1. Introduction
Citrus greening, otherwise known as Huanglongbing (HLB), is one of the most devastating citrus diseases in the world. First discovered in 1920, the disease migrated from Asia to Florida in 2005 and subsequently to other sub-tropical US states, including Texas, Louisiana, and California. It is caused by a difficult-to-culture Gram-negative endophytic bacterium of the
Alphaproteobacteria class called
Candidatum Liberibacter asiaticus, which is transmitted by a Sternorrhynchian insect vector—the Asian citrus psyllid,
Diaphorina citri [
1]. The Asian citrus psyllid is the most heat-tolerant of all three types, which also includes the Americanus and Africanus citrus psyllids, but the Asian type has been found extensively in Florida [
1]. Early symptoms of its infection on leaves include yellowing of the veins and adjacent tissues, followed by asymmetrical chlorosis—symptoms that resemble zinc and other mineral deficiencies. The disease leads to premature defoliation, dieback of twigs, decay of feeder rootlets and lateral roots, decline in vigor, and could ultimately lead to the death of the entire plant. Most affected trees have stunted growth, bear multiple off-season flowers (most of which fall off), and produce small, irregularly shaped fruit with a thick, pale peel that remains green at the bottom and tastes very bitter [
1,
2]. Citrus greening disease (CGD) is prevalent in 80% of groves within the citrus industry in South Florida. Annual losses between 30 and 100% have decimated the 10.8 billion-dollar industry. Since its introduction in 2005 in Miami-Dade, Florida, it has rapidly spread to more than 37 counties and nine other states. The rapid spread was possibly fueled by dysbiosis within the rhizosphere stemming from agricultural management practices that rely on the heavy use of agrochemicals among other factors.
Research on citrus root microbiomes to combat the citrus greening devastation abounds. The use of microbiome-based solutions has produced mixed outcomes. Optimizing these interventions requires an understanding of the functional and taxonomic niches of core plant microbiomes. Understanding why the newly introduced Ca. L. asiaticus pathogen swept rapidly through most Florida Citrus groves is important for proposing solutions for future outbreaks. Knowledge about plant population immunity and the roles played by microbiomes is rapidly accumulating. The translational interpretations and applications of these study outcomes in specific practical situations are critical for sustainable agriculture.
It has long been known that HLB infection causes phloem disruption, sucrose accumulation, and plugging of the sieve pores linked to callose deposition and not HLB aggregates [
3]. The molecular basis of the disease is also well understood. Many plant genes are either upregulated (pathogenesis/stress response, phytohormones, sugar and protein metabolism, and several unknown genes) or downregulated (cell cycle, cell wall and lipid metabolism, and protein kinases) during infection, suggesting an intense perturbation of overall plant metabolism [
4]. Genes, such as the plant defense and disease-resistance response (PR) genes, may provoke a sort of “plant hypersensitive” or “chronic inflammation” damage, which is reflected in callose deposition in and around the phloem. Of greater note, perhaps, is that about 50% of PR genes were downregulated in sweet oranges by Ca.
Libericater sp infection [
5], while inoculating citrus plants with beneficial Burkholderia isolated from healthy citrus plants enhanced the expression of PR genes and salicylic acid-mediated resistance, demonstrating the high potential of biocontrol agents in controlling HLB infection. Further evidence comes from the observation that rhizospheric non-virulent
P. syringae induced the expression of the non-host resistance gene—NHO1 in
Arabidopsis sp., whereas the virulent strains of the same bacterium (Pst DC3000) were able to cause disease by suppressing the expression of the photo-immunity (NH01 plant defense mechanism) via a jasmonic acid (JA) signaling pathway [
6]. Other microorganisms reported to confer pathogen suppression in soils include bacteria (members of the Proteobacteria, Verrucomicrobia, and Firmicutes) and fungi—Ascomycota or
M. circinelloides,
P. oxalicum,
A. niger, and
A. flavus [
7,
8,
9].
Recent advances in disease progression implicate specific microorganisms associated with disease-induced dysbiosis and a microbe-mediated immune response model [
10]. There was a transition from beneficial microorganisms such as
Glomeromycota,
Camptohophora sp., and
Methylobacterium sp. to more competitive and pathogenic species such as
Fusarium sp.,
Giberella sp.,
Streptomyces sp. during heavy infection, and finally, to saprophytic species as the disease progressed to the later stages when the trees had almost died [
11]. Root exudates also provide nourishment for specific microbes, supporting plant immunity through inhibiting the growth of invasive pathogens [
12].
Current control methods for CGD involve the removal of psyllid vectors using insecticides [
13], chemotherapy treatments using antibiotics that target the HLB pathogen [
14], and the disinfection and immediate removal of infected plants. In larger and more commercial citrus groves, these control methods have been insufficient in stemming the spread of the disease [
13]. Genetic transformation of the citrus plant, the HLB pathogen, and the psyllid vector provide high expectations for the development of disease-resistant cultivars and/or amelioration of the bacterial pathogen and insect vector control [
13]. The use of a biocontrol agent—
Tamarixia radiata—has been explored in Texas, also showing great potential, where up to 90% of psyllids were reduced post-treatment [
15]. Genomic solutions are being explored, so solving the growing HLB challenge will entail a concerted multidisciplinary effort.
The plant microbiome, or phytobiome, plays an integral role in the overall health and disease suppression [
8,
16] of different agricultural and non-agricultural plants and is highly sensitive to changes in the environment and varying stress conditions [
17]. The rhizosphere, which consists of soil approximately 1 mm from the plant root surface, is one of the most diverse and enriched habitats for microbial life and consists of many beneficial and antagonistic plant-associated micro-organisms. The pivotal role played by these root microbiomes in plant survival and competition is emerging [
17], and the manipulation of the rhizobiome could have important implications for vegetation health and cover [
18]. Numerous studies using amplicon, metagenomic, and meta-transcriptomic sequencing analyses have assessed the effects of different environmental conditions and diseased states on the microbial community structure of the rhizosphere of a vast array of plants, including invasive plants, Arabidopsis, rice, soybean, and citrus [
8,
19,
20,
21]. As a part of the mandate of the International Citrus Microbiome Consortium, it is crucial to determine the effect of different citrus management strategies (herbicide, fertilizer treatment) on the citrus microbiome and develop methods to engineer the microbiome for sustainable plant production. Recent research has shown a consistent and core microbiome present in citrus varieties across the globe and may provide insights into how these microbiomes can be employed to increase sustainable crop production [
20].
In this study, we leveraged a very rare opportunity to access undisturbed citrus groves of the 1880s to capture potentially unknown microbiomes of citrus. The second goal was to evaluate the impact of routine citrus cultivation practices (which include copious application of agrochemicals) on root microbiomes by contrasting microbial communities on managed groves in the same vicinity with those of the undisturbed terrain. We then compared the profiles of 16S rDNA amplicon sequence variants (ASVs) with the prevalence of greening disease in the plants to infer the effect of dysbiosis on susceptibility to pathogens like Liberibacter asiaticus. Finally, we mined the data from the Global Citrus Consortium to determine the occurrence of published core citrus microbiomes in our study sites. While it might be too late to reverse the damage and rescue the Florida Citrus industry, data from this study would inform policy on soil health and sustainable management practices for other important agricultural crops like sugarcane. To the best of our knowledge, this is one of the first studies of rhizosphere microbiomes of historically undisturbed century-old citrus groves, free of anthropogenic impacts.
2. Materials and Methods
2.1. Site Description
Two varieties of sweet orange (Citrus sinensis)—Valencia and pineapple orange—along with sour orange (Citrus aurantium), grapefruit (Citrus paradisi), and sour lemon (Citrus limon) were assessed visually and at the molecular level for Candidatus L asiaticus infection throughout different groves in Central Florida. East and West Valencia oranges were located in the Hendry County grove (Site 1—26.6105° N, 81.0755° W) and were found to have visual foliar signs of Huanglongbing infection, with the West Valencia oranges showing greater signs of infection than East Valencia. Both East and West Valencia oranges were positive for Ca. L asiaticus. These groves had been cultivated for over 30 years with consistent herbicide and other chemical use (pers comm. McCoquerdale). Herbicide and fertilizer treatments involved the use of glyphosate (Roundupâ) applied at a rate of 0.125–0.37 lbs/acre and a commercial citrus fertilizer (6-4-6 NPK ratio) at an application rate of 120–200 lbs/acre.
Natural stands of grapefruit were located on Site 2, a historical, isolated, and non-cultivated hammock area in Hendry County, with fruits that had no visual symptoms of Ca. L
asiaticus infection. Site 3 was another isolated area in Arcadia, FL (27.2159° N, 81.8584° W), lacking anthropogenic influences with a grove of Greenmount pineapple oranges. On site 4, in Sebring, FL, wild-grown sour orange and sour lemon were found in Highland Hammock State Park and Florida Cracker Trail. These plants exhibited no signs of infection. A sample map of the sampling sites and their respective levels of infection is shown in
Figure 1. Root samples from different plant types were collected using the grab method, in duplicate, and sent immediately to the lab in a cooler with ice between November 2016 and February 2017. The roots were shaken to remove loosely adhering soil, while the rhizosphere soil was scraped off using forceps and used for the study.
2.2. DNA Extraction and Validation
Two grams of total rhizosphere soil sample, instead of 0.75 g (recommended by the manufacturer), were collected from the rhizosphere of each plant along with bulk soil and extracted using the MoBio Power Soil Kit (Mo Bio Laboratories Inc., Carlsbad, CA, USA). This modified in situ technique significantly increased taxa representation and DNA yield from the sample matrix. The two grams of soil samples were vortexed in 10 mL sterile 1X phosphate buffer for 10 min to dislodge bacterial cells, which were then harvested into a pellet at 13,000 rpm for 5 min and subjected to the MoBio Power Soil Kit according to the manufacturer’s instructions.
The DNA concentration and purity of each sample were measured using the Nanodrop 2000c spectrophotometer (Thermo-Fisher Scientific, Waltham, MA, USA). Extracted samples were also run on a 1% agarose gel (1X TAE) at 90 V for 45 min to visually verify the purity of the samples by determining whether DNA bands were intact or degraded. To further confirm the integrity of the extracted DNA, the bacterial 16S rDNA was amplified using the universal primers 1492 Reverse sequences (5′GGTTACCTTGTTACGACTT-3′) and 27 Forward (5′-AGAGTTTGATCCTGGCTCAG-3′) that target the V1-V9 region of the 16S rDNA gene and produce an approximate 1500 base pairs (bp) fragment. Reaction mixtures were incubated for 4 min at 94 °C for denaturation, followed by 35 cycles consisting of 1 min at 94 °C, annealing for 30 s at 45 °C, and extension for 2 min at 72 °C using Taq Polymerase from the 2X Promega PCR master mix (Promega Corp., Madison, WI, USA), along with 0.4 µM of each primer, 10 µg bovine serum albumin (Promega Corp., Madison, WI, USA), 2 mM MgCl2, and approximately 20 ng of DNA template for each sample in a final 25 µL reaction mix. Four (4 µL) microliters of PCR products were run using gel electrophoresis on a 1% agarose (w/v) gel at 90 V for 45 min to view the expected ~1500 bp band size.
2.3. Ca. L asiaticus Detection
Leaf samples were collected from each plant and extracted using the Wizard Magnetic 96 DNA Plant System Instrument (Promega Corp., Madison, WI, USA) according to the manufacturer’s conditions. The qPCR reactions were performed at the University of Florida SWFREC-HLB Laboratory in Immokalee, FL, using the primer/probe set: HLBas (CGAGCGCGTATGCAATACG)/HLBr (CTACCTTTTTCTACGGGATAACGC)/HLBp (AGACGGGTGAGTAACGCG), which amplifies a section of the Las gene on the Applied Biosystems 7500 Fast Real-Time PCR (Applied Biosystems, Foster City, CA, USA) and targets the 16S rDNA. Briefly, qPCR reactions were performed in triplicate in a 25 μL reaction using 2× Quantitect Probe PCR master mix (Qiagen, Valencia, CA, USA), 0.8 μM of each primer, 0.4 μM of probe (IDT, Coralville, IA, USA), and an appropriate amount of template DNA. The PCR conditions were 50 °C for 2 min, 95 °C for 15 min, 45 cycles of each 94 °C for 15 sec, and 60 °C for 1 min. Samples positive for the presence of HLB, resulted in Ct values less than 36, while samples negative for the presence of HLB, resulted in Ct values > or = to 36 (
Table 1). The threshold was automatically assigned by the analysis software. Lower Ct values indicate higher initial template conc in the well. If sample DNA contains
Las DNA, an amplicon will be created in the PCR reaction, and the amount will be reflected in the Ct value. A Ct of 36 or less was considered positive for detection. The range between 32 and 36 Ct was considered putatively positive, and trees tested in this range normally resulted in lower Ct values in subsequent assays [
13].
2.4. Processing of Sequence Data Using QIIME and CosmosID Platform
The forward and reverse reads were paired using the join_paired_ends.py script in QIIME (Quantitative Insights in Microbial Ecology) [
22]. Joined paired-end reads were then quality-filtered using the q30 standard, where the probability of an incorrect base call is 99.9%. Sequences that did not meet these criteria were removed. A second quality control step—chimera check (identify_chimeric_seqs.py)—was performed to identify and remove chimeric sequences. The operational taxonomic unit (OTU) table in ‘biome’ format was then generated by picking OTUs that were aligned against the Greengenes database [
23] for 16S rDNA and also unaligned sequence OTUs using open reference OTU picking at the 97% cut-off. Additional analysis of the OTU tables, using alpha and beta diversity measures along with a taxonomy summary indicating the relative abundances for each sample, was performed using bubble charts and heatmaps from CosmosID. Alpha and Beta analyses were generated by using the QIIME and R software version 4.1.0. Both analyses were rarefied by using the median eigenvalues from the number of reads obtained for all samples. Core OTUs were computed using QIIME version 1.9.1 with the script ‘compute_core_microbiome.py’. The relative abundance of the different phyla observed was calculated from the total number of core OTUs for the rhizosphere of citrus plants from the managed and non-managed sites. The original 16S sequence files were submitted to NCBI with project accession #PRJNA680160 and sample accession numbers SRX9584730-43.
2.5. Statistical Analysis
The statistical significance of the OTUs, Shannon’s diversity index, and observed relative abundance and diversity measures was tested using two-way ANOVAs at the 95% confidence limit, followed by the post hoc Tukey HSD test if significance was found.
4. Discussion
The important role played by soil health and root-resident microbes in pathogen suppression of root-borne diseases is well established. Growing evidence equally emphasizes their involvement in systemic plant immunity against foliar and arthropod-borne diseases through a process called induced systemic resistance (ISR) and more. Plants infested with insects or pathogens release exudates, which attract microbes that trigger ISR. The ISR signals are known to travel from the roots to shoots and leaves, activating enzymes, proteins, and volatile organics in defense pathways that fortify plant immunity [
24]. This process has not been well studied for the Citrus greening disease, whose complex pathogenesis is in part fueled by the plant’s immune response. Nevertheless, data on bacterial community structure and CGD (Ca L. asiaticus) detection in undisturbed soils presented here (
Figure 3,
Figure 4 and
Figure 5) suggest that some poorly known beneficial microbial consortia, depleted in heavily managed groves, could account for the low to no CGD, at least in part. Although the population of the disease insect vector Diaphorina citri was not measured at the managed and undisturbed sites, the close proximity of our study locations (
Figure 1) strongly suggests the ubiquity of the insect in this region.
The plant rhizosphere microbial community is considered one of the most diverse, and many members support the growth of their plant hosts. Many of these microbes are involved in key processes such as nitrogen, phosphorous, and carbon cycling, and help defend plants against soil pathogens through competition and immune response regulation. Distinct shifts in the microbial community structure of the plant rhizosphere will have deleterious effects on the ecosystem function, interfering with nutrient uptake, growth, defense, and overall health. The phytopathogen infection has been shown to cause shifts in plant exudate production [
25] and subsequently the microbial community diversity in many infected plants. This phenomenon has been previously shown in many different plants, including Citrus [
4,
11,
26,
27]. Using 16S rDNA analysis, this study investigated the bacterial community shifts that occur in Citrus plants at various levels of infection and explored, possibly for the first time, the bacterial community structure and abundance of the rhizobiome under historically non-managed citrus groves in Central Florida using 16S rDNA amplicon sequencing. Although the presence of genomic DNA may equate to the microorganisms that are active in the ecosystem, it has been proven on most occasions that the dominant species identified by mRNA are also the most abundant in genomic DNA [
28].
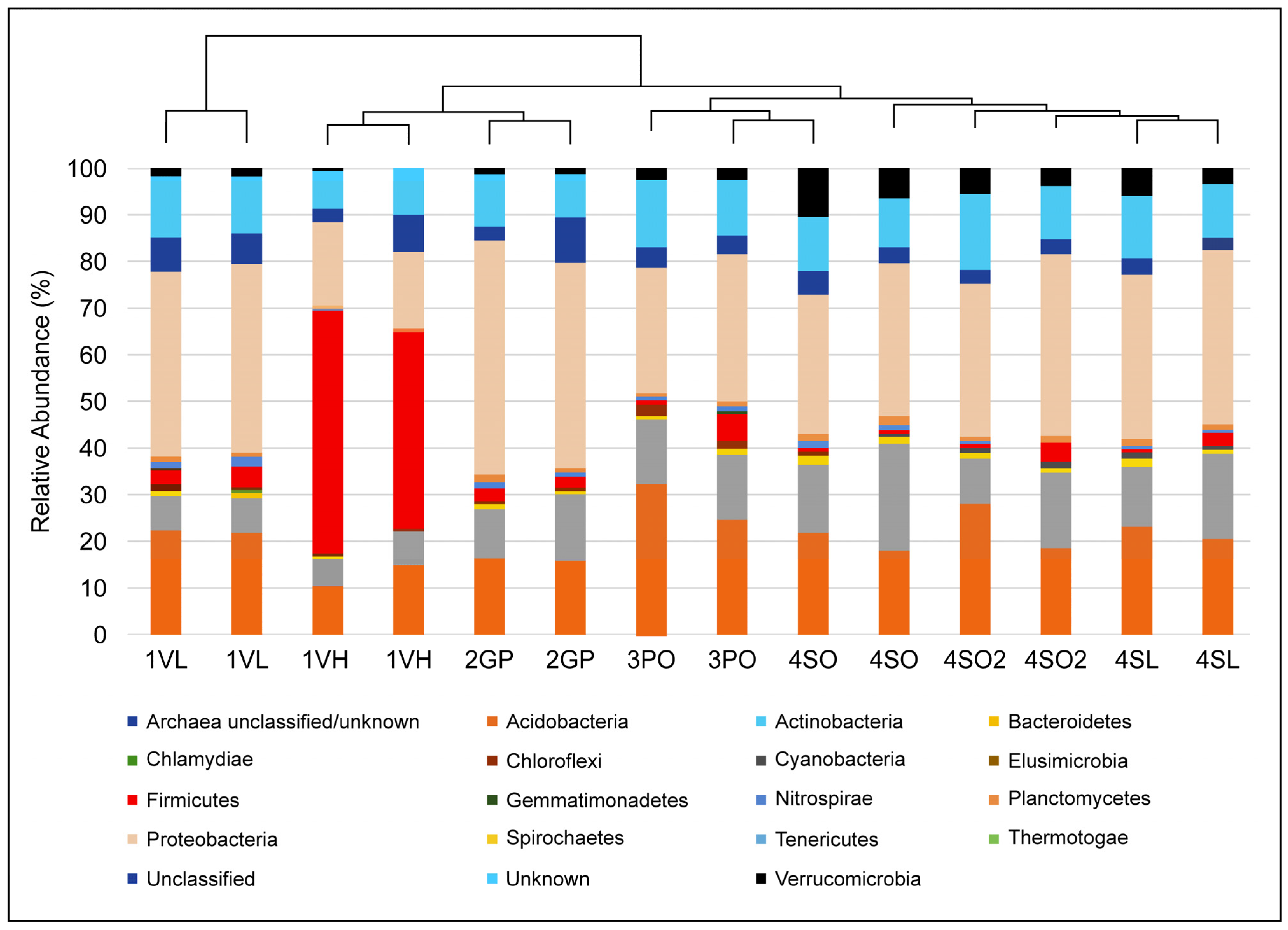
Furthermore, the root exudates of plants differ between varieties and species and could account for differences in 16S amplicon-based community profiles. One of the most intriguing observations in this study is that the microbiome of different types (Lemon, Valencia, Grapefruit) of healthy citrus plants growing in non-managed soils clustered similarly regardless of the site (
Figure 3,
Figure 4 and
Figure 7), implying that major drivers of the bacterial community shifts reported here are external to the plant/microbe interaction: the application of agrochemicals in this case.
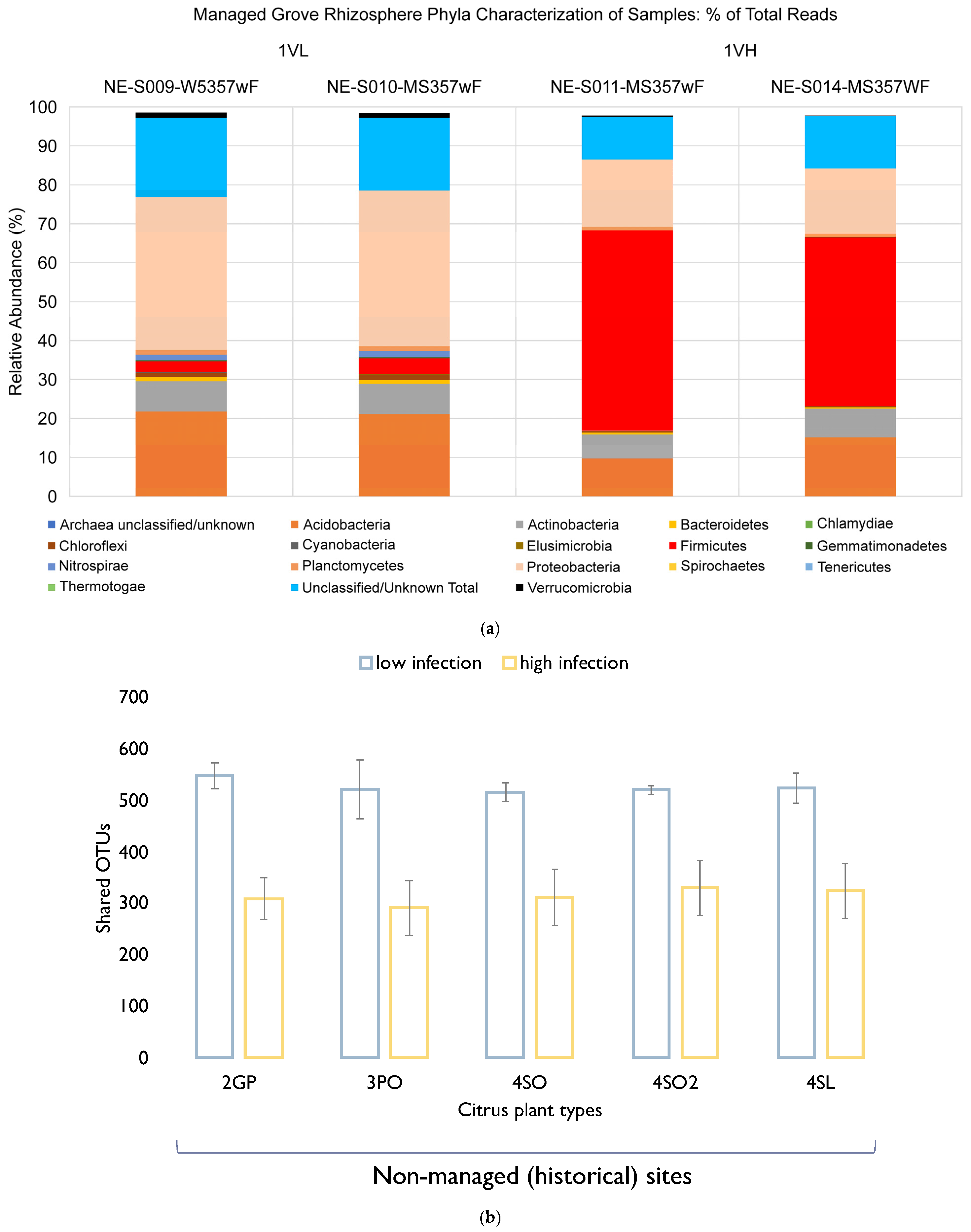
The study showed that a drastic change at the phylum level in the bacterial community structure occurred only after a high level of infection was observed in the West Valencia plants at managed site 1, where there were heavy signs of HLB infection. Remarkably, the East Valencia plant at the same site, also infected, but showing milder symptoms, mimicked the bacterial community structure of the non-infected citrus plants on the non-managed sites, where the Proteobacteria phylum was most prevalent and the Firmicutes had low prevalence. There was an increase in the relative abundance of the Firmicutes phylum and a subsequent decrease in the Proteobacteria phylum (
Figure 5a), similar to what was previously found [
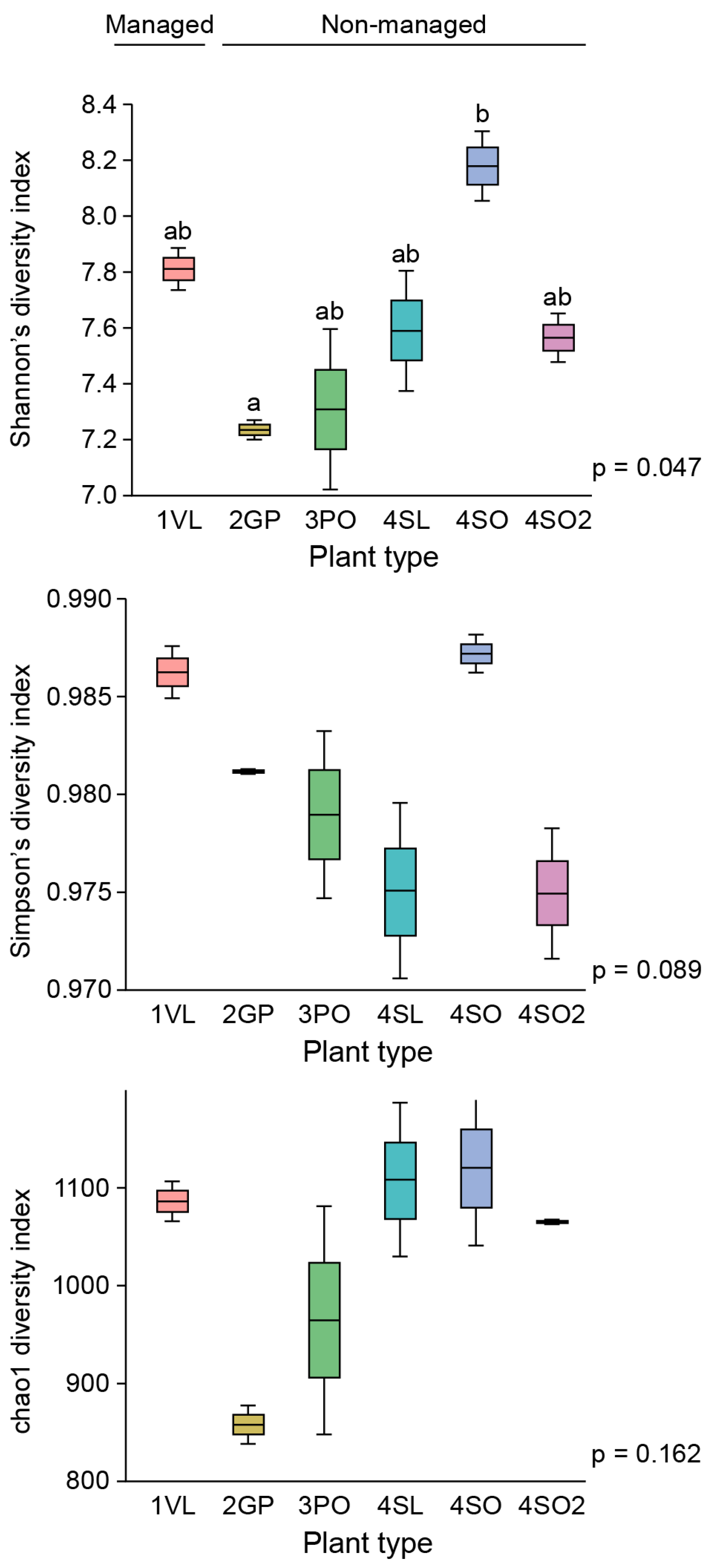
4] for the highly infected West Valencia. There was also a significant decrease in the Shannon’s and Simpson’s diversity indices for the West Valencia plant, while the East Valencia plant at the same site had similar diversity indices to the non-managed uninfected Citrus plants (
Figure 6). The West Valencia plants clustered well away from the other plant types, but similar species from the non-managed Citrus plants clustered with each other (
Figure 7). They likely clustered differently due to the plants being of different citrus species and varieties, where the exudate and bacteria recruitment profiles differ slightly. There was still some variation in the clustering pattern of the East Valencia replicates, likely due to high variations in the relative abundance of some key taxa, such as
Acidobacterium sp.,
Rhodoplanes sp., and other members of the
Alphaproteobacteria class (
Table 1). Infection by the HLB pathogen causes phloem restriction in Citrus plants, preventing the allocation of nutrients throughout the plant [
1], where the roots are eventually starved of nutrients. Bacterial members with high activity, metabolism, and growth will be the first ones to be eliminated, as observed in the Proteobacteria phylum, where members of the
Alphaproteobacteria were reduced under the highly infected West Valencia. Specifically, bacteria involved in nitrogen fixation, such as
Bradyrhizobium sp.,
Rhizobiales order, and
Rhodoplanes sp. (
Table 2), were significantly reduced. There were also other numerous bacterial taxa missing from the rhizosphere of the West Valencia plant but were found in the East Valencia and Citrus plants on the managed sites. When these copiotrophic bacteria are eliminated, the more oligotrophic and slow-growing forms will persist [
29]. In addition, according to a study, a general progression of more pathogenic, competitive, and saprophytic bacteria and fungi will eventually fulfill the niche [
11]. Evidence to support this was seen in the Firmicutes phylum under the infected West Valencia, where there was a 13-fold increase in its prevalence compared to the other Citrus plants, with Bacillus sp. representing close to 50% of the total bacterial reads and 70% of the Firmicutes reads. Bacillus sp. is known to play many roles in the soil ecosystem, being a dominant member of bulk soil and also aiding in plant protection in the rhizosphere. The Bacillus sp. is very resilient and has been shown to survive fluxes in temperature and pH in the plant rhizosphere [
30]. The
Bacillus sp. is also spore-forming, which could be a probable reason for its increased prevalence in the diseased West Valencia plant.
Citrus species at non-managed historical sites devoid of anthropogenic influence (herbicide/fertilizer treatment) have never been fully explored to determine if a stable core rhizobiome exists with potentially beneficial microbes that could be used to improve plant health and productivity. Herbicide treatment, which in this case involves the use of glyphosate (Roundup
®), has been shown to have an overall benign effect on the soil microbial community, only eliciting a significant microbial community shift at application rates higher than recommended [
31]. It is likely, then, to expect that if recommended application rates were followed on the managed citrus grove site, as reported (Dr. D. McCorquodale, Pers Comm, Dec 2017), the microbial community structure would not be substantially affected. The use of commercial fertilizers, however, can greatly affect the abundance, microbial community structure, and function of soil bacteria [
32] at the managed sites, where the number of nitrogen-fixing, phosphate-solubilizing bacteria and fungi may be reduced due to lesser plant dependence on these microbes and niche reduction. Also, it shows that the application of commercial NPK fertilizers reduced members of the phylum
Acidobacteria, Nitrospira, Chloroflexi, and
Planctomycetes while enhancing members such as
Sphingomonas spp. [
33]. Thus, it is imperative that the continued application of fertilizers in managed citrus groves may reduce the formation of a stable core microbiome of beneficial bacteria, unlike the more natural non-managed sites. It is imperative to continue investigating the optimal thresholds of agrochemicals to be employed in current agricultural practices for the purpose of lowering induced dysbiosis. These non-managed sites may contain a richer and more stable core microbiome to be further explored. In our findings, the bacterial community structure of the grapefruit, pineapple orange, sour orange, and sour lemon plant rhizospheres was somewhat consistent with one another (
Figure 7), where the Proteobacteria phylum was the most prevalent, and the Firmicutes and
Verrucomicrobia phyla had the lowest prevalence. They also shared approximately the same relative abundance of Firmicutes with the infected East Valencia, which exhibited mild symptoms. Some of these members were described as putative plant-growth-promoting bacteria in the citrus rhizosphere [
11,
20]. There was also a significant increase in members of the
Verrucomicrobia phylum in a few non-managed sites. Even though many of these members are still unknown, one study has shown that they are extremely sensitive to changes in soil fertility [
33]. The reduction of these and potentially other beneficial organisms could prevent the recovery of these diseased plants under biotic and abiotic stress. A recent study has shown that the inoculation of beneficial plant-associated bacteria, such as
Bacteriodetes sp., can initiate defense responses in plants to help confer resistance. There is a substantial, stable core microbiome that exists in the non-managed citrus rhizosphere to be explored and exploited to create synthetic microbial communities to restore plant health and productivity. Similar studies will have future implications and benefits in improving the sustainability of agricultural and non-agricultural crops as we begin to explore the core microbiome, and the major players are identified in healthy, anthropogenic-free plant crops.



 ,
, 




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}