
Molecular Evolutionary Analyses of the RNA-Dependent RNA Polymerase (RdRp) Region and VP1 Gene in Sapovirus GI.1 and GI.2

 , , ,
, , ,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Strains in This Study
2.2. Maximum Likelihood (ML)
2.3. Time-Scaled Phylogenetic Analyses and Estimation of Evolutionary Rate
2.4. Bayesian Skyline Plot (BSP) Analyses
2.5. Phylogenetic Distance Analyses
2.6. Similarity Plot Analyses
2.7. Construction of the 3D Structure of RdRp and VP1 Proteins
2.8. Conformational B-Cell Epitope Prediction
2.9. Selective Pressure Analyses
3. Results
3.1. The Phylogeny of the RdRp Region and VP1 Gene in HuSaV Genogroups
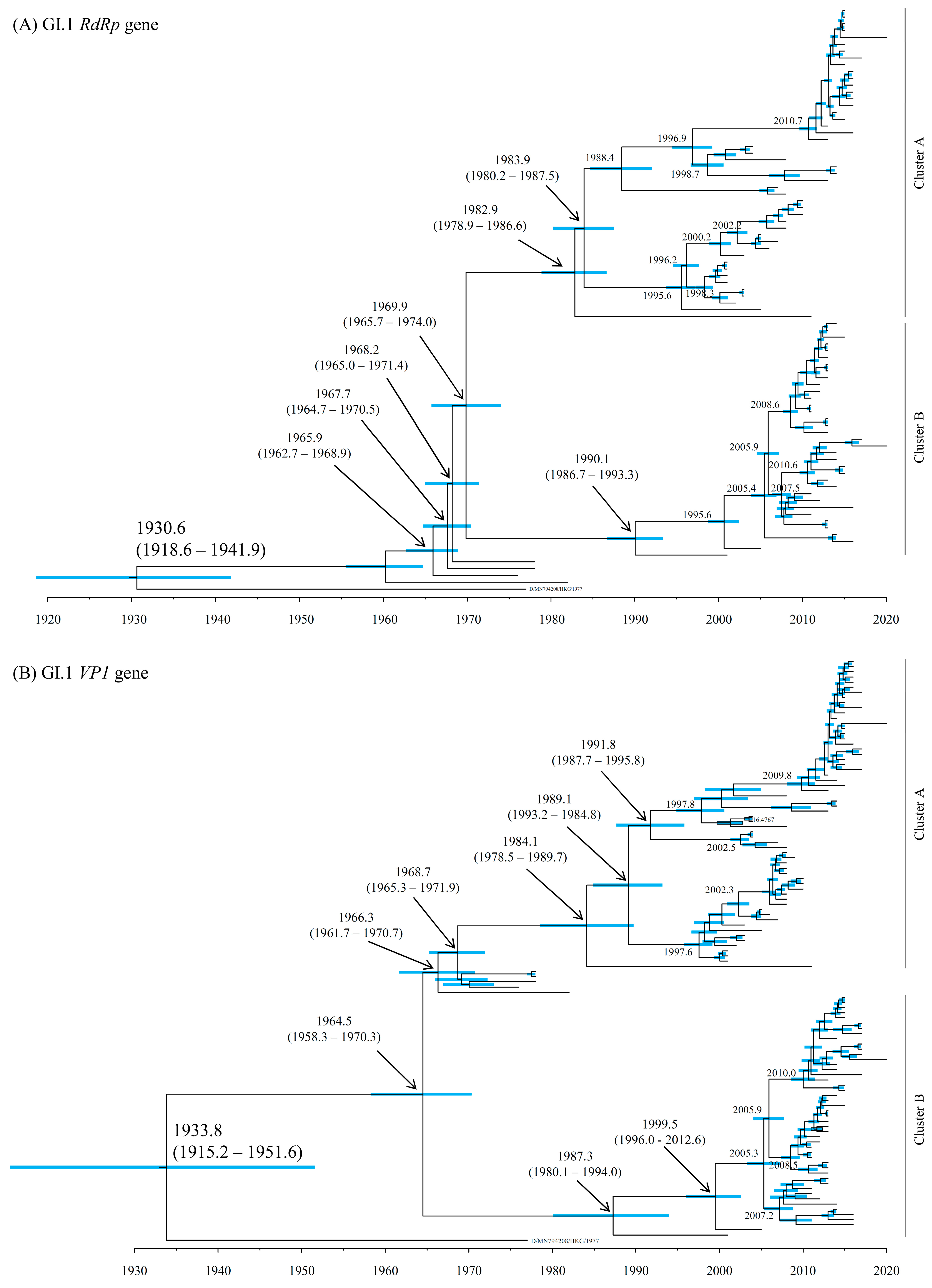
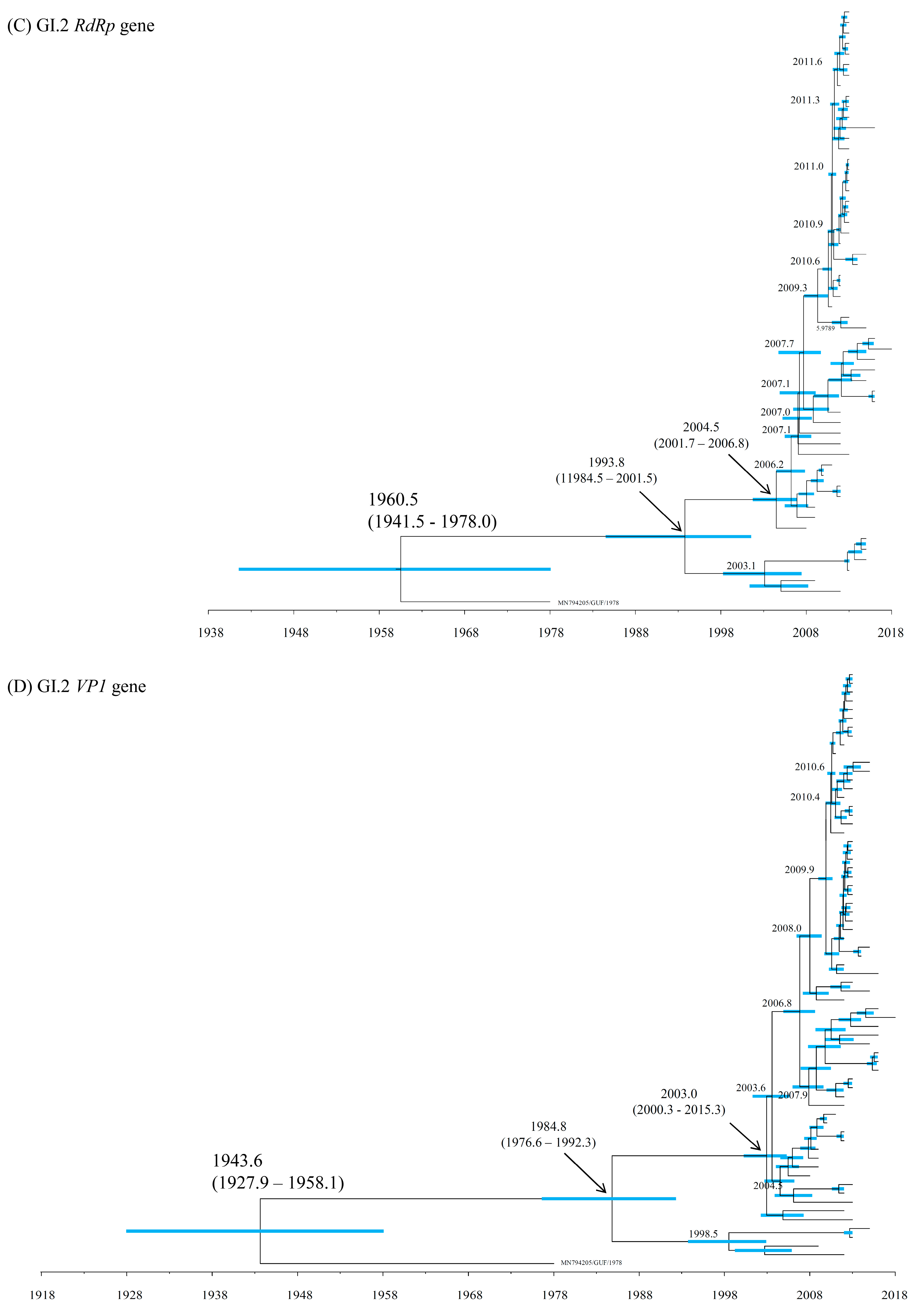
3.2. Time-Scaled Phylogeny
3.3. Evolutionary Rates
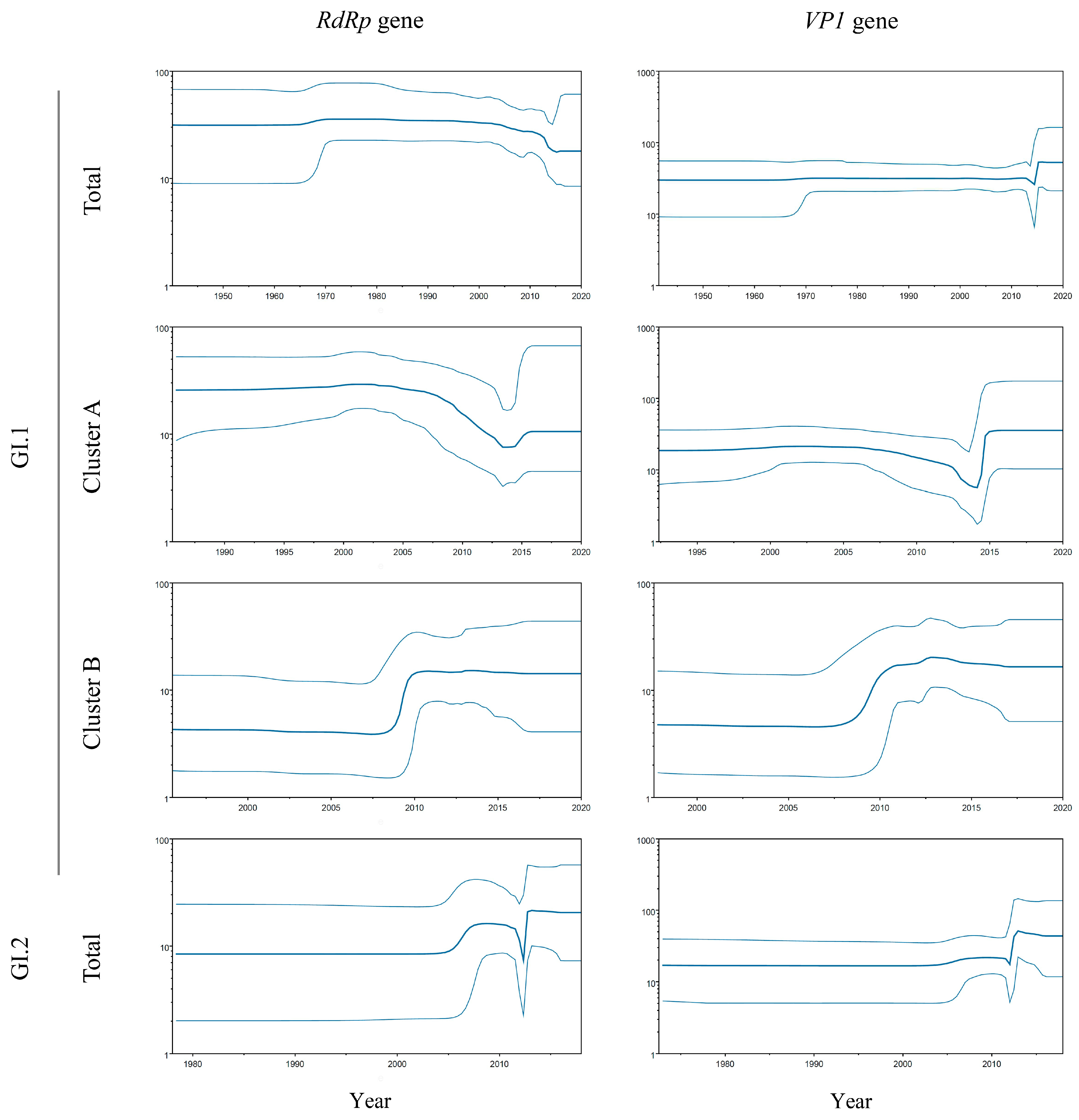
3.4. Phylodynamic Using the BSP Method
3.5. Phylogenetic Distances
3.6. Similarity Plot Analysis
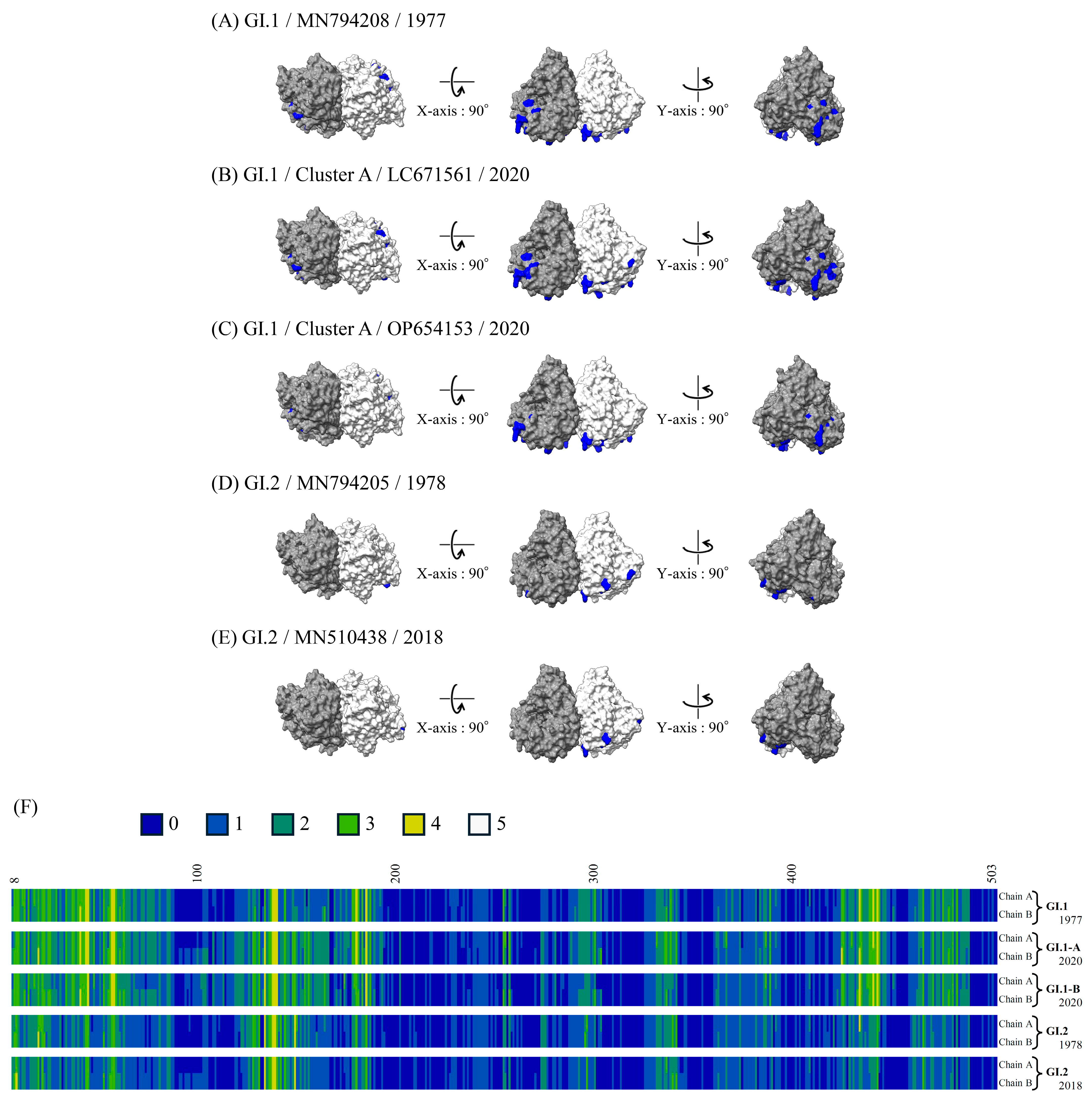
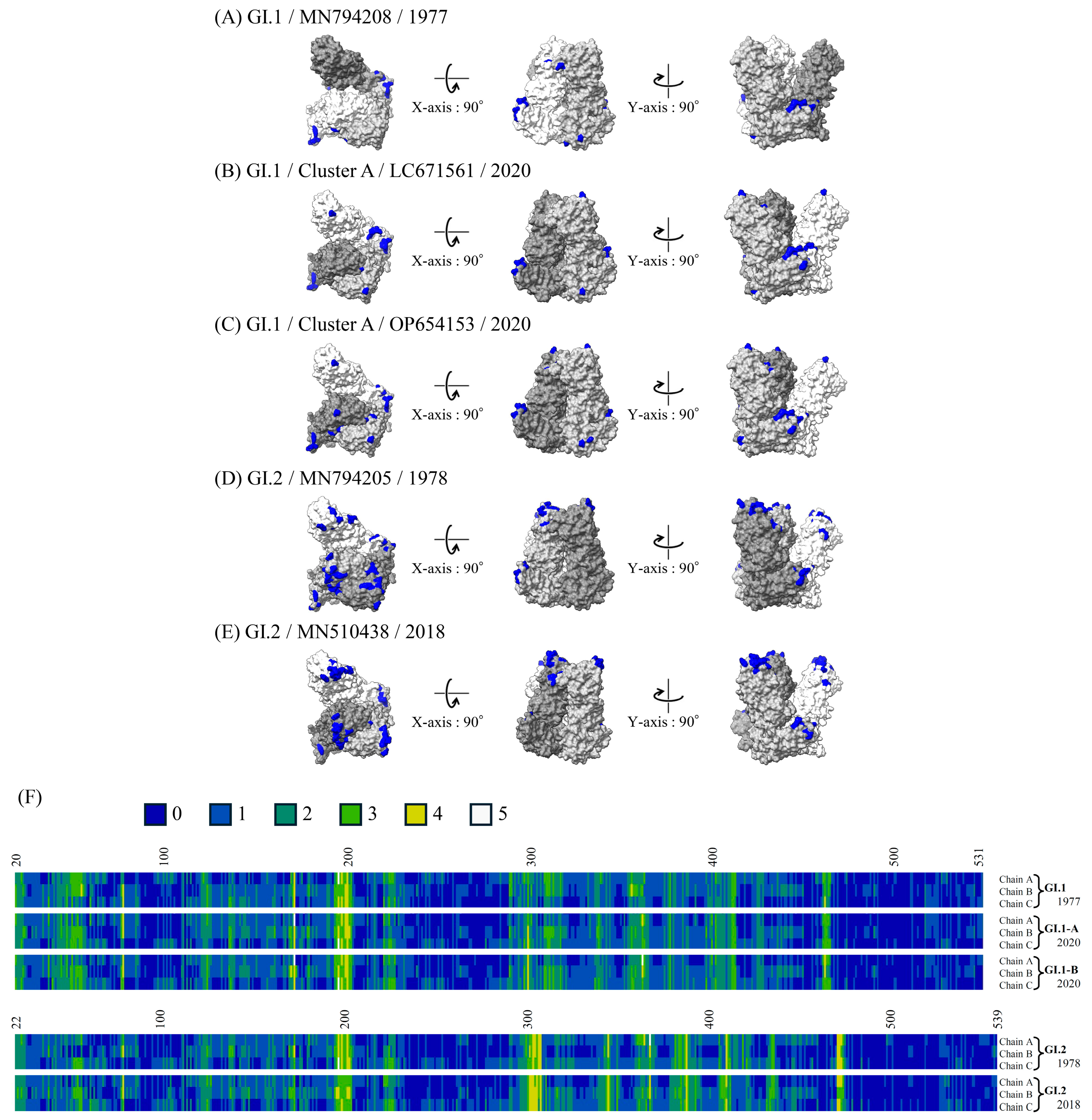
3.7. 3D Mapping of the Conformational Epitopes in the RdRp Dimer and VP1 Trimer Proteins
3.8. Negative and Positive Selection Sites
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Oka, T.; Wang, Q.; Katayama, K.; Saif, L.J. Comprehensive review of human sapoviruses. Clin. Microbiol. Rev. 2015, 28, 32–53. [Google Scholar] [CrossRef] [PubMed]
- Scheuer, K.A.; Oka, T.; Hoet, A.E.; Gebreyes, W.A.; Molla, B.Z.; Saif, L.J.; Wang, Q. Prevalence of porcine noroviruses, molecular characterization of emerging porcine sapoviruses from finisher swine in the United States, and unified classification scheme for sapoviruses. J. Clin. Microbiol. 2013, 51, 2344–2353. [Google Scholar] [CrossRef] [PubMed]
- Oka, T.; Lu, Z.; Phan, T.; Delwart, E.L.; Saif, L.J.; Wang, Q. Genetic Characterization and Classification of Human and Animal Sapoviruses. PLoS ONE 2016, 11, e0156373. [Google Scholar] [CrossRef]
- Becker-Dreps, S.; Gonzalez, F.; Bucardo, F. Sapovirus: An emerging cause of childhood diarrhea. Curr. Opin. Infect. Dis. 2020, 33, 388–397. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Jahuira, H.; Gilman, R.H.; Alva, A.; Cabrera, L.; Okamoto, M.; Xu, H.; Windle, H.J.; Kelleher, D.; Varela, M.; et al. Etiological Role and Repeated Infections of Sapovirus among Children Aged Less than 2 Years in a Cohort Study in a Peri-urban Community of Peru. J. Clin. Microbiol. 2016, 54, 1598–1604. [Google Scholar] [CrossRef] [PubMed]
- Kagning Tsinda, E.; Malasao, R.; Furuse, Y.; Gilman, R.H.; Liu, X.; Apaza, S.; Espetia, S.; Cama, V.; Oshitani, H.; Saito, M. Complete Coding Genome Sequences of Uncommon GII.8 Sapovirus Strains Identified in Diarrhea Samples Collected from Peruvian Children. Genome Announc. 2017, 5, e01137-17. [Google Scholar] [CrossRef] [PubMed]
- Kumthip, K.; Khamrin, P.; Maneekarn, N. Molecular epidemiology and genotype distributions of noroviruses and sapoviruses in Thailand 2000-2016: A review. J. Med. Virol. 2018, 90, 617–624. [Google Scholar] [CrossRef] [PubMed]
- Mann, P.; Pietsch, C.; Liebert, U.G. Genetic Diversity of Sapoviruses among Inpatients in Germany, 2008–2018. Viruses 2019, 11, 726. [Google Scholar] [CrossRef]
- Zhuo, R.; Ding, X.; Freedman, S.B.; Lee, B.E.; Ali, S.; Luong, J.; Xie, J.; Chui, L.; Wu, Y.; Pang, X.; et al. Molecular Epidemiology of Human Sapovirus among Children with Acute Gastroenteritis in Western Canada. J. Clin. Microbiol. 2021, 59, e0098621. [Google Scholar] [CrossRef]
- Doan, Y.H.; Yamashita, Y.; Shinomiya, H.; Motoya, T.; Sakon, N.; Suzuki, R.; Shimizu, H.; Shigemoto, N.; Harada, S.; Yahiro, S.; et al. Distribution of Human Sapovirus Strain Genotypes over the Last Four Decades in Japan: A Global Perspective. Jpn J. Infect. Dis. 2023, 76, 255–258. [Google Scholar] [CrossRef]
- Magwalivha, M.; Kabue, J.P.; Traore, A.N.; Potgieter, N. Prevalence of Human Sapovirus in Low and Middle Income Countries. Adv. Virol. 2018, 2018, 5986549. [Google Scholar] [CrossRef]
- Wang, X.; Li, Y.; Deloria-Knoll, M.; Madhi, S.A.; Cohen, C.; Arguelles, V.L.; Basnet, S.; Bassat, Q.; Brooks, W.A.; Echavarria, M.; et al. Global burden of acute lower respiratory infection associated with human parainfluenza virus in children younger than 5 years for 2018: A systematic review and meta-analysis. Lancet Glob. Health 2021, 9, e1077–e1087. [Google Scholar] [CrossRef]
- Takahashi, T.; Kimura, R.; Shirai, T.; Sada, M.; Sugai, T.; Murakami, K.; Harada, K.; Ito, K.; Matsushima, Y.; Mizukoshi, F.; et al. Molecular Evolutionary Analyses of the RNA-Dependent RNA Polymerase (RdRp) Region and VP1 Gene in Human Norovirus Genotypes GII.P6-GII.6 and GII.P7-GII.6. Viruses 2023, 15, 1497. [Google Scholar] [CrossRef] [PubMed]
- Mizukoshi, F.; Nagasawa, K.; Doan, Y.H.; Haga, K.; Yoshizumi, S.; Ueki, Y.; Shinohara, M.; Ishikawa, M.; Sakon, N.; Shigemoto, N.; et al. Molecular Evolution of the RNA-Dependent RNA Polymerase and Capsid Genes of Human Norovirus Genotype GII.2 in Japan during 2004–2015. Front. Microbiol. 2017, 8, 705. [Google Scholar] [CrossRef] [PubMed]
- Chhabra, P.; Graaf, M.; Parra, G.I.; Chan, M.C.; Green, K.; Martella, V.; Wang, Q.; White, P.A.; Katayama, K.; Vennema, H.; et al. Corrigendum: Updated classification of norovirus genogroups and genotypes. J. Gen. Virol. 2020, 101, 893. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [PubMed]
- Bouckaert, R.; Vaughan, T.G.; Barido-Sottani, J.; Duchene, S.; Fourment, M.; Gavryushkina, A.; Heled, J.; Jones, G.; Kuhnert, D.; De Maio, N.; et al. BEAST 2.5: An advanced software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 2019, 15, e1006650. [Google Scholar] [CrossRef]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef]
- Russel, P.M.; Brewer, B.J.; Klaere, S.; Bouckaert, R.R. Model Selection and Parameter Inference in Phylogenetics Using Nested Sampling. Syst. Biol. 2019, 68, 219–233. [Google Scholar] [CrossRef]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [PubMed]
- Fourment, M.; Gibbs, M.J. PATRISTIC: A program for calculating patristic distances and graphically comparing the components of genetic change. BMC Evol. Biol. 2006, 6, 1. [Google Scholar] [CrossRef]
- Demšar, J.; Curk, T.; Erjavec, A.; Gorup, Č.; Hočevar, T.; Milutinovič, M.; Možina, M.; Polajnar, M.; Toplak, M.; Starič, A.; et al. Orange: Data Mining Toolbox in Python. J. Mach. Learn. Res. 2013, 14, 2349–2353. [Google Scholar]
- Lole, K.S.; Bollinger, R.C.; Paranjape, R.S.; Gadkari, D.; Kulkarni, S.S.; Novak, N.G.; Ingersoll, R.; Sheppard, H.W.; Ray, S.C. Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J. Virol. 1999, 73, 152–160. [Google Scholar] [CrossRef]
- Mirdita, M.; Schutze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold: Making protein folding accessible to all. Nat. Methods 2022, 19, 679–682. [Google Scholar] [CrossRef] [PubMed]
- Meng, E.C.; Goddard, T.D.; Pettersen, E.F.; Couch, G.S.; Pearson, Z.J.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Tools for structure building and analysis. Protein Sci. 2023, 32, e4792. [Google Scholar] [CrossRef] [PubMed]
- Hoie, M.H.; Gade, F.S.; Johansen, J.M.; Wurtzen, C.; Winther, O.; Nielsen, M.; Marcatili, P. DiscoTope-3.0: Improved B-cell epitope prediction using inverse folding latent representations. Front. Immunol. 2024, 15, 1322712. [Google Scholar] [CrossRef] [PubMed]
- Ponomarenko, J.; Bui, H.H.; Li, W.; Fusseder, N.; Bourne, P.E.; Sette, A.; Peters, B. ElliPro: A new structure-based tool for the prediction of antibody epitopes. BMC Bioinform. 2008, 9, 514. [Google Scholar] [CrossRef] [PubMed]
- da Silva, B.M.; Myung, Y.; Ascher, D.B.; Pires, D.E.V. epitope3D: A machine learning method for conformational B-cell epitope prediction. Brief. Bioinform. 2022, 23, bbab423. [Google Scholar] [CrossRef]
- Zhou, C.; Chen, Z.; Zhang, L.; Yan, D.; Mao, T.; Tang, K.; Qiu, T.; Cao, Z. SEPPA 3.0-enhanced spatial epitope prediction enabling glycoprotein antigens. Nucleic Acids Res. 2019, 47, W388–W394. [Google Scholar] [CrossRef] [PubMed]
- Shashkova, T.I.; Umerenkov, D.; Salnikov, M.; Strashnov, P.V.; Konstantinova, A.V.; Lebed, I.; Shcherbinin, D.N.; Asatryan, M.N.; Kardymon, O.L.; Ivanisenko, N.V. SEMA: Antigen B-cell conformational epitope prediction using deep transfer learning. Front. Immunol. 2022, 13, 960985. [Google Scholar] [CrossRef]
- Weaver, S.; Shank, S.D.; Spielman, S.J.; Li, M.; Muse, S.V.; Kosakovsky Pond, S.L. Datamonkey 2.0: A Modern Web Application for Characterizing Selective and Other Evolutionary Processes. Mol. Biol. Evol. 2018, 35, 773–777. [Google Scholar] [CrossRef] [PubMed]
- Kosakovsky Pond, S.L.; Frost, S.D. Not so different after all: A comparison of methods for detecting amino acid sites under selection. Mol. Biol. Evol. 2005, 22, 1208–1222. [Google Scholar] [CrossRef]
- Murrell, B.; Moola, S.; Mabona, A.; Weighill, T.; Sheward, D.; Kosakovsky Pond, S.L.; Scheffler, K. FUBAR: A fast, unconstrained bayesian approximation for inferring selection. Mol. Biol. Evol. 2013, 30, 1196–1205. [Google Scholar] [CrossRef] [PubMed]
- Murrell, B.; Wertheim, J.O.; Moola, S.; Weighill, T.; Scheffler, K.; Kosakovsky Pond, S.L. Detecting individual sites subject to episodic diversifying selection. PLoS Genet. 2012, 8, e1002764. [Google Scholar] [CrossRef]
- Wu, F.T.; Oka, T.; Kuo, T.Y.; Doan, Y.H.; Tzu-Chi Liu, L. Sapoviruses detected from acute gastroenteritis outbreaks and hospitalized children in Taiwan. J. Formos. Med. Assoc. 2021, 120, 1591–1601. [Google Scholar] [CrossRef]
- Luo, X.; Deng, J.K.; Mu, X.P.; Yu, N.; Che, X. Detection and characterization of human astrovirus and sapovirus in outpatients with acute gastroenteritis in Guangzhou, China. BMC Gastroenterol. 2021, 21, 455. [Google Scholar] [CrossRef] [PubMed]
- Desselberger, U. Caliciviridae Other Than Noroviruses. Viruses 2019, 11, 286. [Google Scholar] [CrossRef]
- Katayama, K.; Miyoshi, T.; Uchino, K.; Oka, T.; Tanaka, T.; Takeda, N.; Hansman, G.S. Novel recombinant sapovirus. Emerg. Infect. Dis. 2004, 10, 1874–1876. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.H.; Han, M.G.; Funk, J.A.; Bowman, G.; Janies, D.A.; Saif, L.J. Genetic diversity and recombination of porcine sapoviruses. J. Clin. Microbiol. 2005, 43, 5963–5972. [Google Scholar] [CrossRef] [PubMed]
- Tohma, K.; Kulka, M.; Coughlan, S.; Green, K.Y.; Parra, G.I. Genomic Analyses of Human Sapoviruses Detected over a 40-Year Period Reveal Disparate Patterns of Evolution among Genotypes and Genome Regions. Viruses 2020, 12, 516. [Google Scholar] [CrossRef] [PubMed]
- Saito, M.; Tsukagoshi, H.; Ishigaki, H.; Aso, J.; Ishii, H.; Okayama, K.; Ryo, A.; Ishioka, T.; Kuroda, M.; Saruki, N.; et al. Molecular evolution of the capsid (VP1) region in human norovirus genogroup II genotype 3. Heliyon 2020, 6, e03835. [Google Scholar] [CrossRef] [PubMed]
- Motoya, T.; Nagasawa, K.; Matsushima, Y.; Nagata, N.; Ryo, A.; Sekizuka, T.; Yamashita, A.; Kuroda, M.; Morita, Y.; Suzuki, Y.; et al. Molecular Evolution of the VP1 Gene in Human Norovirus GII.4 Variants in 1974-2015. Front. Microbiol. 2017, 8, 2399. [Google Scholar] [CrossRef] [PubMed]
- Matsushima, Y.; Mizukoshi, F.; Sakon, N.; Doan, Y.H.; Ueki, Y.; Ogawa, Y.; Motoya, T.; Tsukagoshi, H.; Nakamura, N.; Shigemoto, N.; et al. Evolutionary Analysis of the VP1 and RNA-Dependent RNA Polymerase Regions of Human Norovirus GII.P17-GII.17 in 2013-2017. Front. Microbiol. 2019, 10, 2189. [Google Scholar] [CrossRef] [PubMed]
- Winder, N.; Gohar, S.; Muthana, M. Norovirus: An Overview of Virology and Preventative Measures. Viruses 2022, 14, 2811. [Google Scholar] [CrossRef] [PubMed]
- Nagasawa, K.; Matsushima, Y.; Motoya, T.; Mizukoshi, F.; Ueki, Y.; Sakon, N.; Murakami, K.; Shimizu, T.; Okabe, N.; Nagata, N.; et al. Genetic Analysis of Human Norovirus Strains in Japan in 2016–2017. Front. Microbiol. 2018, 9, 1. [Google Scholar] [CrossRef]
- Groenen, M.A. A decade of pig genome sequencing: A window on pig domestication and evolution. Genet. Sel. Evol. 2016, 48, 23. [Google Scholar] [CrossRef]
- Kobayashi, M.; Matsushima, Y.; Motoya, T.; Sakon, N.; Shigemoto, N.; Okamoto-Nakagawa, R.; Nishimura, K.; Yamashita, Y.; Kuroda, M.; Saruki, N.; et al. Molecular evolution of the capsid gene in human norovirus genogroup II. Sci. Rep. 2016, 6, 29400. [Google Scholar] [CrossRef] [PubMed]
- Luchs, A.; Timenetsky Mdo, C. Group A rotavirus gastroenteritis: Post-vaccine era, genotypes and zoonotic transmission. Einstein 2016, 14, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Tomori, O.; Oluwayelu, D.O. Domestic Animals as Potential Reservoirs of Zoonotic Viral Diseases. Annu. Rev. Anim. Biosci. 2023, 11, 33–55. [Google Scholar] [CrossRef] [PubMed]
- Bank-Wolf, B.R.; Konig, M.; Thiel, H.J. Zoonotic aspects of infections with noroviruses and sapoviruses. Vet. Microbiol. 2010, 140, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Amin, M.R.; Siddiqui, M.S.; Ahmed, D.; Ahmed, F.; Hossain, A. B- and T-cell epitope mapping of human sapovirus capsid protein: An immunomics approach. Int. J. Bioinform. Res. Appl. 2011, 7, 287–298. [Google Scholar] [CrossRef] [PubMed]
- Ji, X.; Guo, C.; Dai, Y.; Chen, L.; Chen, Y.; Wang, S.; Sun, Y. Genomic Characterization and Molecular Evolution of Sapovirus in Children under 5 Years of Age. Viruses 2024, 16, 146. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mizukoshi, F.; Kimura, R.; Shirai, T.; Hirata-Saito, A.; Hiraishi, E.; Murakami, K.; Doan, Y.H.; Tsukagoshi, H.; Saruki, N.; Tsugawa, T.; et al. Molecular Evolutionary Analyses of the RNA-Dependent RNA Polymerase (RdRp) Region and VP1 Gene in Sapovirus GI.1 and GI.2. Microorganisms 2025, 13, 322. https://doi.org/10.3390/microorganisms13020322
Mizukoshi F, Kimura R, Shirai T, Hirata-Saito A, Hiraishi E, Murakami K, Doan YH, Tsukagoshi H, Saruki N, Tsugawa T, et al. Molecular Evolutionary Analyses of the RNA-Dependent RNA Polymerase (RdRp) Region and VP1 Gene in Sapovirus GI.1 and GI.2. Microorganisms. 2025; 13(2):322. https://doi.org/10.3390/microorganisms13020322
Chicago/Turabian StyleMizukoshi, Fuminori, Ryusuke Kimura, Tatsuya Shirai, Asumi Hirata-Saito, Eri Hiraishi, Kosuke Murakami, Yen Hai Doan, Hiroyuki Tsukagoshi, Nobuhiro Saruki, Takeshi Tsugawa, and et al. 2025. "Molecular Evolutionary Analyses of the RNA-Dependent RNA Polymerase (RdRp) Region and VP1 Gene in Sapovirus GI.1 and GI.2" Microorganisms 13, no. 2: 322. https://doi.org/10.3390/microorganisms13020322
APA StyleMizukoshi, F., Kimura, R., Shirai, T., Hirata-Saito, A., Hiraishi, E., Murakami, K., Doan, Y. H., Tsukagoshi, H., Saruki, N., Tsugawa, T., Kidera, K., Suzuki, Y., Sakon, N., Katayama, K., Kageyama, T., Ryo, A., & Kimura, H. (2025). Molecular Evolutionary Analyses of the RNA-Dependent RNA Polymerase (RdRp) Region and VP1 Gene in Sapovirus GI.1 and GI.2. Microorganisms, 13(2), 322. https://doi.org/10.3390/microorganisms13020322