
Distinct Effects of Lactiplantibacillus plantarum HNU082 on Microbial Single-Nucleotide Variants in Large Intestine and Small Intestine

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Methods and Materials
2.1. Preparation of Strains for Gavage
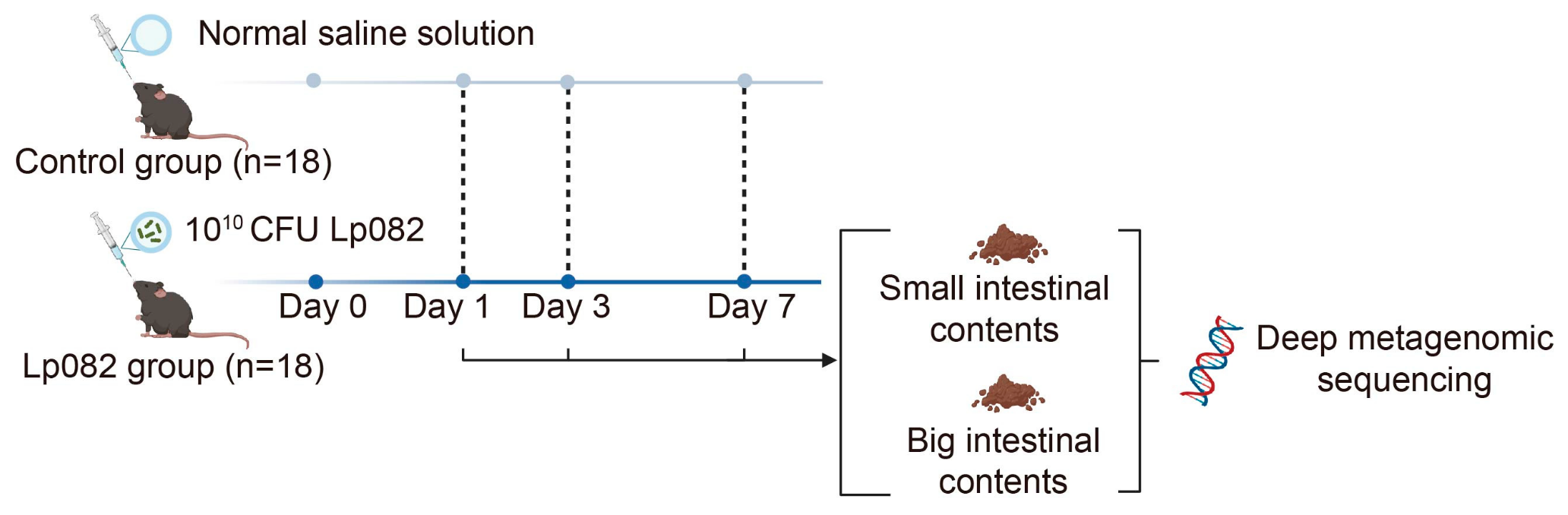
2.2. Experimental Design
2.3. Metagenomic Sequencing and Annotation of Gut Microbiota
2.4. Single-Nucleotide Mutation Annotation and Statistics Analysis
3. Results
3.1. The Response of the Indigenous Microbiota in Different Intestinal Segments to Lp082
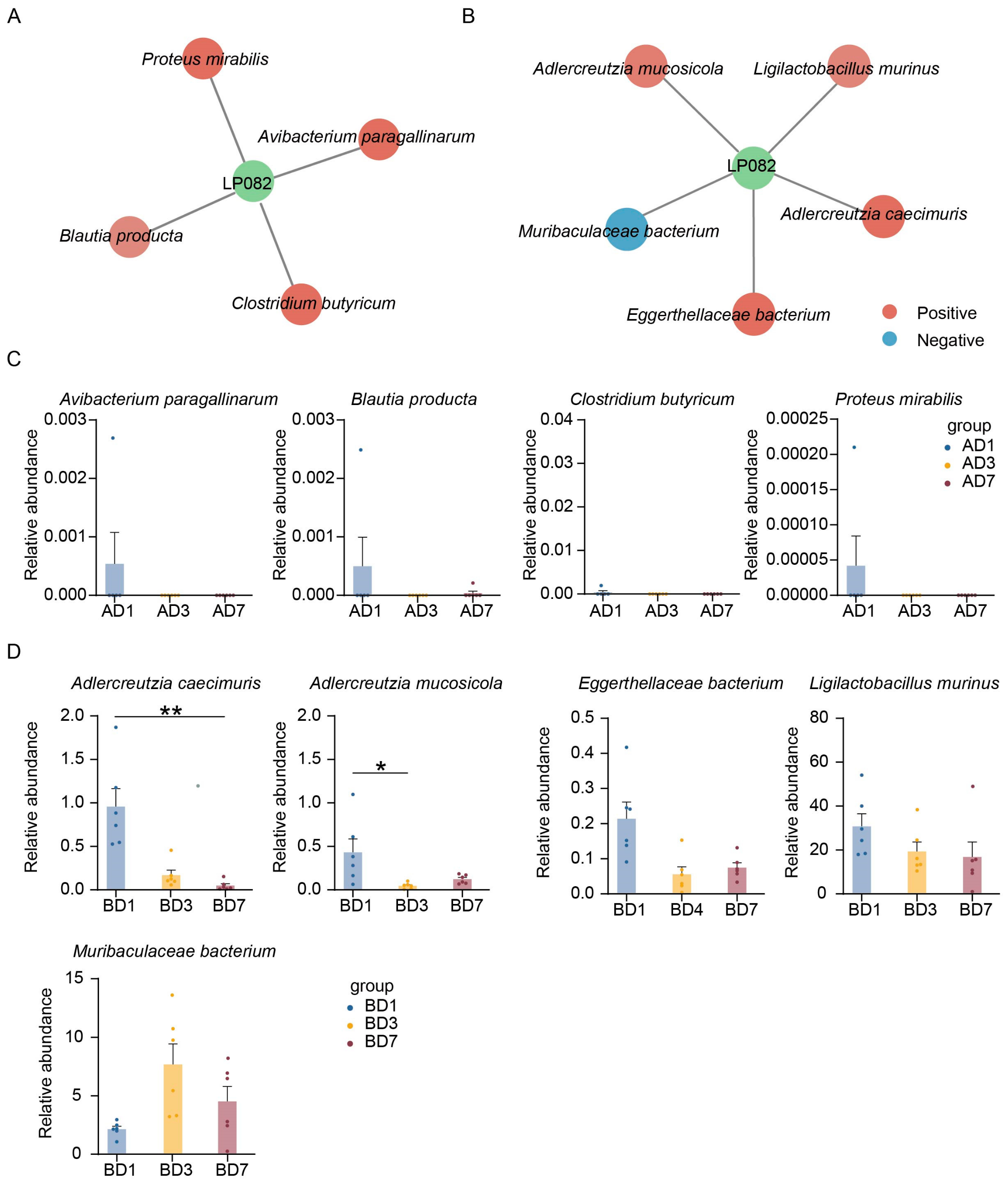
3.2. Annotation and Identification of Lp082-Related Microbiota in Different Intestinal Segments
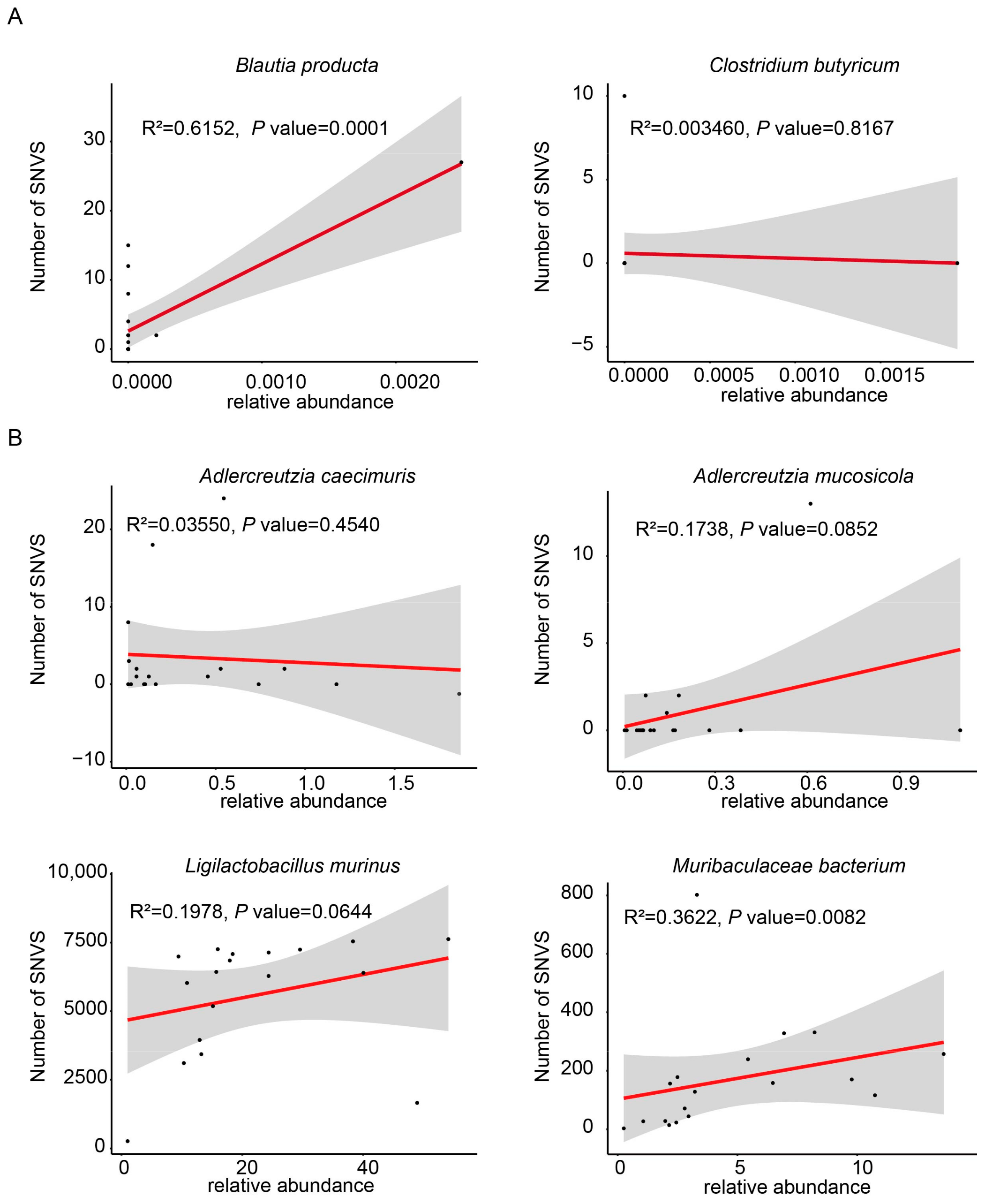
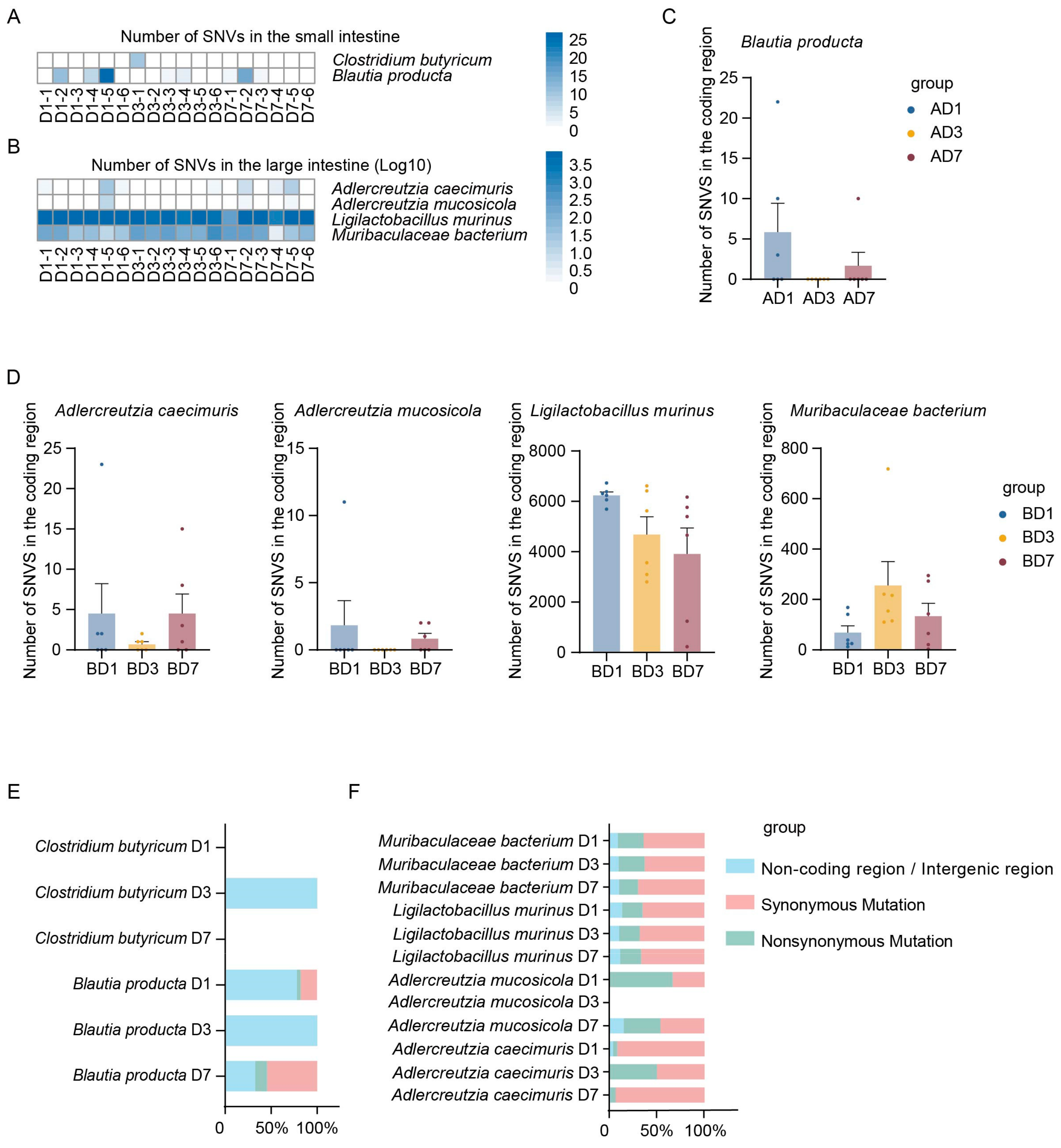
3.3. Annotation of Mutation Number in the Indigenous Microbial Populations Across Different Intestinal Segments
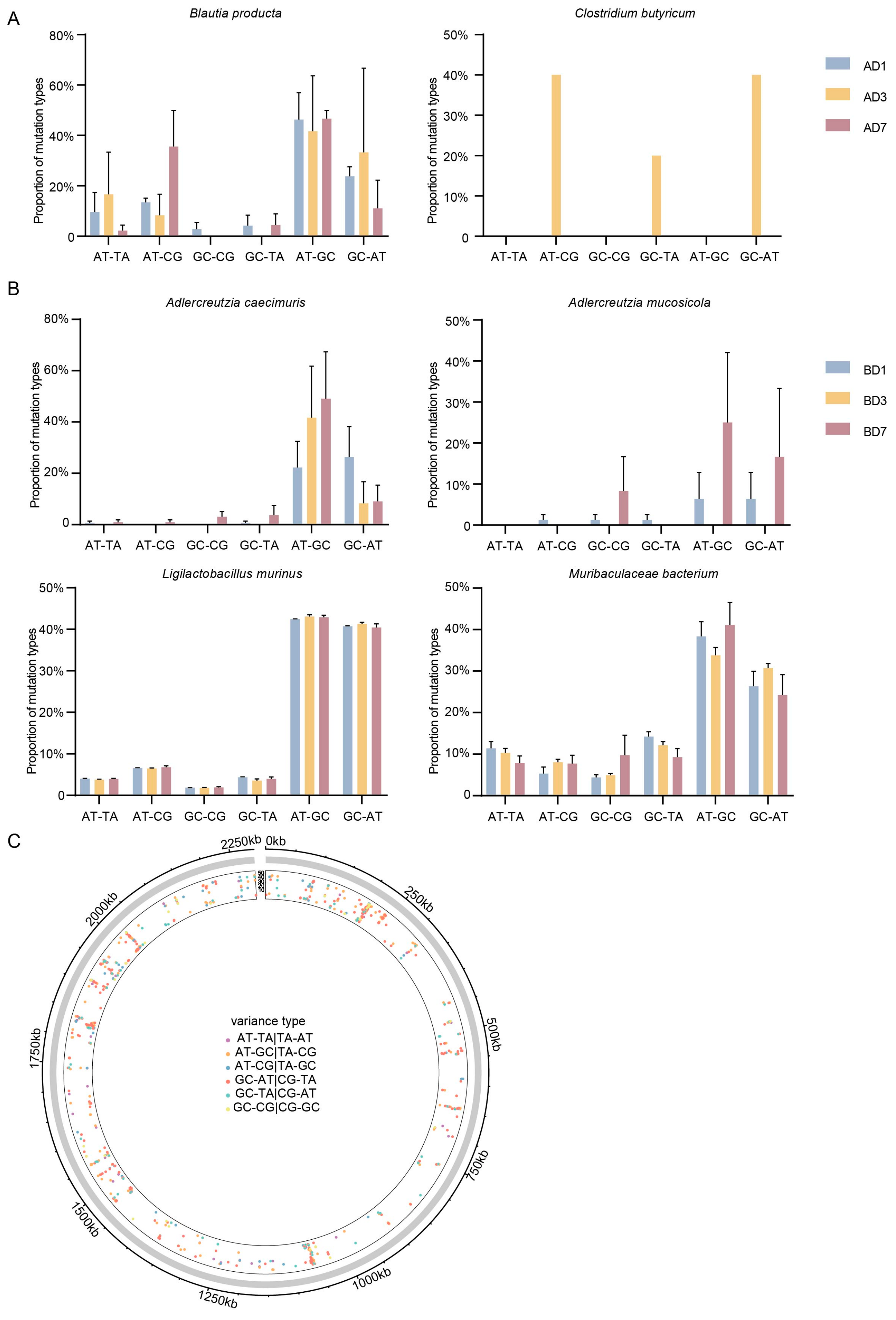
3.4. Annotation of Mutation Type in the Indigenous Microbial Populations Across Different Intestinal Segments
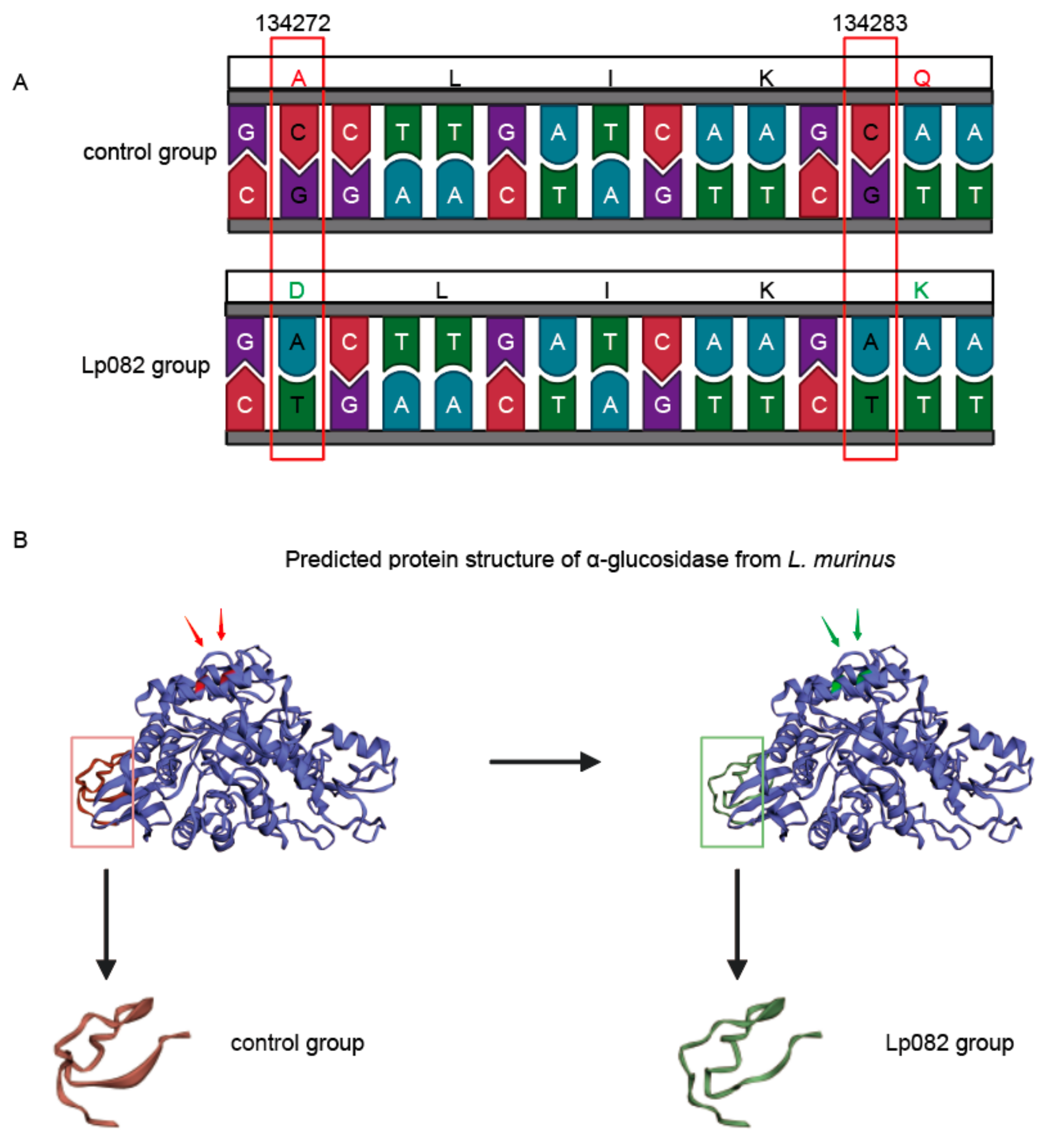
3.5. Functional Annotation of SNVs in Different Intestinal Segments
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhang, X.; Li, L.; Butcher, J.; Stintzi, A.; Figeys, D. Advancing functional and translational microbiome research using meta-omics approaches. Microbiome 2019, 7, 154. [Google Scholar] [CrossRef]
- Yuan, S.; Wang, K.; Meng, H.; Hou, X.; Xue, J.; Liu, B.; Cheng, W.; Li, J.; Zhang, H.; Nan, J.X.; et al. The gut microbes in inflammatory bowel disease: Future novel target option for pharmacotherapy. Biomed. Pharmacother. 2023, 165, 114893. [Google Scholar] [CrossRef]
- de Vos, W.M.; Tilg, H.; Van Hul, M.; Cani, P.D. Gut microbiome and health: Mechanistic insights. Gut 2022, 71, 1020–1032. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Chen, X.; Huo, D.; Arifuzzaman, M.; Qiao, S.; Jin, W.; Shi, H.; Li, X.V.; Iliev, I.D.; Artis, D.; et al. Microbiota metabolism of intestinal amino acids impacts host nutrient homeostasis and physiology. Cell Host Microbe 2024, 32, 661–675.e610. [Google Scholar] [CrossRef]
- Al-Rashidi, H.E. Gut microbiota and immunity relevance in eubiosis and dysbiosis. Saudi J. Biol. Sci. 2022, 29, 1628–1643. [Google Scholar] [CrossRef]
- Dinan, T.G.; Stilling, R.M.; Stanton, C.; Cryan, J.F. Collective unconscious: How gut microbes shape human behavior. J. Psychiatr. Res. 2015, 63, 1–9. [Google Scholar] [CrossRef]
- Fang, J.; Wang, H.; Zhou, Y.; Zhang, H.; Zhou, H.; Zhang, X. Slimy partners: The mucus barrier and gut microbiome in ulcerative colitis. Exp. Mol. Med. 2021, 53, 772–787. [Google Scholar] [CrossRef]
- Espey, M.G. Role of oxygen gradients in shaping redox relationships between the human intestine and its microbiota. Free Radic. Biol. Med. 2013, 55, 130–140. [Google Scholar] [CrossRef]
- Kastl, A.J.; Terry, N.A.; Wu, G.; Albenberg, L.G. The Structure and Function of the Human Small Intestinal Microbiota: Current Understanding and Future Directions. Cell. Mol. Gastroenterol. Hepatol. 2020, 9, 33–45. [Google Scholar] [CrossRef]
- Hill, C.; Guarner, F.; Reid, G.; Gibson, G.R.; Merenstein, D.J.; Pot, B.; Morelli, L.; Canani, R.B.; Flint, H.J.; Salminen, S.; et al. Expert consensus document. The International Scientific Association for Probiotics and Prebiotics consensus statement on the scope and appropriate use of the term probiotic. Nat. Rev. Gastroenterol. Hepatol. 2014, 11, 506–514. [Google Scholar] [CrossRef]
- Nagpal, R.; Wang, S.H.; Ahmadi, S.; Hayes, J.; Gagliano, J.; Subashchandrabose, S.; Kitzman, D.W.; Becton, T.; Read, R.; Yadav, H. Human-origin probiotic cocktail increases short-chain fatty acid production via modulation of mice and human gut microbiome. Sci. Rep. 2018, 8, 12649. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Zhang, C.; Han, Z.; Ma, W.; Wang, S.; Huo, D.; Cui, W.; Zhai, Q.; Huang, S.; Zhang, J. Native microbiome dominates over host factors in shaping the probiotic genetic evolution in the gut. NPJ Biofilms Microbiomes 2023, 9, 80. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Zhang, Z.; Shen, S.; Li, J.; Su, S.; Du, L.; Ma, W.; Zhang, J. Leuconostoc citreum enhances the colonization ability of Lactiplantibacillus plantarum in the gut by synergistically modifying the intestinal niche. Gut Microbes 2025, 17, 2447819. [Google Scholar] [CrossRef]
- Huang, S.; Jiang, S.; Huo, D.; Allaband, C.; Estaki, M.; Cantu, V.; Belda-Ferre, P.; Vazquez-Baeza, Y.; Zhu, Q.; Ma, C.; et al. Candidate probiotic Lactiplantibacillus plantarum HNU082 rapidly and convergently evolves within human, mice, and zebrafish gut but differentially influences the resident microbiome. Microbiome 2021, 9, 151. [Google Scholar] [CrossRef]
- Gao, W.; Gao, X.; Zhu, L.; Gao, S.; Sun, R.; Feng, Z.; Wu, D.; Liu, Z.; Zhu, R.; Jiao, N. Multimodal metagenomic analysis reveals microbial single nucleotide variants as superior biomarkers for early detection of colorectal cancer. Gut Microbes 2023, 15, 2245562. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, X.; Huo, D.; Li, W.; Hu, Q.; Xu, C.; Liu, S.; Li, C. Metagenomic approach reveals microbial diversity and predictive microbial metabolic pathways in Yucha, a traditional Li fermented food. Sci. Rep. 2016, 6, 32524. [Google Scholar] [CrossRef]
- Shao, Y.; Huo, D.; Peng, Q.; Pan, Y.; Jiang, S.; Liu, B.; Zhang, J. Lactobacillus plantarum HNU082-derived improvements in the intestinal microbiome prevent the development of hyperlipidaemia. Food Funct. 2017, 8, 4508–4516. [Google Scholar] [CrossRef]
- Yu, S.; Wang, Y.; Li, X.; Yu, F.; Li, W. The factors affecting the reproducibility of micro-volume DNA mass quantification in Nanodrop 2000 spectrophotometer. Opt.-Int. J. Light Electron Opt. 2017, 145, 555–560. [Google Scholar] [CrossRef]
- Jin, H.; Quan, K.; He, Q.; Kwok, L.Y.; Ma, T.; Li, Y.; Zhao, F.; You, L.; Zhang, H.; Sun, Z. A high-quality genome compendium of the human gut microbiome of Inner Mongolians. Nat. Microbiol. 2023, 8, 150–161. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 15 October 2024).
- Nousias, O.; Montesanto, F. Metagenomic profiling of host-associated bacteria from 8 datasets of the red alga Porphyra purpurea with MetaPhlAn3. Mar. Genom. 2021, 59, 100866. [Google Scholar] [CrossRef]
- Raju, S.C.; Molinaro, A.; Awoyemi, A.; Jørgensen, S.F.; Braadland, P.R.; Nendl, A.; Seljeflot, I.; Ueland, P.M.; McCann, A.; Aukrust, P.; et al. Microbial-derived imidazole propionate links the heart failure-associated microbiome alterations to disease severity. Genome Med. 2024, 16, 27. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Sepulveda, J.L. Using R and Bioconductor in Clinical Genomics and Transcriptomics. J. Mol. Diagn. 2020, 22, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Olm, M.R.; Crits-Christoph, A.; Bouma-Gregson, K.; Firek, B.A.; Morowitz, M.J.; Banfield, J.F. inStrain profiles population microdiversity from metagenomic data and sensitively detects shared microbial strains. Nat. Biotechnol. 2021, 39, 727–736. [Google Scholar] [CrossRef]
- Tybout, A.; Sternthal, B.; Verducci, J.; Meyers-Levy, J.; Barnes, J.; Maxwell, S.; Allenby, G.; Steenkamp, J.B.; Gupta, S. Analysis of Variance. J. Consum. Psychol. 2001, 10, 5–35. [Google Scholar]
- Li, X.; Wu, Y.; Wei, M.; Guo, Y.; Yu, Z.; Wang, H.; Li, Z.; Fan, H. A novel index of functional connectivity: Phase lag based on Wilcoxon signed rank test. Cogn. Neurodyn. 2021, 15, 621–636. [Google Scholar] [CrossRef]
- Yvonne, C.; Walmsley, R.P. Learning and Understanding the Kruskal-Wallis One-Way Analysis-of-Variance-by-Ranks Test for Differences Among Three or More Independent Groups. Phys. Ther. 1997, 77, 1755–1762. [Google Scholar]
- Cornea-Cipcigan, M.; Pamfil, D.; Sisea, C.R.; Margaoan, R. Characterization of Cyclamen genotypes using morphological descriptors and DNA molecular markers in a multivariate analysis. Front. Plant Sci. 2023, 14, 1100099. [Google Scholar] [CrossRef]
- Ginestet, C. ggplot2: Elegant Graphics for Data Analysis. J. R. Stat. Soc. 2011, 174, 245–246. [Google Scholar] [CrossRef]
- Simpson, G.L. Permute: Functions for Generating Restricted Permutations of Data. Available online: https://CRAN.R-project.org/package=permute (accessed on 15 October 2024).
- Lo, T.T.; Van Lissa, C.J.; Verhagen, M.; Hoemann, K.; Erbas, Y.; Maciejewski, D.F. A theory-informed emotion regulation variability index: Bray-Curtis dissimilarity. Emotion 2024, 24, 1273–1285. [Google Scholar] [CrossRef] [PubMed]
- Anastasia, N.; Richard, M.; Kwiatkowski, D.P.; Udalova, I.A. Comparing the fine specificity of DNA binding by NF-kappaB p50 and p52 using principal coordinates analysis. Nucleic Acids Res. 2003, 31, 1497–1501. [Google Scholar] [CrossRef]
- Motulsky, H. Analyzing Data with GraphPad Prism; GraphPad Software Incorporated: La Jolla, CA, USA, 1999. [Google Scholar]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Kelley, L.A.; Sternberg, M.J. Protein structure prediction on the Web: A case study using the Phyre server. Nat. Protoc. 2009, 4, 363–371. [Google Scholar] [CrossRef]
- Ma, C.; Zhang, C.; Chen, D.; Jiang, S.; Shen, S.; Huo, D.; Huang, S.; Zhai, Q.; Zhang, J. Probiotic consumption influences universal adaptive mutations in indigenous human and mouse gut microbiota. Commun. Biol. 2021, 4, 1198. [Google Scholar] [CrossRef]
- Daniel, N.; Lécuyer, E.; Chassaing, B. Host/microbiota interactions in health and diseases—Time for mucosal microbiology! Mucosal Immunol. 2021, 14, 1006–1016. [Google Scholar] [CrossRef]
- Sanders, M.E. Impact of probiotics on colonizing microbiota of the gut. J. Clin. Gastroenterol. 2011, 45, S115. [Google Scholar] [CrossRef]
- Fassarella, M.; Blaak, E.E.; Penders, J.; Nauta, A.; Smidt, H.; Zoetendal, E.G. Gut microbiome stability and resilience: Elucidating the response to perturbations in order to modulate gut health. Gut 2021, 70, 595–605. [Google Scholar] [CrossRef]
- Arumugam, M.; Raes, J.; Pelletier, E.; Le Paslier, D.; Yamada, T.; Mende, D.R.; Fernandes, G.R.; Tap, J.; Bruls, T.; Batto, J.M.; et al. Enterotypes of the human gut microbiome. Nature 2011, 473, 174–180. [Google Scholar] [CrossRef]
- Ratzke, C.; Barrere, J.; Gore, J. Strength of species interactions determines biodiversity and stability in microbial communities. Nat. Ecol. Evol. 2020, 4, 376–383. [Google Scholar] [CrossRef]
- Craven, D.; Eisenhauer, N.; Pearse, W.D.; Hautier, Y.; Isbell, F.; Roscher, C.; Bahn, M.; Beierkuhnlein, C.; Bönisch, G.; Buchmann, N.; et al. Multiple facets of biodiversity drive the diversity-stability relationship. Nat. Ecol. Evol. 2018, 2, 1579–1587. [Google Scholar] [CrossRef] [PubMed]
- Eisenhauer, N.; Scheu, S.; Jousset, A. Bacterial diversity stabilizes community productivity. PLoS ONE 2012, 7, e34517. [Google Scholar] [CrossRef] [PubMed]
- Swain, A.; Fussell, L.; Fagan, W.F. Higher-order effects, continuous species interactions, and trait evolution shape microbial spatial dynamics. Proc. Natl. Acad. Sci. USA 2022, 119, e2020956119. [Google Scholar] [CrossRef] [PubMed]
- Chu, N.D.; Crothers, J.W.; Nguyen, L.T.T.; Kearney, S.M.; Smith, M.B.; Kassam, Z.; Collins, C.; Xavier, R.; Moses, P.L.; Alm, E.J. Dynamic Colonization of Microbes and Their Functions after Fecal Microbiota Transplantation for Inflammatory Bowel Disease. mBio 2021, 12, e0097521. [Google Scholar] [CrossRef]
- Martinez-Guryn, K.; Leone, V.; Chang, E.B. Regional Diversity of the Gastrointestinal Microbiome. Cell Host Microbe 2019, 26, 314–324. [Google Scholar] [CrossRef]
- Zhu, X.; Qin, J.; Tan, C.; Ning, K. The seasonal changes of the gut microbiome of the population living in traditional lifestyles are represented by characteristic species-level and functional-level SNP enrichment patterns. BMC Genom. 2021, 22, 83. [Google Scholar] [CrossRef]
- Saber, M.M.; Donner, J.; Levade, I.; Acosta, N.; Parkins, M.D.; Boyle, B.; Levesque, R.C.; Nguyen, D.; Shapiro, B.J. Single nucleotide variants in Pseudomonas aeruginosa populations from sputum correlate with baseline lung function and predict disease progression in individuals with cystic fibrosis. Microb. Genom. 2023, 9, 000981. [Google Scholar] [CrossRef]
- Xie, J.; Guo, G.; Wang, Y.; Hu, T.; Wang, L.; Li, J.; Qiu, D.; Li, Y.; Wu, Q.; Lu, P.; et al. A rare single nucleotide variant in Pm5e confers powdery mildew resistance in common wheat. New Phytol. 2020, 228, 1011–1026. [Google Scholar] [CrossRef]
- Wang, J.; Chen, L.; Zhao, N.; Xu, X.; Xu, Y.; Zhu, B. Of genes and microbes: Solving the intricacies in host genomes. Protein Cell 2018, 9, 446–461. [Google Scholar] [CrossRef]
- Almeida, A.; Nayfach, S.; Boland, M.; Strozzi, F.; Beracochea, M.; Shi, Z.J.; Pollard, K.S.; Sakharova, E.; Parks, D.H.; Hugenholtz, P.; et al. A unified catalog of 204,938 reference genomes from the human gut microbiome. Nat. Biotechnol. 2021, 39, 105–114. [Google Scholar] [CrossRef]
- Chen, J.; Yang, H.; Feng, Y.; Shi, Q.; Li, Z.; Tao, Z.; Fan, J.; Jin, Y.; Li, S.; Cheng, J.; et al. A single nucleotide mutation drastically increases the expression of tumor-homing NGR-TNFalpha in the E. coli M15-pQE30 system by improving gene transcription. Appl. Microbiol. Biotechnol. 2021, 105, 1447–1460. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Zhang, Y.; Jiang, S.; Teng, F.; Huang, S.; Zhang, J. Cross-cohort single-nucleotide-variant profiling of gut microbiota suggests a novel gut-health assessment approach. mSystems 2023, 8, e0082823. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Chen, D.; Ma, C.; Liu, H.; Huang, S.; Zhang, J. Establishing a novel inflammatory bowel disease prediction model based on gene markers identified from single nucleotide variants of the intestinal microbiota. iMeta 2022, 1, e40. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Li, Y.J.; Shao, J.; Fu, C.; Li, Y.S.; Cui, Z.N. 2,5-Disubstituted furan derivatives containing imidazole, triazole or tetrazole moiety as potent α-glucosidase inhibitors. Bioorganic Chem. 2023, 131, 106298. [Google Scholar] [CrossRef]
- Ma, C.; Wasti, S.; Huang, S.; Zhang, Z.; Mishra, R.; Jiang, S.; You, Z.; Wu, Y.; Chang, H.; Wang, Y.; et al. The gut microbiome stability is altered by probiotic ingestion and improved by the continuous supplementation of galactooligosaccharide. Gut Microbes 2020, 12, 1785252. [Google Scholar] [CrossRef]
- Mazhar, M.; Zhu, Y.; Qin, L. The Interplay of Dietary Fibers and Intestinal Microbiota Affects Type 2 Diabetes by Generating Short-Chain Fatty Acids. Foods 2023, 12, 1023. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, W.; Han, Z.; Liu, X.; Cui, W.; Zhen, D.; Zhou, X.; Song, Y.; Jiang, S. Distinct Effects of Lactiplantibacillus plantarum HNU082 on Microbial Single-Nucleotide Variants in Large Intestine and Small Intestine. Microorganisms 2025, 13, 731. https://doi.org/10.3390/microorganisms13040731
Ma W, Han Z, Liu X, Cui W, Zhen D, Zhou X, Song Y, Jiang S. Distinct Effects of Lactiplantibacillus plantarum HNU082 on Microbial Single-Nucleotide Variants in Large Intestine and Small Intestine. Microorganisms. 2025; 13(4):731. https://doi.org/10.3390/microorganisms13040731
Chicago/Turabian StyleMa, Wenyao, Zhe Han, Xinlei Liu, Weipeng Cui, Dongyu Zhen, Xiaolu Zhou, Yuan Song, and Shuaiming Jiang. 2025. "Distinct Effects of Lactiplantibacillus plantarum HNU082 on Microbial Single-Nucleotide Variants in Large Intestine and Small Intestine" Microorganisms 13, no. 4: 731. https://doi.org/10.3390/microorganisms13040731
APA StyleMa, W., Han, Z., Liu, X., Cui, W., Zhen, D., Zhou, X., Song, Y., & Jiang, S. (2025). Distinct Effects of Lactiplantibacillus plantarum HNU082 on Microbial Single-Nucleotide Variants in Large Intestine and Small Intestine. Microorganisms, 13(4), 731. https://doi.org/10.3390/microorganisms13040731