | 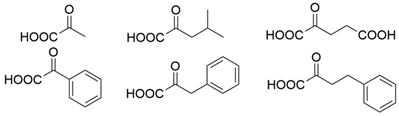 | F113L/V60A/S214A | [51] |
| OaTA | 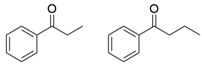 | L57A/W58A | [52] |
| LsTA |  | V37A | [53] |
| PpTA | 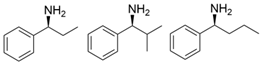 | M78F/W82A/I284F/T440Q | [54] |
| AcTA | 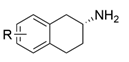 | M46T/D48G/Y60C/Y164F/Y185C/N186S/P195S/M197T/C205Y/A242V/A245T/I252V/F255I/N268S/T409R/K424E/V436A | [55] |
| PjTA | 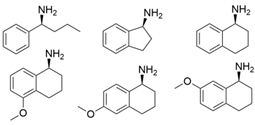 | W58G and W58M/F86L/R417L | [56] |
| RhTA | 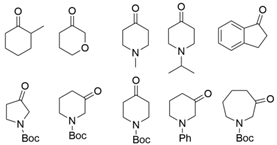 | Y125A/I6A/L7A/L158V | [57] |
| 3FCR | 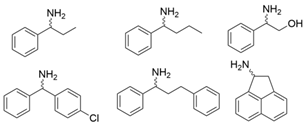 | Y59W/Y87F/Y152F/T231A | [58] |
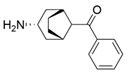 | Y59L/S86A/Y87F/Y152F/T231A/I234M/L382M | [59] | |
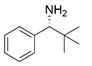 | Y59W/Y87L/T231A/L382M/G429A | [60] | |
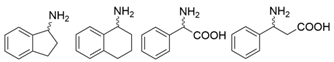 | Y59W/Y87F/T231A/S19W Y59W/Y87F/T231A/Y152F F91F/T231A/Y59W F91L/S19W/T231L/Y59W | [61] | |
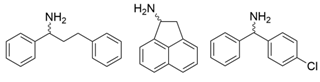 | Y59W/Y87F/T231A/S155A Y59W/Y87F/T231A/F167Y Y59W/Y87F/T231A/F168W Y59W/Y87L/T231A/Y152F Y59W/Y87F/T231A/Y152F/L382M Y59W/Y87F/T231A/Y152F/F168W | [62] |
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| PLP | pyridoxal-5′-phosphate |
| VfTA | (S)-ω-transaminase from Vibrio fluvialis JS17 |
| CvTA | (S)-ω-transaminase from Chromobacterium violaceum |
| AtTA | (R)-ω-transaminase from Aspergillus terreus |
| ExTA | (R)-ω-transaminase from Exophiala xenobiotica |
| ArTA | (R)-ω-transaminase from Arthrobacter sp. |
| HeTA | (S)-ω-transaminase from Halomonas elongata |
| MvTA | (R)-ω-transaminase from Mycobacterium vanbaalenii |
| OaTA | (S)-ω-transaminase from Ochrobactrum anthropic |
| CcTA | (S)-ω-transaminase from Caulobacter crescentus |
| BvTA | (S)-ω-transaminase from Burkholderia vietnamiensis |
| PdTA | (S)-ω-transaminase from Paracoccus denitrificans |
| GzTA | (R)-ω-transaminase from Gibberella zeae |
| OaTA | (S)-ω-transaminase from Ochrobactrum anthropic |
| LsTA | (R)-ω-transaminase from Luminiphilus syltensis |
| PpTA | (S)-ω-transaminase from Paraburkholderia phymatum |
| AcTA | (S)-ω-transaminase from Athrobacter citreus |
| PjTA | (S)-ω-transaminase from Pseudomonas jessenii |
| RhTA | (R)-ω-transaminase from Rhodobacter sp. |
References
- Wang, J.; Li, Y.; Nie, W.; Chang, Z.; Yu, Z.; Zhao, Y.; Lu, X.; Fu, Y. Catalytic asymmetric reductive hydroalkylation of enamides and enecarbamates to chiral aliphatic amines. Nat. Commun. 2021, 12, 1313. [Google Scholar] [CrossRef] [PubMed]
- Cabré, A.; Verdaguer, X.; Riera, A. Recent advances in the enantioselective synthesis of chiral amines via transition metal-catalyzed asymmetric hydrogenation. Chem. Rev. 2022, 122, 269–339. [Google Scholar] [CrossRef] [PubMed]
- Aleku, G.A.; France, S.P.; Man, H.; Mangas-Sanchez, J.; Montgomery, S.L.; Sharma, M.; Leipold, F.; Hussain, S.; Grogan, G.; Turner, N.J. A reductive aminase from Aspergillus oryzae. Nat. Chem. 2017, 9, 961–969. [Google Scholar] [CrossRef] [PubMed]
- Qian, D.; Bera, S.; Hu, X. Chiral alkyl amine synthesis via catalytic enantioselective hydroalkylation of enecarbamates. J. Am. Chem. Soc. 2021, 143, 1959–1967. [Google Scholar] [CrossRef]
- Hanefeld, U.; Hollmann, F.; Paul, C.E. Biocatalysis making waves in organic chemistry. Chem. Soc. Rev. 2022, 51, 594–627. [Google Scholar] [CrossRef]
- Gomm, A.; O’Reilly, E. Transaminases for chiral amine synthesis. Curr. Opin. Chem. Biol. 2018, 43, 106–112. [Google Scholar] [CrossRef]
- Savile, C.K.; Janey, J.M.; Mundorff, E.C.; Moore, J.C.; Tam, S.; Jarvis, W.R.; Colbeck, J.C.; Krebber, A.; Fleitz, F.J.; Brands, J.; et al. Biocatalytic asymmetric synthesis of chiral amines from ketones applied to sitagliptin manufacture. Science 2010, 329, 305–309. [Google Scholar] [CrossRef]
- Bornscheuer, U.T.; Huisman, G.W.; Kazlauskas, R.J.; Lutz, S.; Moore, J.C.; Robins, K. Engineering the third wave of biocatalysis. Nature 2012, 485, 185–194. [Google Scholar] [CrossRef]
- Mathew, S.; Yun, H. ω-Transaminases for the production of optically pure amines and unnatural amino acids. ACS Catal. 2012, 2, 993–1001. [Google Scholar] [CrossRef]
- Calvelage, S.; Dörr, M.; Höhne, M.; Bornscheuer, U.T. A systematic analysis of the substrate scope of (S)- and (R)-selective amine transaminases. Adv. Synth. Catal. 2017, 359, 4235–4243. [Google Scholar] [CrossRef]
- Ramírez-Palacios, C.; Wijma, H.J.; Thallmair, S.; Marrink, S.J.; Janssen, D.B. Computational prediction of ω-transaminase specificity by a combination of docking and molecular dynamics simulations. J. Chem. Inf. Model. 2021, 61, 5569–5580. [Google Scholar] [PubMed]
- Yang, L.; Zhang, K.; Xu, M.; Xie, Y.; Meng, X.; Wang, H.; Wei, D. Mechanism-guided computational design of ω-transaminase by reprograming of high-energy-barrier steps. Angew. Chem. Int. Ed. 2022, 61, e202212555. [Google Scholar]
- Gao, X.; Wei, P. Advances in molecular modification of ω-transaminase. Chin. J. Biotechnol. 2018, 34, 1057–1068. [Google Scholar]
- Andrzej, Ł.; Christian, G.; Steinkellner, G.; Martin, S.; Helmut, S.; Karl, G.; Kerstin, S. Crystal Structure of an (R)-Selective ω-Transaminase from Aspergillus terreus. PLoS ONE 2014, 9, e87350. [Google Scholar]
- Jang, T.H.; Kim, B.; Park, O.K.; Bae, J.Y.; Kim, B.G.; Yun, H.; Park, H.H. Crystallization and preliminary X-ray crystallographic studies of omega-transaminase from Vibrio fluvialis JS17. Acta Crystallogr. F 2010, 66, 923–925. [Google Scholar]
- Cassimjee, K.E.; Humble, M.S.; Miceli, V.; Colomina, C.G.; Berglund, P. Active site quantification of an ω-transaminase by performing a half transamination reaction. ACS Catal. 2011, 1, 1051–1055. [Google Scholar]
- Slabu, I.; Galman, J.L.; Lloyd, R.C.; Turner, N.J. Discovery, engineering, and synthetic application of transaminase biocatalysts. ACS Catal. 2017, 7, 8263–8284. [Google Scholar]
- Guo, F.; Berglund, P. Transaminase biocatalysis: Optimization and application. Green. Chem. 2017, 19, 333–360. [Google Scholar]
- Skalden, L.; Thomsen, M.; Höhne, M.; Bornscheuer, U.T.; Hinrichs, W. Structural and biochemical characterization of the dual substrate recognition of the (R)-selective amine transaminase from Aspergillus fumigatus. FEBS J. 2015, 282, 407–415. [Google Scholar]
- Guan, L.J.; Ohtsuka, J.; Okai, M.; Miyakawa, T.; Mase, T.; Zhi, Y.; Hou, F.; Ito, N.; Iwasaki, A.; Yasohara, Y.; et al. A new target region for changing the substrate specificity of amine transaminases. Sci. Rep. 2015, 5, 10753. [Google Scholar]
- Shin, J.S.; Kim, B.G. Comparison of the ω-transaminases from different microorganisms and application to production of chiral amines. Biosci. Biotechnol. Biochem. 2001, 65, 1782–1788. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.S.; Yun, H.; Jang, J.W.; Park, I.; Kim, B.G. Purification, characterization, and molecular cloning of a novel amine:pyruvate transaminase from Vibrio fluvialis JS17. Appl. Microbiol. Biotechnol. 2003, 61, 463–471. [Google Scholar] [CrossRef]
- Kaulmann, U.; Smithies, K.; Smith, M.E.B.; Hailes, H.C.; Ward, J.M. Substrate spectrum of ω-transaminase from Chromobacterium violaceum DSM30191 and its potential for biocatalysis. Enzym. Microb. Technol. 2007, 41, 628–637. [Google Scholar] [CrossRef]
- Yamada, Y.; Iwasaki, A.; Kizaki, N.; Ikenaka, Y.; Ogura, M.; Hasegawa, J. Process for Producing Optically Active Amino Compounds. U.S. Patent 2001031487, 18 October 2001. [Google Scholar]
- Höhne, M.; Schätzle, S.; Jochens, H.; Robins, K.; Bornscheuer, U.T. Rational assignment of key motifs for function guides in silico enzyme identification. Nat. Chem. Biol. 2010, 6, 807–813. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Chen, X.; Zhang, D.; Wu, Q.; Zhu, D. Characterization of (R)-selective amine transaminases identified by in silico motif sequence blast. Appl. Microbiol. Biotechnol. 2015, 99, 2613–2621. [Google Scholar]
- Telzerow, A.; Paris, J.; Håkansson, M.; González-Sabín, J.; Ríos-Lombardía, N.; Schürmann, M.; Gröger, H.; Morís, F.; Kourist, R.; Schwab, H.; et al. Amine transaminase from Exophiala Xenobiotica-crystal structure and engineering of a fold IV transaminase that naturally converts biaryl ketones. ACS Catal. 2019, 9, 1140–1148. [Google Scholar] [CrossRef]
- Kim, E.M.; Park, J.H.; Kim, B.G.; Seo, J.H. Identification of (R)-selective ω-aminotransferases by exploring evolutionary sequence space. Enzym. Microb. Technol. 2018, 110, 46–52. [Google Scholar]
- Tang, X.; Zhang, N.; Ye, G.; Zheng, Y. Efficient biosynthesis of (R)-3-amino-1-butanol by a novel (R)-selective transaminase from Actinobacteria sp. J. Biotechnol. 2019, 295, 49–54. [Google Scholar] [CrossRef]
- Yun, H.; Hwang, B.Y.; Lee, J.H.; Kim, B.G. Use of enrichment culture for directed evolution of the Vibrio fluvialis JS17 ω-transaminase, which is resistant to product inhibition by aliphatic ketones. Appl. Environ. Microbiol. 2005, 71, 4220–4224. [Google Scholar] [CrossRef]
- Cho, B.K.; Park, H.Y.; Seo, J.H.; Kim, J.; Kang, T.J.; Lee, B.S.; Kim, B.G. Redesigning the substrate specificity of ω-aminotransferase for the kinetic resolution of aliphatic chiral amines. Biotechnol. Bioeng. 2008, 99, 275–284. [Google Scholar] [CrossRef]
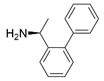
- Dourado, D.F.A.R.; Pohle, S.; Carvalho, A.T.P.; Dheeman, D.S.; Caswell, J.M.; Skvortsov, T.; Miskelly, I.; Brown, R.T.; Quinn, D.J.; Allen, C.C.R.; et al. Rational design of a (S)-selective-transaminase for asymmetric synthesis of (1S)-1-(1,1′-biphenyl-2-yl)ethanamine. ACS Catal. 2016, 6, 7749–7759. [Google Scholar]
- Contente, M.L.; Planchestainer, M.; Molinari, F.; Paradisi, F. Stereoelectronic effects in the reaction of aromatic substrates catalysed by Halomonas elongata transaminase and its mutants. Org. Biomol. Chem. 2016, 14, 9306–9311. [Google Scholar] [PubMed]
- Cheng, F.; Chen, X.; Xiang, C.; Liu, Z.; Wang, Y.; Zheng, Y. Fluorescence-based high-throughput screening system for R-ω-transaminase engineering and its substrate scope extension. Appl. Microbiol. Biot. 2020, 104, 2999–3009. [Google Scholar]
- Han, S.W.; Park, E.S.; Dong, J.Y.; Shin, J.S. Mechanism-guided engineering of ω-transaminase to accelerate reductive amination of ketones. Adv. Synth. Catal. 2015, 357, 1732–1740. [Google Scholar]
- Genz, M.; Vickers, C.; Van den Bergh, T.; Joosten, H.J.; Dörr, M.; Höhne, M.; Bornscheuer, U.T. Alteration of the donor/acceptor spectrum of the (S)-amine transaminase from Vibrio fluvialis. Int. J. Mol. Sci. 2015, 16, 26953–26963. [Google Scholar] [CrossRef]
- Gao, X.; Zhang, X.; Zhu, N.; Mou, Y.; Zhang, H.; Liu, X.; Wei, P. Reshaping the substrate binding region of (R)-selective ω-transaminase for asymmetric synthesis of (R)-3-amino-1-butanol. Appl. Microbiol. Biot. 2020, 104, 3959–3969. [Google Scholar] [CrossRef]
- Hwang, B.Y.; Kim, B.G. High-throughput screening method for the identification of active and enantioselective ω-transaminases. Enzym. Microb. Technol. 2004, 34, 429–436. [Google Scholar]
- Midelfort, K.S.; Kumar, R.; Han, S.; Karmilowicz, M.J.; McConnell, K.; Gehlhaar, D.K.; Mistry, A.; Chang, J.S.; Anderson, M.; Villalobos, A.; et al. Redesigning and characterizing the substrate specificity and activity of Vibrio fluvialis aminotransferase for the synthesis of imagabalin. Protein Eng. Des. Sel. 2013, 26, 25–33. [Google Scholar]
- Nobili, A.; Steffen-Munsberg, F.; Kohls, H.; Trentin, I.; Schulzke, C.; Höhne, M.; Bornscheuer, U.T. Engineering the active site of the amine transaminase from Vibrio fluvialis for the asymmetric synthesis of aryl-alkyl amines and amino alcohols. ChemCatChem 2015, 7, 757–760. [Google Scholar]
- Genz, M.; Melse, O.; Schmidt, S.; Vickers, C.; Dörr, M.; Van Den Bergh, T.; Joosten, H.J.; Bornscheuer, U.T. Engineering the amine transaminase from Vibrio fluvialis towards branched-chain substrates. ChemCatChem 2016, 8, 3199–3202. [Google Scholar]
- Novick, S.J.; Dellas, N.; Garcia, R.; Ching, C.; Bautista, A.; Homan, D.; Alvizo, O.; Entwistle, D.; Kleinbeck, F.; Schlama, T.; et al. Engineering an amine transaminase for the efficient production of a chiral sacubitril precursor. ACS Catal. 2021, 11, 3762–3770. [Google Scholar] [CrossRef]
- Voss, M.; Das, D.; Genz, M.; Kumar, A.; Kulkarni, N.; Kustosz, J.; Kumar, P.; Bornscheuer, U.T.; Höhne, M. In silico based engineering approach to improve transaminases for the conversion of bulky substrates. ACS Catal. 2018, 8, 11524–11533. [Google Scholar] [CrossRef]
- Land, H.; Ruggieri, F.; Szekrenyi, A.; Fessner, W.-D.; Berglund, P. Engineering the active site of an (S)-selective amine transaminase for acceptance of doubly bulky primary amines. Adv. Synth. Catal. 2020, 362, 812–821. [Google Scholar] [CrossRef]
- Sheludko, Y.V.; Slagman, S.; Gittings, S.; Charnock, S.J.; Land, H.; Berglund, P.; Fessner, W.D. Enantioselective synthesis of pharmaceutically relevant bulky arylbutylamines using engineered transaminases. Adv. Synth. Catal. 2022, 364, 2972–2981. [Google Scholar] [CrossRef]
- Ma, Y.; Jiao, X.; Wang, Z.; Mu, H.; Sun, K.; Li, X.; Zhao, T.; Liu, X.; Zhang, N. Engineering a transaminase for the efficient synthesis of a key intermediate for rimegepant. Org. Process Res. Dev. 2022, 26, 1971–1977. [Google Scholar] [CrossRef]
- Desai, A.A. Sitagliptin manufacture: A compelling tale of green chemistry, process intensification, and industrial asymmetric catalysis. Angew. Chem. Int. Ed. 2011, 50, 1974–1976. [Google Scholar] [CrossRef]
- Hwang, B.Y.; Ko, S.H.; Park, H.Y.; Seo, J.H.; Lee, B.S.; Kim, B.G. Identification of ω-aminotransferase from Caulobacter crescentus and site-directed mutagenesis to broaden substrate specificity. J. Microbiol. Biotechnol. 2008, 18, 48–54. [Google Scholar]
- Wang, Y.; Feng, J.; Dong, W.; Chen, X.; Yao, P.; Wu, Q.; Zhu, D. Improving catalytic activity and reversing enantio-specificity of ω-transaminase by semi-rational engineering en route to chiral bulky β-amino esters. ChemCatChem 2021, 13, 3396–3400. [Google Scholar] [CrossRef]
- Park, E.S.; Park, S.R.; Han, S.W.; Dong, J.Y.; Shin, J.S. Structural determinants for the non-canonical substrate specificity of the ω-transaminase from Paracoccus denitrificans. Adv. Synth. Catal. 2014, 356, 212–220. [Google Scholar] [CrossRef]
- Jia, D.X.; Peng, C.; Li, J.L.; Wang, F.; Liu, Z.Q.; Zheng, Y.G. Redesign of (R)-omega-transaminase and its application for synthesizing amino acids with bulky side chain. Appl. Biochem. Biotechnol. 2021, 193, 3624–3640. [Google Scholar] [CrossRef]
- Han, S.W.; Kim, J.; Cho, H.-S.; Shin, J.S. Active site engineering of ω-transaminase guided by docking orientation analysis and virtual activity screening. ACS Catal. 2017, 7, 3752–3762. [Google Scholar]
- Konia, E.; Chatzicharalampous, K.; Drakonaki, A.; Muenke, C.; Ermler, U.; Tsiotis, G.; Pavlidis, I.V. Rational engineering of Luminiphilus syltensis (R)-selective amine transaminase for the acceptance of bulky substrates. Chem. Commun. 2021, 57, 12948–12951. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Xu, F.; Yang, L.; Liu, H.; Xu, X.; Wang, H.; Wei, D. Engineering the large pocket of an (S)-selective transaminase for asymmetric synthesis of (S)-1-amino-1-phenylpropane. Catal. Sci. Technol. 2021, 11, 2461–2470. [Google Scholar]
- Abraham, R.M.; Rocco, D.; Irina, P.; Sanjay, K.; David, S.; Sachin, P. Improved activity and thermostability of (S)-aminotransferase by error-prone polymerase chain reaction for the production of a chiral amine. Biochem. Eng. J. 2007, 37, 246–255. [Google Scholar]
- Meng, Q.; Ramírez-Palacios, C.; Capra, N.; Hooghwinkel, M.E.; Thallmair, S.; Rozeboom, H.J.; Thunnissen, A.M.W.H.; Wijma, H.J.; Marrink, S.J.; Janssen, D.B. Computational redesign of an ω-transaminase from Pseudomonas jessenii for asymmetric synthesis of enantiopure bulky amines. ACS Catal. 2021, 11, 10733–10747. [Google Scholar]
- Li, F.; Du, Y.; Liang, Y.; Wei, Y.; Zheng, Y.; Yu, H. Redesigning an (R)-selective transaminase for the efficient synthesis of pharmaceutical N-heterocyclic amines. ACS Catal. 2023, 13, 422–432. [Google Scholar]
- Pavlidis, I.V.; Weiß, M.S.; Genz, M.; Spurr, P.; Hanlon, S.P.; Wirz, B.; Iding, H.; Bornscheuer, U.T. Identification of (S)-selective transaminases for the asymmetric synthesis of bulky chiral amines. Nat. Chem. 2016, 8, 1076–1082. [Google Scholar]
- Weiß, M.S.; Pavlidis, I.V.; Spurr, P.; Hanlon, S.P.; Wirz, B.; Iding, H.; Bornscheuer, U.T. Protein-engineering of an amine transaminase for the stereoselective synthesis of a pharmaceutically relevant bicyclic amine. Org. Biomol. Chem. 2016, 14, 10249–10254. [Google Scholar]
- Weiß, M.S.; Pavlidis, I.V.; Spurr, P.; Hanlon, S.P.; Wirz, B.; Iding, H.; Bornscheuer, U.T. Amine transaminase engineering for spatially bulky substrate acceptance. ChemBioChem 2017, 18, 1022–1026. [Google Scholar]
- Ao, Y.F.; Pei, S.; Xiang, C.; Menke, M.J.; Shen, L.; Sun, C.; Dörr, M.; Born, S.; Höhne, M.; Bornscheuer, U.T. Structure- and data-driven protein engineering of transaminases for improving activity and stereoselectivity. Angew. Chem. Int. Ed. 2023, 62, e202301660. [Google Scholar]
- Menke, M.J.; Ao, Y.F.; Bornscheuer, U.T. Practical machine learning-assisted design protocol for protein engineering: Transaminase engineering for the conversion of bulky substrates. ACS Catal. 2024, 14, 6462–6469. [Google Scholar]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, X.; He, Q.; Chen, H.; Cai, W.; Xu, L.; Zhang, X.; Zhu, N.; Feng, S. Advances in the Molecular Modification of Microbial ω-Transaminases for Asymmetric Synthesis of Bulky Chiral Amines. Microorganisms 2025, 13, 820. https://doi.org/10.3390/microorganisms13040820
Gao X, He Q, Chen H, Cai W, Xu L, Zhang X, Zhu N, Feng S. Advances in the Molecular Modification of Microbial ω-Transaminases for Asymmetric Synthesis of Bulky Chiral Amines. Microorganisms. 2025; 13(4):820. https://doi.org/10.3390/microorganisms13040820
Chicago/Turabian StyleGao, Xinxing, Qingming He, Hailong Chen, Wangshui Cai, Long Xu, Xin Zhang, Nianqing Zhu, and Shoushuai Feng. 2025. "Advances in the Molecular Modification of Microbial ω-Transaminases for Asymmetric Synthesis of Bulky Chiral Amines" Microorganisms 13, no. 4: 820. https://doi.org/10.3390/microorganisms13040820
APA StyleGao, X., He, Q., Chen, H., Cai, W., Xu, L., Zhang, X., Zhu, N., & Feng, S. (2025). Advances in the Molecular Modification of Microbial ω-Transaminases for Asymmetric Synthesis of Bulky Chiral Amines. Microorganisms, 13(4), 820. https://doi.org/10.3390/microorganisms13040820