
The Role of Membrane-Bound Extracellular Vesicles During Co-Stimulation and Conjugation in the Ciliate Tetrahymena thermophila

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Methods
2.1. Cell Culture
2.2. Pairing Assay
2.3. Preparation for Thin-Section TEM
2.4. Freeze-Substitution TEM
2.5. Electron Tomography
2.6. Negative Staining
2.7. TEM on Pelleted EMVs
2.8. EMV Isolation by Differential Ultra-Centrifugation
2.9. EMV Size Analysis
2.10. Mass Spectrometry of EMV Contents
2.10.1. Protein Extraction
2.10.2. In-Solution Proteolytic Digestion
2.10.3. Mass Spectrometry
2.10.4. Database Search
2.11. RNA Isolation and Size Analysis for cEMVs from Wild-Type Co-Stimulated Cells
2.12. RNA Isolation and Size Analysis for jEMVs from HAP2D 4 h Mating Pairs
2.13. Dynasore Treatment for Pairing Assay
3. Results
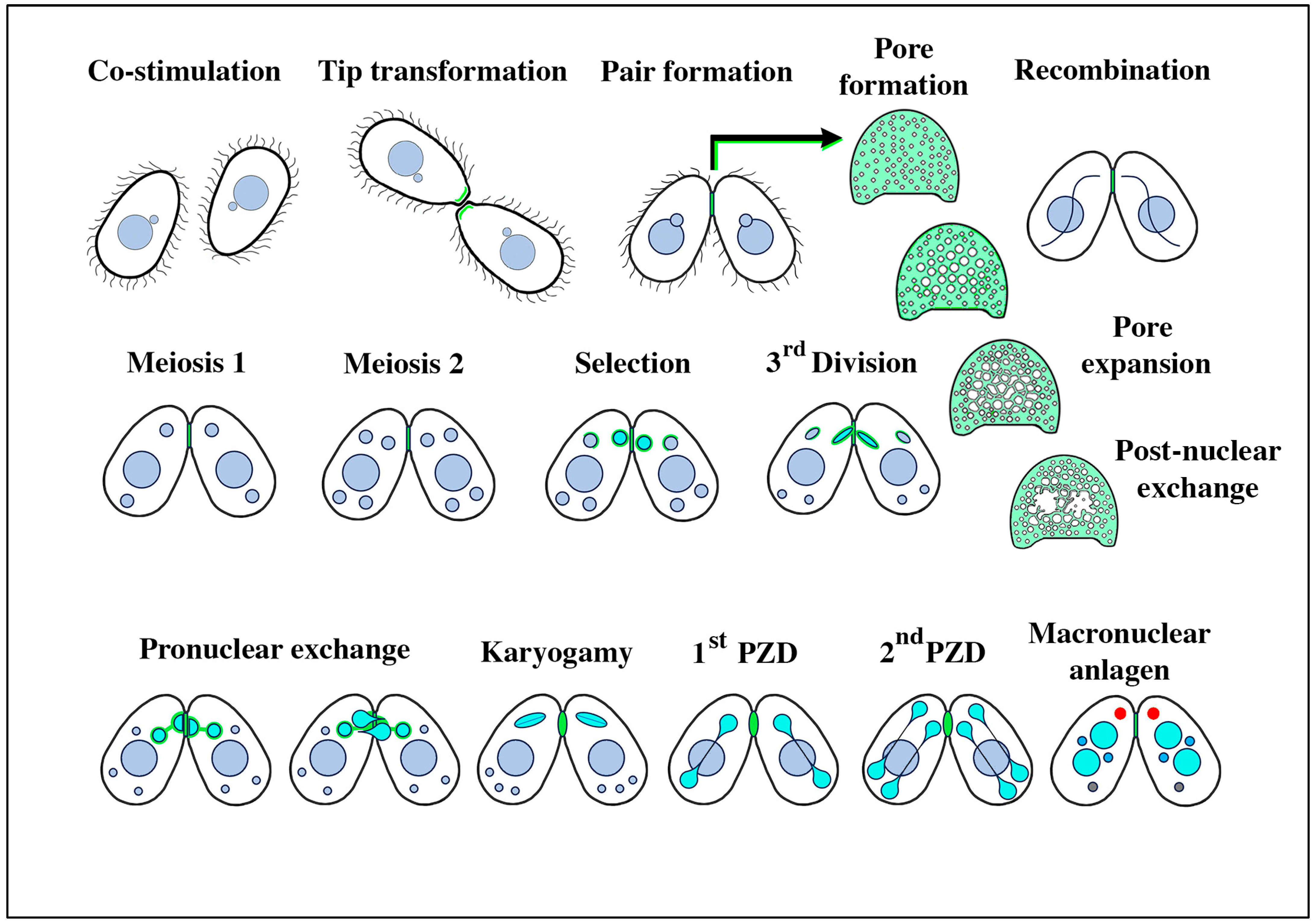
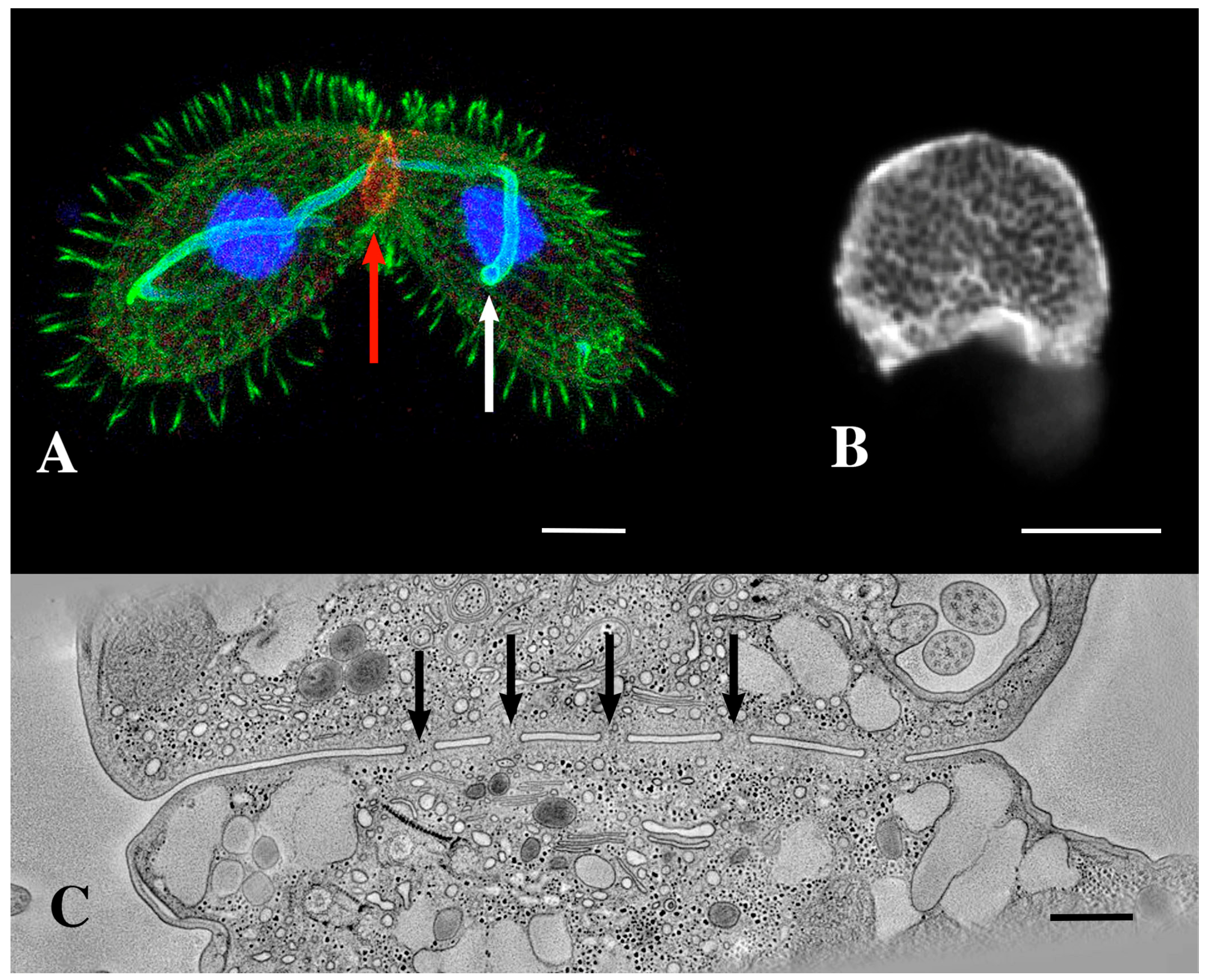
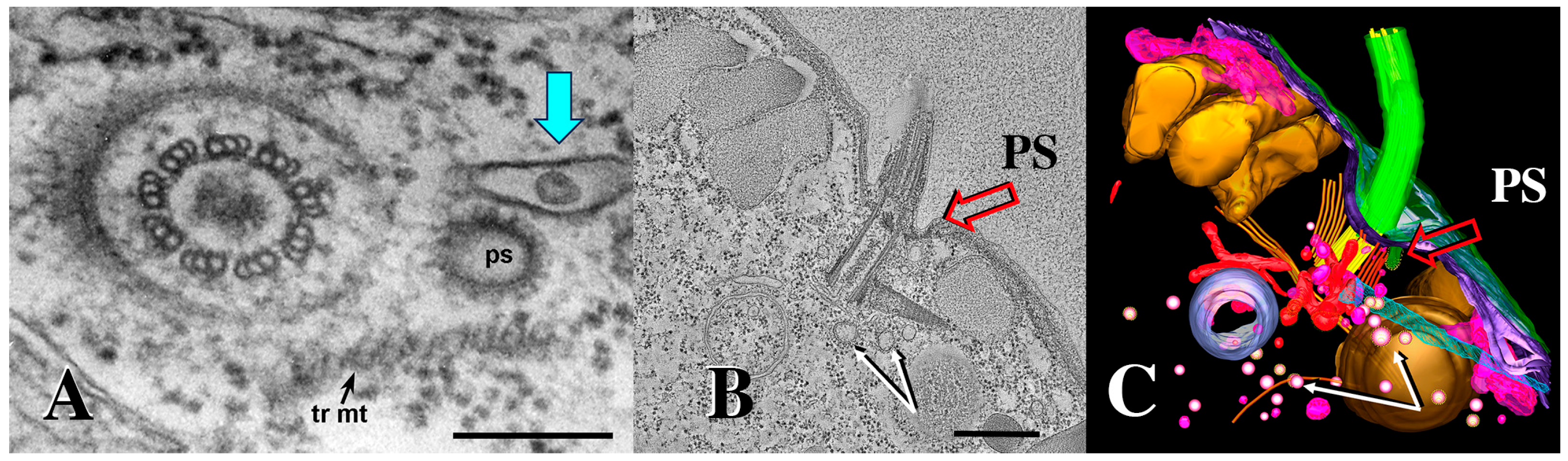
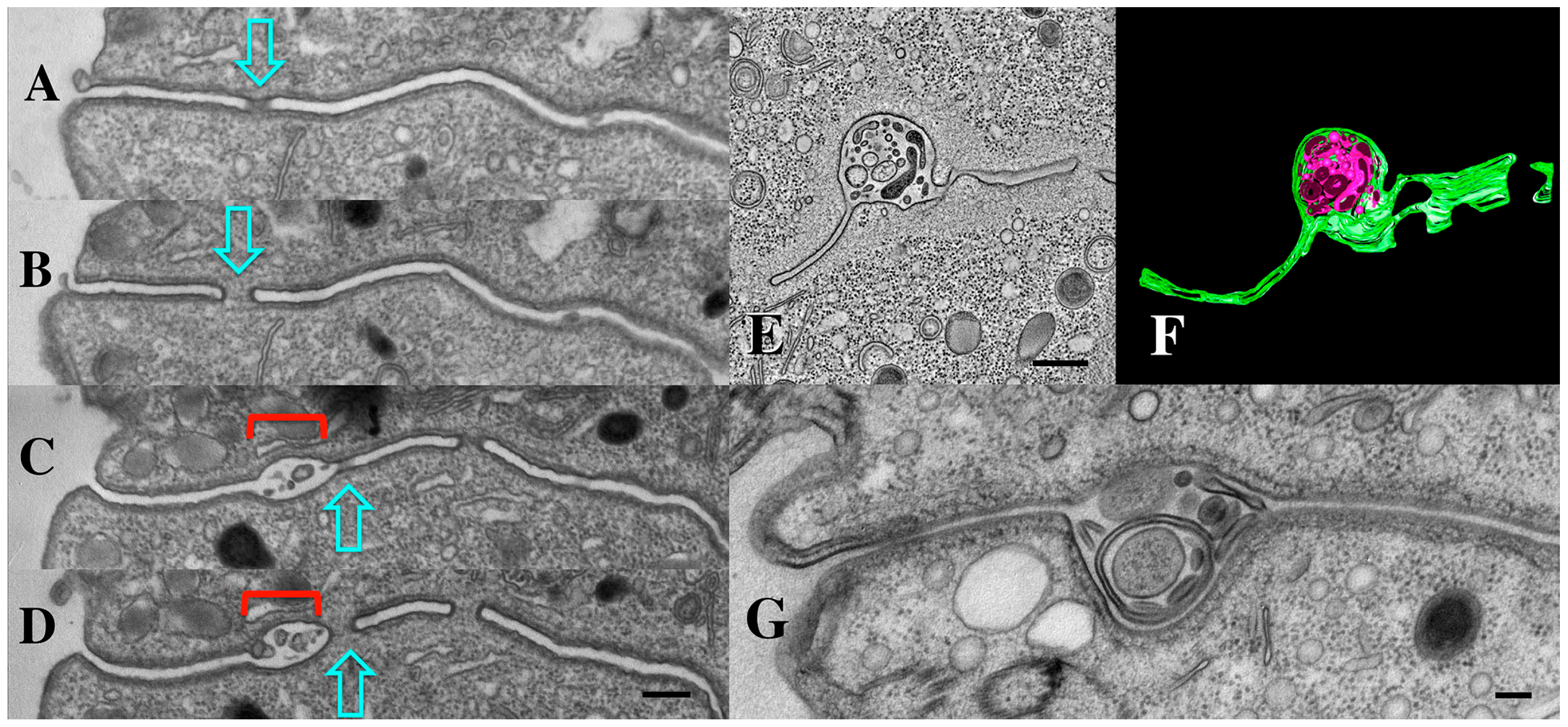
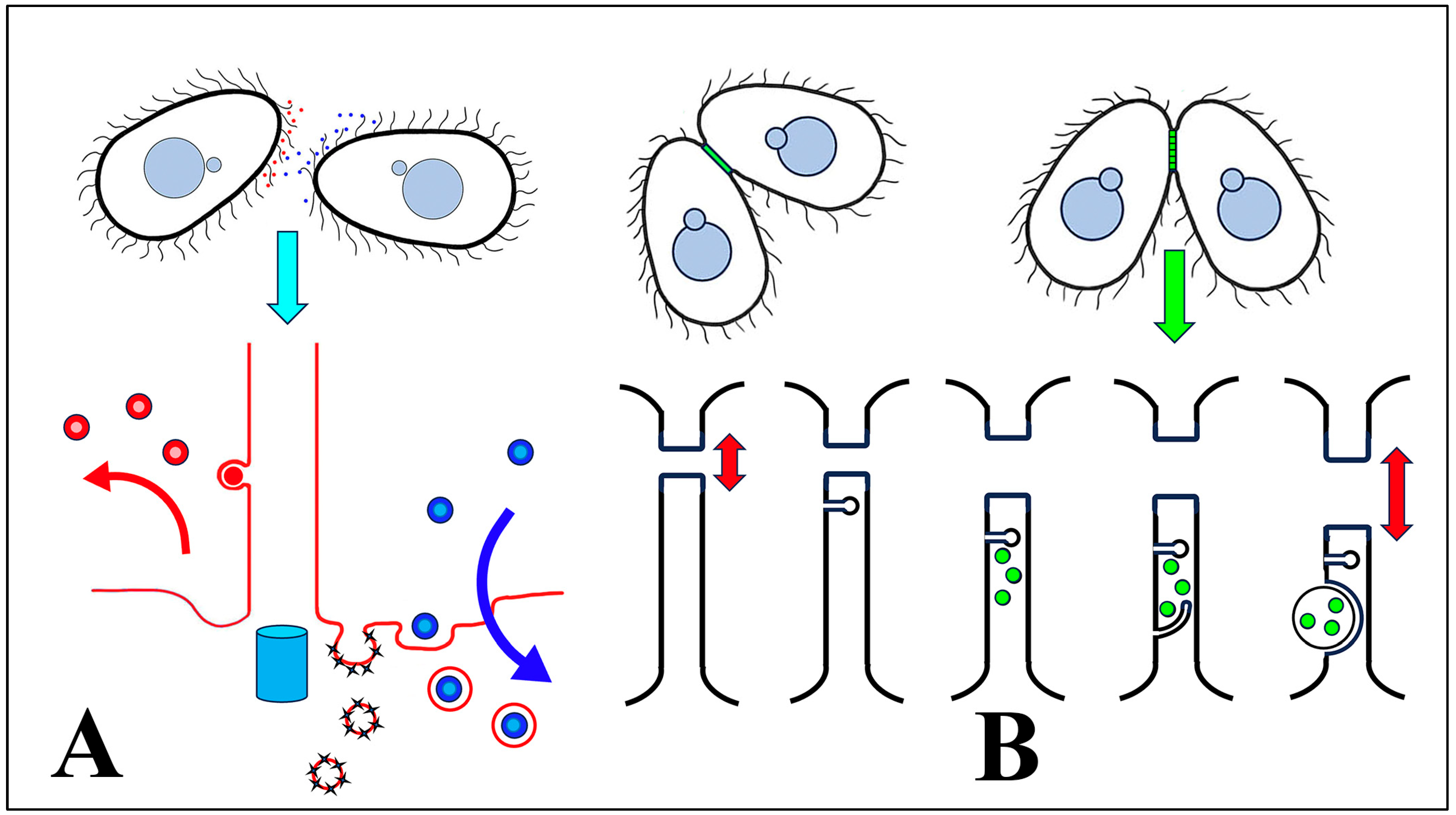
3.1. Membrane-Bound Vesicles Are Shed from Cilia During Co-Stimulation and into the Mating Junction During Conjugation and Membrane Remodeling
3.2. The Wild-Type cEMV Proteome and the HAP2Δ jEMV Transcriptome
3.3. EMVs Harvested from Both Co-Stimulating Cells and Mating Meiotic Cells Are Rich in RNA
3.4. EMVs Are Active in Conjugation
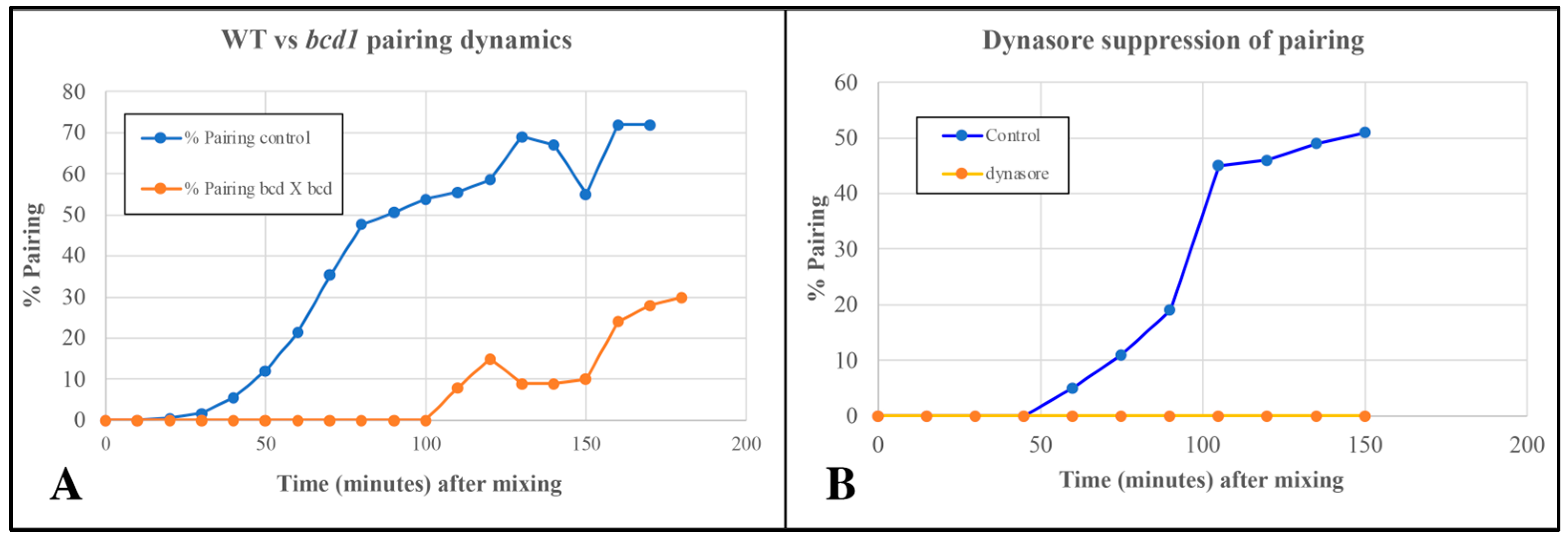
3.5. Endocytosis May Be Necessary for Pairing in Tetrahymena
3.6. EMVs Become Enriched in the ‘Ciliary Pockets’ in bcd1 Mutants Undergoing Conjugation
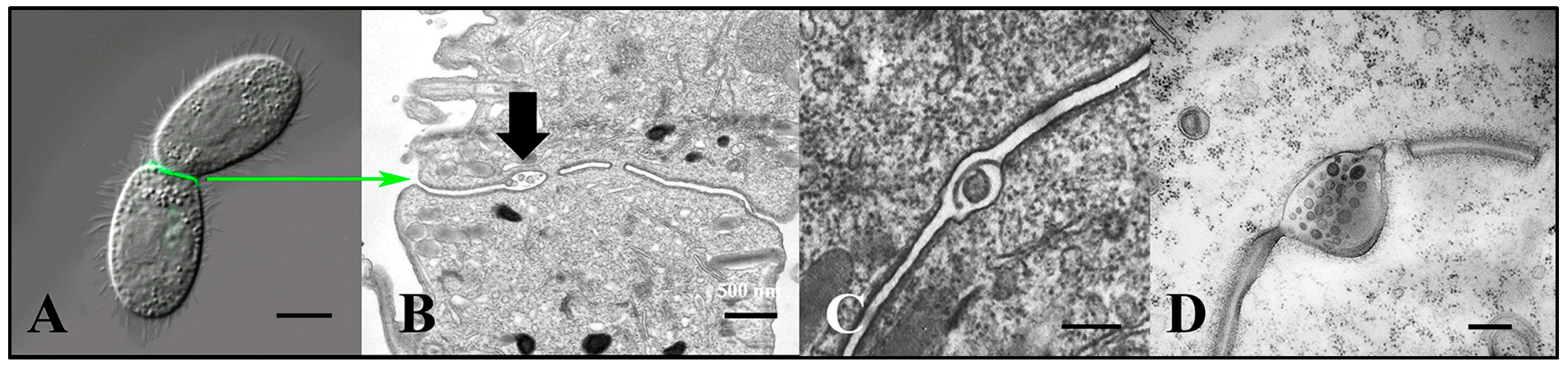
3.7. Tetrahymena Shed Membrane Bound Vesicles Directly into the Mating Junction Forming ‘Junction EMVs’ (jEMVs) That May Then Be Internalized by Macro-Pinocytosis
3.8. Endocytosis and Membrane Fusion Influence Vesicle Shedding at the Mating Junction
4. Discussion
4.1. Membrane Vesicle Shedding Occurs in Ciliates
4.2. Shed Micro-Vesicle Contents from Tetrahymena Resemble Micro-Vesicle Proteomes from Other (Distantly Related) Phyla
4.3. Proteins Involved in ‘Ectosome’ Formation Are Present in the Tetrahymena EMV Proteome
4.4. A Role for the Proteasome
4.5. Membrane Vesicles Trigger Pairing in Ciliates
4.6. Extracellular Micro-Vesicles Shed from Tetrahymena Cilia (cEMVs) Resemble the ‘Factor Activate in Conjugation’ (FAC) Described by Jason Wolfe
4.7. cEMVs Serve as Mobile Platforms, Expanding Surface Presentation of MT Proteins During Brush-By Encounters
4.8. Tetrahymena Conjugation Requires Active Endocytosis
4.9. A Potential Role for sRNA in cEMV Signaling
4.10. A Possible Role for jEMVs During Membrane Remodeling
4.11. A Possible Role for jEMVs in scnRNA Elimination
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Guttery, D.S.; Zeeshan, M.; Holder, A.A.; Tromer, E.C.; Tewari, R. Meiosis in Plasmodium: How does it work? Trends Parasitol. 2023, 39, 812–821. [Google Scholar] [CrossRef] [PubMed]
- Figueroa, R.I.; Howe-Kerr, L.I.; Correa, A.M.S. Direct evidence of sex and a hypothesis about meiosis in Symbiodiniaceae. Sci. Rep. 2021, 11, 18838. [Google Scholar] [CrossRef] [PubMed]
- Orias, E. Ciliate conjugation. In The Molecular Biology of Ciliated Protozoa; Gall, J.G., Ed.; Academic Press: London, UK, 1986; pp. 45–84. [Google Scholar]
- Cole, E.; Sugai, T. Developmental progression of Tetrahymena through the cell cycle and conjugation. Methods Cell Biol. 2012, 109, 177–236. [Google Scholar]
- Ruehle, M.D.; Orias, E.; Pearson, C.G. Tetrahymena as a unicellular model eukaryote: Genetic and genomic tools. Genetics 2016, 203, 649–665. [Google Scholar] [CrossRef] [PubMed]
- Prescott, D.M. The DNA of ciliated protozoa. Microbiol. Rev. 1994, 58, 233–267. [Google Scholar] [CrossRef]
- Cervantes, M.D.; Hamilton, E.P.; Xiong, J.; Lawson, M.J.; Yuan, D.; Hadjithomas, M.; Miao, W.; Orias, E. Selecting One of Several Mating Types through Gene Segment Joining and Deletion in Tetrahymena thermophila. PLoS Biol. 2013, 11, e1001518. [Google Scholar] [CrossRef]
- Bruns, P.J.; Brussard, T.B. Pair formation in Tetrahymena pyriformis, an inducible developmental system. J. Exp. Zool. 1974, 188, 337–344. [Google Scholar] [CrossRef]
- Bruns, P.J.; Palestine, R.F. Costimulation in Tetrahymena pyriformis: A developmental interaction between specially prepared cells. Develop. Biol. 1975, 42, 75–83. [Google Scholar] [CrossRef]
- Finley, M.J.; Bruns, P.J. Costimulation in Tetrahymena. II. A non-specific response to heterotypic cell-cell interactions. Dev. Biol. 1980, 79, 81–94. [Google Scholar] [CrossRef]
- Takahashi, M. Does fluid have any function for mating in Tetrahymena pyriformis. In Science Reports of the Tohoku University; Fourth Series (Biology) Maruzen Company Ltd.: Tokyo/Sendai, Japan, 1973; Volume 36, pp. 223–229. [Google Scholar]
- Miyake, A. Cell interaction in conjugation of ciliates. Curr. Top. Microbiol. Immunol. 1974, 64, 49–77. [Google Scholar]
- Wolfe, J.; Grimes, G.W. Tip transformation in Tetrahymena: A morphogenetic response to interactions between mating types. J. Protozool. 1979, 26, 82–89. [Google Scholar] [CrossRef]
- Allewell, J.M.; Oles, J.; Wolfe, J. A physicochemical analysis of conjugation in Tetrahymena pyriformis. Exp. Cell Res. 1976, 97, 394–405. [Google Scholar] [PubMed]
- Allewell, N.M.; Wolfe, J. A kinetic analysis of the memory of a developmental interaction: Mating interactions in Tetrahymena Pyriformis. Exp. Cell Res. 1977, 109, 15–24. [Google Scholar]
- Adair, W.S.; Wolfe, J. Induced alteration in uptake properties of Tetrahymena and its association with the development of mating competency. J. Cell. Physiol. 1977, 92, 77–90. [Google Scholar]
- Adair, W.S.; Barker, R.; Turner, R.S., Jr.; Wolfe, J. Demonstration of a cell-free factor involved in cell interactions during mating in Tetrahymena. Nature 1978, 274, 54–55. [Google Scholar]
- Wolfe, J.; Turner, R., Jr.; Barker, R.; Adair, W.S. The need for an extracellular component for cell pairing in Tetrahymena. Exp. Cell Res. 1979, 121, 27–30. [Google Scholar]
- Wolfe, J.; Meal, K.J.; Soiffer, R. Limiting conditions for conjugation in Tetrahymena: Cellular development and factor active in conjugation. J. Exp. Zool. 1980, 212, 37–46. [Google Scholar] [CrossRef]
- Cole, E.S.; Maier, W.; Joachimiak, E.; Jiang, Y.Y.; Lee, C.; Collet, E.; Chmelik, C.; Romero, D.P.; Chalker, D.; Alli, N.K.; et al. The Tetrahymena bcd1 mutant implicates endosome trafficking in ciliate, cortical pattern formation. Mol. Biol. Cell 2023, 34, ar82. [Google Scholar] [CrossRef]
- Snell, W.J.; Kroop, S.A.; Rosenbau, J.L. Characterization of adhesive substances on surface of Chlamydomonas gamete flagella. J. Cell Biol. 1973, 59, A327. [Google Scholar]
- Bergman, K.; Goodenough, U.W.; Goodenough, D.A.; Jawitz, J.; Martin, H. Gametic differentiation in Chlamydomonas reinhardtii. II. Flagellar membranes and the agglutination reaction. J. Cell Biol. 1975, 67, 606–622. [Google Scholar]
- Cao, M.; Ning, J.; Hernandez-Lara, C.I.; Belzile, O.; Wang, Q.; Dutcher, S.K.; Liu, Y.; Snell, W.J. Uni-directional ciliary membrane protein trafficking by a cytoplasmic retrograde IFT motor and ciliary ectosome shedding. eLife 2015, 4, e05242. [Google Scholar] [CrossRef] [PubMed]
- Wood, C.R.; Rosenbaum, J.L. Ciliary ectosomes: Transmissions from the cell’s antenna. Trends Cell Biol. 2015, 25, 276–285. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, J. The conjugation junction of Tetrahymena: Its structure and development. J. Morphol. 1982, 172, 159–178. [Google Scholar] [CrossRef] [PubMed]
- Chiba, Y.; Takenaka, Y.; Haga, N. Identification and Characterization of the Gene Responsible for the O3 Mating Type Substance in Paramecium caudatum. Microorganisms 2024, 12, 588. [Google Scholar] [CrossRef]
- Yan, G.; Ma, Y.; Wang, Y.; Zhang, J.; Cheng, H.; Tan, F.; Wang, S.; Zhang, D.; Xiong, J.; Yin, P.; et al. A seven-sex species recognizes self and non-self mating-type via a novel protein complex. eLife 2024, 13, RP93770. [Google Scholar] [CrossRef]
- Janetopoulos, C.; Cole, E.; Smothers, J.F.; Allis, C.D.; Aufderheide, K.J. The conjusome: A novel structure in Tetrahymena found only during sexual reorganization. J. Cell Sci. 1999, 112, 1003–1011. [Google Scholar] [CrossRef]
- Ma, Y.; Yan, G.; Han, X.; Zhang, J.; Xiong, J.; Miao, W. Sexual cell cycle initiation is regulated by CDK19 and CYC9 in Tetrahymena thermophila. J. Cell Sci. 2020, 133, jcs235721. [Google Scholar] [CrossRef]
- Ma, Y.; Yan, G.; Zhang, J.; Xiong, J.; Miao, W. Cip1, a CDK regulator, determines heterothallic mating or homothallic selfing in a protist. Proc. Natl. Acad. Sci. USA 2024, 121, e2315531121. [Google Scholar] [CrossRef]
- Cole, E.S.; Cassidy-Hanley, D.; Pinello, J.F.; Zeng, H.; Hsueh, M.; Kolbin, D.; Ozzello, C.; Giddings, T.; Winey, M.; Clark, T.G. Function of the Male-Gamete-Specific Fusion Protein HAP2 in a Seven-Sexed Ciliate. Curr. Biol. 2014, 24, 2168–2173. [Google Scholar] [CrossRef]
- Cole, E.S.; Dmytrenko, O.; Chmelik, C.J.; Li, M.; Christensen, T.A.; Macon, E.P.; Nilsson, H.J.; Blower, R.J.; Reuter, T.G.; Beckman, J.P.; et al. Restoration of cellular integrity following ‘ballistic’ pronuclear exchange during Tetrahymena conjugation. Dev. Biol. 2018, 444, 33–40. [Google Scholar] [CrossRef]
- Jiang, Y.-Y.; Maier, W.; Baumeister, R.; Joachimiak, E.; Ruan, Z.; Kannan, N.; Clarke, D.; Louka, P.; Guha, M.; Frankel, J.; et al. Two antagonistic hippo signaling circuits set the division plane at the medial position in the ciliate tetrahymena. Genetics 2019, 211, 651–663. [Google Scholar] [PubMed]
- Cole, E.S.; Giddings, T.H., Jr.; Ozzello, C.; Winey, M.; O’Toole, E.; Orias, J.; Hamilton, E.; Guerrier, S.; Ballard, A.; Aronstein, T. Membrane dynamics at the nuclear exchange junction during early mating (one to four hours) in the ciliate Tetrahymena thermophila. Eukaryot. Cell 2015, 14, 116–127. [Google Scholar] [PubMed]
- Pinello, J.F.; Lai, A.L.; Millet, J.K.; Cassidy-Hanley, D.; Freed, J.H.; Clark, T.G. Structure-Function Studies Link Class II Viral Fusogens with the Ancestral Gamete Fusion Protein HAP2. Curr. Biol. 2017, 27, 651–660. [Google Scholar] [CrossRef]
- Pinello, J.F.; Clark, T.G. HAP2-mediated gamete fusion: Lessons from the world of unicellular eukaryotes. Front. Cell Dev. Biol. 2022, 9, 807313. [Google Scholar] [CrossRef]
- Pinello, J.F.; Loidl, J.; Seltzer, E.S.; Cassidy-Hanley, D.; Kolbin, D.; Abdelatif, A.; Rey, F.A.; An, R.; Newberger, N.J.; Bisharyan, Y.; et al. Novel requirements for HAP2/GCS1-mediated gamete fusion in Tetrahymena. iScience 2024, 27, 110146. [Google Scholar] [CrossRef]
- Wong, J.L.; Johnson, M.A. Is HAP2-GCS1 an ancestral gamete fusogen? Trends Cell Biol. 2010, 20, 134–141. [Google Scholar]
- Clark, T. HAP2/GCS1: Mounting evidence of our true biological EVE? PLoS Biol. 2018, 16, e3000007. [Google Scholar]
- Mulcahy, L.A.; Pink, R.C.; Carter, D.R.F. Routes and mechanisms of extracellular vesicle uptake. J. Extracell. Vesicles 2014, 3, 24641. [Google Scholar]
- Giddings, T.H.; Meehl, J.B.; Pearson, C.G.; Winey, M. Electron Tomography and Immuno-labeling of Tetrahymena thermophila Basal Bodies. Method. Cell Biol. 2010, 96, 117–141. [Google Scholar]
- Meehl, J.B.; Giddings, T.H., Jr.; Winey, M. High Pressure Freezing, Electron Microscopy, and Immuno-Electron Microscopy of Tetrahymena thermophila Basal Bodies. In Cytoskeleton Methods and Protocols; Gavin, R.H., Ed.; Humana Press Inc.: Totowa, NJ, USA, 2009; pp. 227–241. [Google Scholar]
- Mastronarde, D.N. Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. 2005, 152, 36–51. [Google Scholar] [CrossRef]
- Hacker, C.; Asadi, J.; Pliotas, C.; Ferguson, S.; Sherry, L.; Marius, P.; Tello, J.; Jackson, D.; Naismith, J.; Lucocq, J.M. Nanoparticle suspensions enclosed in methylcellulose: A new approach for quan-tifying nanoparticles in transmission electron microscopy. Sci. Rep. 2016, 6, 25275. [Google Scholar]
- Thery, C.; Amigorena, S.; Raposo, G.; Clayton, A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr. Protoc. Cell Biol. 2006, 30, 3–22. [Google Scholar]
- Dobin, A.; Davis, C.A.; Schlesigner, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR:ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar]
- Wang, L.; Wang, S.; Li, W. RSeQC: Quality control of RNA-seq experiments. Bioinformatics 2012, 28, 2184–2185. [Google Scholar]
- Koppen, T.; Weckmann, A.; Muller, S.; Staubach, S.; Bloch, W.; Dohmen, R.J.; Schwientek, T. Proteomics analyses of microvesicles released by Drosophila Kc167 and S2 cells. Proteomics 2011, 11, 4397–4410. [Google Scholar]
- Kim, D.K.; Kang, B.; Kim, O.Y.; Choi, D.S.; Lee, J.; Kim, S.R.; Go, G.; Yoon, Y.J.; Kim, J.H.; Jang, S.C.; et al. EVpedia: An integrated database of high-throughput data for systemic analyses of extracellular vesicles. J. Extracell. Vesicles 2013, 2, 20384. [Google Scholar]
- Choi, D.S.; Kim, D.K.; Kim, Y.K.; Gho, Y.S. Proteomics of extracellular vesicles: Exosomes and ectosomes. Mass. Spectrom. Rev. 2015, 34, 474–490. [Google Scholar] [CrossRef]
- Dovrat, S.; Caspi, M.; Zilberberg, A.; Lahav, L.; Firsow, A.; Gur, H.; Rosin-Arbesfeld, R. 14-3-3 and β-catenin are secreted on extracellular vesicles to activate the oncogenic Wnt pathway. Mol. Oncol. 2014, 8, 894–911. [Google Scholar]
- Andreu, Z.; Yáñez-Mó, M. Tetraspanins in extracellular vesicle formation and function. Front. Immunol. 2014, 5, 442. [Google Scholar] [CrossRef] [PubMed]
- Jankovičová, J.; Sečová, P.; Michalková, K.; Antalíková, J. Tetraspanins, more than markers of extracellular vesicles in reproducetion. Int. J. Mol. Sci. 2020, 21, 7568. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.R.; Collins, K. Starvation-induced cleavage of the tRNA anticodon loop in Tetrahymena thermophila. J. Biol. Chem. 2005, 280, 42744–42749. [Google Scholar] [CrossRef]
- Lee, S.R.; Collins, K. Two classes of endogenous small RNAs in Tetrahymena thermophila. Genes Dev. 2006, 20, 28–33. [Google Scholar] [CrossRef]
- Dentler, W. Ciliary and Flagellar Membrane Vesicle (Ectosome) Purification. Bio-Protocol 2014, 4, e1156. [Google Scholar] [CrossRef]
- Caron, F.; Meyer, E. Molecular basis of surface antigen variation in Paramecia. Annu. Rev. Microbiol. 1989, 43, 23–42. [Google Scholar] [CrossRef]
- Smith, D.L.; Berkowitz, M.S.; Potoczak, D.; Krause, M.; Raab, C.; Quinn, F.; Doerder, F.P. Characterization of the T, L, I, S, M and P cell surface (immobilization) antigens of Tetrahymena thermophila: Molecular weights, isoforms, and cross-reactivity of antisera. J. Protozool. 1992, 39, 420–428. [Google Scholar] [CrossRef]
- Clark, T.G.; Gao, Y.; Gaertig, J.; Wang, X.; Cheng, G. The I-antigens of Ichthyophthirius multifiliis are GPI-anchored proteins. J. Eukaryot. Microbiol. 2001, 48, 332–337. [Google Scholar] [CrossRef]
- Doerder, F.P. Differential expression of immobilization antigens in Tetrahymena thermophila. I. Genetic and epistatic relationships among recessive mutations which alter normal expression of i-antigens. Immunogenetics 1979, 9, 551–562. [Google Scholar] [CrossRef]
- Doerder, F.P. Differential expression of immobilization antigen genes in Tetrahymena thermophila, II. Reciprocal and non-reciprocal transfer of i-antigen during conjugation and expression of i-antigen genes during macronuclear development. Cell Differ. 1981, 10, 299–307. [Google Scholar] [CrossRef]
- Bisharyan, Y.; Clark, T.G. Calcium-dependent mitochondrial extrusion in ciliated protozoa. Mitochondrion 2011, 11, 909–918. [Google Scholar] [PubMed]
- Bisharyan, Y.T.C. Signaling Through GPI-Anchored Surface Antigens in Ciliates. In Biocommunication of Ciliates; Witzany, G., Nowacki, M., Eds.; Springer: Cham, Switzerland, 2016; pp. 139–157. [Google Scholar]
- Ko, Y.-G.; Thompson, G.A., Jr. Immobilization antigens from Tetrahymena thermophila are glycosyl-phosphatidylinositol-linked proteins. J. Protozool. 1992, 39, 719–723. [Google Scholar] [PubMed]
- Evans-Osses, I.; Reichembach, L.H.; Ramirez, M.I. Exosomes or microvesicles? Two kinds of extracellular vesicles with different routes to modify protozoan-host cell interaction. Parasitol. Res. 2015, 114, 3567–3575. [Google Scholar]
- Rojas, A.; Regev-Rudzki, N. Biogenesis of extracellular vesicles from the pathogen perspective: Trans-kingdom strategies for delivering messages. Curr. Opin. Cell Biol. 2024, 88, 102366. [Google Scholar] [CrossRef]
- Nievas, Y.R.; Coceres, V.M.; Midlej, V.; de Souza, W.; Benchimol, M.; Pereira-Neves, A.; Vashisht, A.A.; Wohlschlegel, J.A.; Johnson, P.J.; de Miguel, N. Membrane-shed vesicles from the parasite Trichomonas vaginalis: Characterization and their association with cell interaction. Cell. Mol. Life Sci. 2018, 75, 2211–2226. [Google Scholar]
- Twu, O.; de Miguel, N.; Lustig, G.; Stevens, G.C.; Vashisht, A.A.; Wohlschlegel, J.A.; Johnson, P.J. Trichomonas vaginalis exosomes deliver cargo to host cells and mediate hostratioparasite interactions. PLoS Pathog. 2013, 9, e1003482. [Google Scholar]
- Abdi, A.; Yu, L.; Goulding, D.; Rono, M.K.; Bejon, P.; Choudhary, J.; Rayner, J. Proteomic analysis of extracellular vesicles from a Plasmodium falciparum Kenyan clinical isolate defines a core parasite secretome. Wellcome Open Res. 2017, 2, 50. [Google Scholar]
- Evans-Osses, I.; Mojoli, A.; Monguió-Tortajada, M.; Marcilla, A.; Aran, V.; Amorim, M.; Inal, J.; Borràs, F.E.; Ramirez, M.I. Microvesicles released from Giardia intestinalis disturb host-pathogen response in vitro. J. Cell Biol. 2017, 96, 131–142. [Google Scholar] [CrossRef]
- Goodenough, U.W.; Heuser, J.E. Deep-etch Analysis of Adhesion Complexes Between Gametic Flagellar Membranes of Chlamydomonas reinhardtii (Chlorophyceae). J. Phycol. 1999, 35, 756–767. [Google Scholar] [CrossRef]
- Hugel, B.; Martinez, M.C.; Kunzelmann, C.; Freyssinet, J.M. Membrane microparticles: Two sides of the coin. Physiology 2005, 20, 22–27. [Google Scholar]
- Xu, P.; Baldridge, R.D.; Chi, R.J.; Burd, C.G.; Graham, T.R. Phosphatidylserine flipping enhances membrane curvature and negative charge required for vesicular transport. J. Cell Biol. 2013, 202, 875–886. [Google Scholar] [CrossRef] [PubMed]
- Kalra, H.; Drummen, G.P.; Mathivanan, S. Focus on Extracellular Vesicles: Introducing the Next Small Big Thing. Int. J. Mol. Sci. 2016, 17, 170. [Google Scholar] [CrossRef] [PubMed]
- Pap, E.; Pallinger, E.; Pasztoi, M.; Falus, A. Highlights of a new type of intercellular communication: Microvesicle-based information transfer. Inflamm. Res. 2009, 58, 1–8. [Google Scholar] [PubMed]
- Cocucci, E.; Meldolesi, J. Ectosomes and exosomes: Shedding the confusion between extracellular vesicles. Trends Cell Biol. 2015, 25, 364–372. [Google Scholar]
- Nabhan, J.F.; Hu, R.; Oh, R.S.; Cohen, S.N.; Lu, Q. Formation and release of arrestin domain-containing protein 1-mediated microvesicles (ARMMs) at plasma membrane by recruitment of TSG101 protein. Proc. Natl. Acad. Sci. USA 2012, 109, 4146–4151. [Google Scholar] [CrossRef]
- Lai, R.C.; Tan, S.S.; Teh, B.J.; Sze, S.K.; Arslan, F.; de Kleijn, D.P.; Choo, A.; Lim, S.K. Proteolytic Potential of the MSC Exosome Proteome: Implications for an Exosome-Mediated Delivery of Therapeutic Proteasome. Int. J. Proteom. 2012, 2012, 971907. [Google Scholar]
- Bochmann, I.; Ebstein, F.; Lehmann, A.; Wohlschlaeger, J.; Sixt, S.U.; Kloetzel, P.M.; Dahlmann, B. T lymphocytes export proteasomes by way of microparticles: A possible mechanism for generation of extracellular proteasomes. J. Cell. Mol. Med. 2014, 18, 59–68. [Google Scholar]
- Zimmerman, S.W.; Manandhar, G.; Yi, Y.J.; Gupta, S.K.; Sutovsky, M.; Odhiambo, J.F.; Powell, M.D.; Miller, D.J.; Sutovsky, P. Sperm proteasomes degrade sperm receptor on the egg zona pellucida during mammalian fertilization. PLoS ONE 2011, 6, e17256. [Google Scholar]
- Sawada, H.; Sakai, N.; Abe, Y.; Tanaka, E.; Takahashi, Y.; Fujino, J.; Kodama, E.; Takizawa, S.; Yokosawa, H. Extracellular ubiquitination and proteasome-mediated degradation of the ascidian sperm receptor. Proc. Natl. Acad. Sci. USA 2002, 99, 1223–1228. [Google Scholar] [CrossRef]
- Sakai, N.; Sawada, M.T.; Sawada, H. Non-traditional roles of ubiquitin-proteasome system in fertilization and gametogenesis. Int. J. Biochem. Cell Biol. 2004, 36, 776–784. [Google Scholar]
- Metz, C.B. Induction of meiosis and “pseudo-selfing” in Paramecium aurelia of one mating type by dead animals of opposite type. Anat. Rec. 1946, 96, 35–36. [Google Scholar]
- Metz, C.B. Induction of pseudo selfing and meiosis in Paramecium aurelia by formalin killed animals of opposite mating type. J. Exp. Zool. 1947, 105, 115–139. [Google Scholar] [PubMed]
- Metz, C.B. Mating substances and the physiology of fertilization in ciliates. In Sex in Microorganisms; Wenrich, D.H., Ed.; AAAS: Washington, DC, USA, 1954; pp. 284–334. [Google Scholar]
- Hiwatashi, K. Paramecium. In Fertilization; Academic Press: New York, NY, USA, 1969; Volume 2, pp. 255–293. [Google Scholar]
- Fukushi, T.; Hiwatashi, K. Preparation of mating reactive cilia from Paramecium caudatum by MnCl2. J. Protozool. 1970, 17, 1. [Google Scholar]
- Miyake, A. Induction of conjugation by cell-free preparations in Paramecium multimicronucleatum. Science 1964, 146, 1583–1585. [Google Scholar] [CrossRef]
- Kitamura, A.; Hiwatashi, K. Mating-reactive membrane-vesicles from cilia of Paramecium caudatum. J. Cell Biol. 1976, 69, 736–740. [Google Scholar] [CrossRef]
- Kitamura, A.; Hiwatashi, K. Reconstitution of mating active membrane-vesicles in Paramecium. Exp. Cell Res. 1980, 125, 486–489. [Google Scholar]
- Watanabe, T. Ciliary membranes and mating substances in Paramecium caudatum. J. Protozool. 1977, 24, 426–429. [Google Scholar] [CrossRef]
- Cole, E.S. Cell-Cell Interactions Leading to Establishment of a Mating Junction in Tetrahymena and Paramecium, Two “Contact-Mediated” Mating Systems. In Biocommunication of Ciliates; Witzany, G., Nowacki, M., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 195–220. [Google Scholar]
- Love, B.; Rotheim, M.B. Cell surface interactions in conjugation—Tetrahymena ciliary membrane vesicles. Mol. Cell. Biol. 1984, 4, 681–687. [Google Scholar]
- Wang, S. P-Type Atpases in Tetrahymena thermophila; The Ohio State University: Columbus, OH, USA, 1996. [Google Scholar]
- Wang, S.; Takeyasu, K. Primary structure and evolution of the ATP-binding domains of the P-type ATPases in Tetrahymena thermophila. Am. J. Physiol. Cell Physiol. 1997, 272, C715–C728. [Google Scholar]
- Elde, N.C.; Morgan, G.; Winey, M.; Sperling, L.; Turkewitz, A.P. Elucidation of clathrin-mediated endocytosis in Tetrahymena reveals an evolutionarily convergent recruitment of dynamin. PLoS Genet 2005, 1, e52. [Google Scholar]
- Rotheim, M.B.; Love, B. Role for endocytosis in conjugation in Tetrahymena. Mol. Cell. Biol. 1982, 2, 633–637. [Google Scholar] [PubMed]
- Couvillion, M.T.; Lee, S.R.; Hogstad, B.; Malone, C.D.; Tonkin, L.A.; Sachidanandam, R.; Hannon, G.J.; Collins, K. Sequence, biogenesis, and function of diverse small RNA classes bound to the Piwi family proteins of Tetrahymena thermophila. Genes Dev. 2009, 23, 2016–2032. [Google Scholar] [PubMed]
- Howard-Till, R.A.; Yao, M.-C. Induction of gene silencing by hairpin RNA expression in Tetrahymena thermophila reveals a second small RNA pathway. Mol. Cell. Biol. 2006, 26, 8731–8742. [Google Scholar]
- Cole, E.S. Conjugal blocks in Tetrahymena pattern mutants and their cytoplasmic rescue: I. broadened cortical domains (bcd). Dev. Biol. 1991, 148, 403–419. [Google Scholar] [CrossRef]
- Mochizuki, K.; Gorovsky, M.A. Studies on an RNAi-like mechanism of germline DNA elimination in Tetrahymena thermophila. Dev. Biol. 2005, 283, 576. [Google Scholar]
- Chalker, D.L.; Yao, M.C. DNA elimination in ciliates: Transposon domestication and genome surveil-lance. Annu. Rev. Genet. 2011, 45, 227–246. [Google Scholar]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cole, E.S.; Dmytrenko, O.; Li, M.; Krishnan, N.; Thorp, J.; Higgins, L.; Markowski, T.; Morgan, G.; O’Toole, E. The Role of Membrane-Bound Extracellular Vesicles During Co-Stimulation and Conjugation in the Ciliate Tetrahymena thermophila. Microorganisms 2025, 13, 803. https://doi.org/10.3390/microorganisms13040803
Cole ES, Dmytrenko O, Li M, Krishnan N, Thorp J, Higgins L, Markowski T, Morgan G, O’Toole E. The Role of Membrane-Bound Extracellular Vesicles During Co-Stimulation and Conjugation in the Ciliate Tetrahymena thermophila. Microorganisms. 2025; 13(4):803. https://doi.org/10.3390/microorganisms13040803
Chicago/Turabian StyleCole, Eric S., Oleksandr Dmytrenko, Mark Li, Neetij Krishnan, Josh Thorp, LeeAnn Higgins, Todd Markowski, Garry Morgan, and Eileen O’Toole. 2025. "The Role of Membrane-Bound Extracellular Vesicles During Co-Stimulation and Conjugation in the Ciliate Tetrahymena thermophila" Microorganisms 13, no. 4: 803. https://doi.org/10.3390/microorganisms13040803
APA StyleCole, E. S., Dmytrenko, O., Li, M., Krishnan, N., Thorp, J., Higgins, L., Markowski, T., Morgan, G., & O’Toole, E. (2025). The Role of Membrane-Bound Extracellular Vesicles During Co-Stimulation and Conjugation in the Ciliate Tetrahymena thermophila. Microorganisms, 13(4), 803. https://doi.org/10.3390/microorganisms13040803