Abstract
Terpenes are the largest category of specialised metabolites. Aerobic endospore-forming bacteria (AEFB), a diverse group of microorganisms, can thrive in various habitats and produce specialised metabolites, including terpenes. This study investigates the potential for terpene biosynthesis in 10 AEFB strain whole-genome sequences by performing a bioinformatics analyses to identify genes associated with these isoprene biosynthesis pathways. Specifically, we focused on the sequences coding for enzymes in the methylerythritol-phosphate (MEP) pathway and the polyprenyl synthase family, which play crucial roles in synthesising terpene precursors together with terpene synthases. A comparative analysis revealed the unique genetic architecture of these biosynthetic gene clusters (BGCs). Our results indicated that some strains possessed the complete genetic machinery required to produce terpenes such as squalene, hopanoids, and carotenoids. We also reconstructed phylogenetic trees based on the amino acid sequences of terpene synthases, which aligned with the phylogenetic relationships inferred from the whole-genome sequences, suggesting that the production of terpenes is an ancestor property in AEFB. Our findings highlight the importance of genome mining as a powerful tool for discovering new biological activities. Furthermore, this research lays the groundwork for future investigations to enhance our understanding of terpene biosynthesis in AEFB and the potential applications of these Brazilian environmental strains.
1. Introduction
Specialised or secondary metabolites are not essential for growth. Nevertheless, they play a relevant ecological role by providing nutrients in competitive environments and offering adaptive advantages to the producing organism [1,2]. Terpenes are hydrocarbons of linked five-carbon units of isoprene [3], and they are the largest category of specialised metabolites, with over 80,000 known structures [4]. These molecules present a wide range of applications in pharmaceuticals, food, and cosmetics, among other relevant industries [3,4].
Terpenes are fundamental in the protection against diseases in plants. This property resulting from the antimicrobial activity of these molecules prevents the reproduction and development of phytopathogenic microorganisms [5]. In addition, these specialised metabolites can act as hormones, protective pigments responsible for the colours of various tissues, and odours, which protect plants against herbivory [6,7]. The social and bioeconomic importance of terpenes has increased their relevance in the pharmaceutical and cosmetics industries. Terpenes also play a prominent role in human health due to their excellence as antitumor, antimicrobial, anti-allergic, anti-inflammatory, and antioxidant agents, among other applications [5,8]. Likewise, terpenes are suitable for the food and beverage industries as preservatives or aromatic additives [9]. Additionally, they are recognised as possible energy-transition agents in biofuel formulations.
Terpenes were believed to be produced exclusively by plants, but it is now understood that their synthesis occurs in bacteria, fungi, protozoa, and invertebrates [6,10,11,12]. Various biochemical pathways are involved in terpene production, which can be independent or integrated [13]. The synthesis of these isoprenes starts from the precursor isopentenyl pyrophosphate (IPP), produced via either the mevalonate (MVA) or the methylerythritol-phosphate (MEP) pathways [14].
The enzyme isopentenyl pyrophosphate isomerase (IDI) converts IPP into dimethylallyl pyrophosphate (DMAPP), an IPP isomer. Subsequently, geranyl pyrophosphate synthase (GPPS) condenses these two isomers to produce geranyl pyrophosphate (GPP), which serves as monoterpenes’ precursor. GPPS belongs to the enzyme family polyprenyl synthase (PPS), which includes farnesyl pyrophosphate synthase (FPPS) and geranyl-geranyl pyrophosphate synthase (GGPPS). FPPS is responsible for sesquiterpenes’ and triterpenes’ syntheses, and GGPPS is involved in diterpene and tetraterpene precursors. An overview of the substrates for the MEP pathway, the enzymes of the PPS family, and their corresponding products is provided in Table 1.

Table 1.
Enzymes involved in terpene biosynthesis. Profile of MEP pathway and polyprenyl synthase family molecules.
Plants and fungi utilise the MVA and MEP pathways for terpene synthesis. Indeed, both pathways are involved in the biosynthesis of gibberellins, a diterpene plant hormone class, crucial for various developmental processes in Arabidopsis thaliana [15]. Pardo et al. [16] expressed a geraniol synthase gene from Ocimum basilicum (sweet basil) in a Saccharomyces cerevisiae wine strain. This construction changed the terpene profile of wine, because these self-aromatising recombinant yeasts overproduce these plant metabolites in wines de novo.
Specialised metabolites exhibit diverse biological activities within fungal communities, which play vital roles in adaptation, defence, competition, and communication [17]. Comparative genomics and metabolomics were used to investigate the high diversity of terpene BGCs potentially involved in the fungal competition and communication of three Suillus species [18]. Though the functionality of these BGCs could not be understood adequately during this study, these authors observed that terpenes were significantly more abundant in co-culture conditions. The mycelium of wood-rotting fungi Polyporus brumalis produces two sesquiterpenes by the MVA and MEP pathways [19]. Even if the co-expression of genes involved in these pathways can help elucidate these sesquiterpenes’ synthesis, these observations do not fully explain the production mechanisms.
Most bacteria natively possess the MEP pathway for terpene synthesis [14]. It includes species of Bacillus and related genera [20,21] called aerobic endospore-forming bacteria (AEFB). In contrast other Gram-positive cocci and Lactobacillus spp. exclusively rely on the MVA pathway [21]. Conversely, Listeria spp. and a subset of Actinobacteria, such as Streptomyces spp., utilise one or the other to produce these isoprenes. Many terpenes, such as the antimicrobial albaflavenone [22], germacrene D, and pentalenene [6], have been observed in bacteria, notably strains of Actinomicetales and Gram-positives. However, terpenes’ physiological and ecological roles in these organisms remain largely unknown.
The genus Streptomyces is considered a model for terpene studies and heterologous expression. However, recent advancements in the bioproduction of lycopene are well documented, showcasing engineered Escherichia coli strains bearing modified MEP pathways with the potential to develop monoterpene-enhanced wines [23]. Despite the recognisable potential for expressing BGCs, the heterologous biosynthesis of terpenes on alternative backgrounds, such as Bacillus species, is still scarce, lagging behind hosts such as E. coli, Streptomyces spp. and S. cerevisiae [21,24].
Species allocated to the genus Bacillus and related genera—whether assigned to the same or different orders and families—are designated aerobic endospore-forming bacteria (AEFB), and soil is considered their primary reservoir [25,26,27]. To acquire knowledge on AEFB and gain insights into their potential as sources of novel bioactive substances, including terpenes, we previously isolated 312 strains through heat-shocking soil samples collected in random areas of the Federal District, Midwest region of Brazil. These environmental strains are designated SDF0001-SDF0312 and are deposited at the Coleção de Bactérias aeróbias formadoras de endósporos (CBafes, or AEFB Collection–AEFBC). The CBafes is hosted at the University of Brasilia and is currently undergoing taxonomic classification using a polyphasic approach [28,29,30,31].
Endosporulation is an outstanding differentiation mechanism that evolved to help some bacteria survive adverse conditions [32,33]. The resulting dormant spore remains sensitive to environmental changes and can germinate to return to active metabolism and reproduction [33]. The ability to form spores has been observed only inside the phylum Firmicutes, recently renamed Bacillota [34]. This phylum allocates low G+C bacteria, most of which have Gram-positive cell wall structures, distributed in eight classes [35].
Spore formation is not a universal characteristic inside Bacillota, but endospore-formers share a minimal homologous gene set involved in this event [35]. Spore formation is widespread in the two Bacillota major classes—Bacilli, which are aerobic or facultative, and Clostridia, which are anaerobic [34,35].
Besides their remarkable spore resistance, AEFB can thrive across large temperatures and pH levels and exhibit metabolic diversity. These traits contribute to their ubiquity. These prokaryotic cells are known to synthesise a diverse array of specialised metabolites, which include terpenes [20,26,36,37,38]. Researchers extensively study these exceptional characteristics in various industrial contexts [20]. These metabolites’ ecological and socioeconomic significance is well recognised, as they can promote plant growth, manage insect pests and disease vectors, and possess immunosuppressive, antimicrobial, and antitumor activities [20,38,39,40]. Despite their potential importance, the terpene biosynthetic pathways in AEFB remain largely unexplored.
Genome prediction offers a powerful means for identifying and characterising the genetic basis of terpene production in bacteria. Here, we used a genome mining approach to investigate the potential for terpene biosynthesis in the whole genome of 10 SDF strains (Table 2). We focused on identifying the key genes involved in the MEP pathway and the enzyme of the PPS family by examining 16 BGCs we previously identified in these genomes employing the antiSMASH in silico pipeline [41]. We also sought to identify genes encoding terpene synthases (TSs), the enzymes responsible for the final steps in terpene biosynthesis. Finally, we reconstructed phylogenetic trees based on corresponding amino acid sequences of the TSs found to resemble phylogenetic relationships based on the whole genomes of the respective SDF strains. Our findings provide new insights into the diversity and evolution of terpene biosynthesis in AEFB and highlight the potential of these environmental strains as a source of novel terpenes with valuable applications.
Table 2.
Comparative genomic features of the ten SDF strains analysed.
2. Materials and Methods
2.1. Bacterial Strains
The 10 SDF strains evaluated in this study (Table 2) are deposited at the Coleção de Bactérias aeróbias formadoras de endósporos (CBafes, or Aerobic Endospore-Forming Bacteria—AEFB Collection), hosted at the University of Brasilia, Brazil. Six genomes were explicitly sequenced for this study, plus four previously sequenced genomes from the same culture collection, accessible at the NCBI. They were isolated from Brazilian soils, preserved as dry spores in filter paper, and stored at room temperature, as described in Orem et al. [28] and Cavalcante et al. [29]. It is important to note that the isolation of SDF strains and the related studies did not intend to explore ecological interactions of all kinds among soil and/or any microbial community type. Furthermore, the location for soil collection was randomly chosen.
2.2. Ethics Statement
The specific permissions required to collect the SDF strains used in this study were endorsed by the Federal Brazilian Authority (CNPq; Authorisation of Access and Sample of Genetic Patrimony n° 010439/2015-3). Sampling did not involve endangered or protected species.
2.3. Sequencing, Assembly, Annotation, and Data Availability
The total DNA of the six SDF strains sequenced explicitly for this study (Table 2) was extracted and purified using the Wizard genomic kit (Promega, Madison, WI, USA) following the manufacturer’s instructions and sequenced using an Illumina Miseq PE (San Diego, CA, CA, USA) (150 bp) platform at the Catholic University of Brasilia (Brazil). MiSeq reads were evaluated for quality control using FASTQC 0.12.0 (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/, accessed on 10 May 2016), followed by trimming trichromatics (a phred quality score threshold of 33) and assembly into contigs/scaffolds using the A5-mise pipeline [42]. This pipeline automatically processes adapter trimming, quality filtering, error correction, contig, scaffold generation, and misassembly detections. The genomes were deposited at the NCBI (Table 2). Gene annotation was performed using the NCBI prokaryotic genome annotation pipeline [43].
2.4. Whole Genome-Based Features and Phylogeny
A whole genome-based phylogeny analysis was performed using the OrthologSorter tool [44], available at https://git.facom.ufms.br/bioinfo/orthologsorter (accessed on 25 February 2025). Orthologsorter generates, among other data, protein families shared across all genomes (core genome). Orhtologsorter employs BLASTp [45], using as parameters a BLOSUM62 substitution matrix, gap opening penalty of 11, gap extension penalty of 1, and an E-value cutoff of 1 × 10−5, and OrthoMCL [46] tools with default parameters (inflation value of 1.5 for the Markov clustering algorithm, a BLASTp E-value cutoff of 1 × 10−5, a minimum per cent match length of 50%, and a per cent identity cutoff of 30%) to determine orthology. For our set of the 10 SDF strain genomes, plus the included outgroup Staphylococcus pseudintermedius, 918 core families have been found. These families were aligned, and, after removing poorly aligned positions and divergent regions using GBlocks [47], the resulting whole alignment was used to build the phylogenetic tree with RAxML [48] with a PROTCATJTT substitution model, rapid bootstrapping (1000 replicates), and a subsequent maximum likelihood search.
2.5. BGC Predictions
The antiSMASH 6.0 bacterial standalone version [49] optimised for prokaryotic sequences (https://antismash.secondarymetabolites.org/#!/start, accessed on 4 July 2023) was run on the 10 genomes of the SDF strains to identify BGCs linked to terpene synthesis (Table 2). The accuracy parameter of the detected clusters was relaxed (full-featured run) with algorithms provided by the antiSMASH platform (KnownClusterBlast, ActiveSiteFinder, ClusterPfam, ClusterBlast, and Pfam-based GO term annotation). The BGC similarity level (0–100%) reported for a specific metabolite was obtained by crossing over data available in the Minimum Information about a Biosynthetic Gene cluster (MiBig) platform (https://mibig.secondarymetabolites.org, accessed on 1 March 2023). The percentage index indicates the number of the gene sequences within a BGC that have a hit to any gene in a particular BGC in the MiBiG’s reference strain related to terpene production.
2.6. MEP Pathway Reconstruction
Pathway Tools 26.5, a systems-biology software—associated with the BioCyc Pathway/Genome Database Collection (http://bioinformatics.ai.sri.com/ptools/, accessed on 1 March 2023), was used to predict the gene sequences coding for the MEP pathway catalysts (Table 1). The algorithm PathoLogic was used to create a Pathway/Genome Database (PGDB) containing the predicted metabolic pathways of the respective strains. The PGDB was built using a cutoff score of 0.15 and the inference tools Transport Inference Parser, Pathway Hole Filler, Operon Predictor, and Protein Complex Predictor in the activated mode. The Omics Dashboard tool was used to orient metabolomic data to create one diagram showing an aggregated system-oriented view of the metabolic routes of the 10 SDF strains.
2.7. Detection of Polyprenyl Synthase Enzymes
Data from the NCBI platform (https://ncbi.nlm.nih.gov, accessed on 1 March 2023) were used to investigate the presence of sequences coding for PPS enzymes (Table 1) in the 10 SDF strains studied (Table 2). To this end, a database containing 6146 files of amino acid sequences (.fasta) of polyprenyl synthases—obtained from species allocated to the four orders explored in this study and deposited at NCBI—was built. The extraction of protein sequences from each SDF strain genome (.gbk) was accomplished using the script available in Bogdanove et al. [50]. The alignment and comparison of the amino acid sequences between the database created and SDF genome sequences were achieved using BLASTp amino acid sequences (https://blast.ncbi.nlm.nih.gov/Blast.cgi?PAGE=Proteins, accessed on 4 July 2023) where the highest hits were considered to detect the enzyme presence in the SDF strains.
2.8. Similarity of the Enzyme Set for Terpene Production
The putative SDF producers, the enzymes of the MEP pathway and PPS family (Table 1), and the TSs detected for each strain were arranged in heatmaps [51] using the software R (https://www.r-project.org, accessed on 27 February 2025). The dichotomous values 0 (for the absence of catalyst) and 1 (for the presence of catalyst) were taken as binary variables representing these associations. Pearson’s correlation was employed to cluster the SDF strains, taking a similar set of enzyme results [52].
2.9. Phylogenetic Tree Reconstruction Based on Terpene Synthase Content
The amino acid sequences of the TSs identified were extracted from the corresponding BGC obtained by antiSMASH 6.0 [49] and using an in-house script in the Biopython programming language (http://biopython.org/DIST/docs/tutorial/Tutorial.html, accessed on 1 March 2023). Inside MEGA software version 11, the amino acid sequences (.fasta) obtained were aligned using ClustalW with default parameters (https://www.megasoftware.net/ClustalW, accessed on 18 October 2024). The file generated (.mas) was used to reconstruct phylogenetic trees employing the maximum likelihood statistical method based on 1000 bootstrap replicates.
3. Results
3.1. SDF Strain Genome Features
This study describes genomic resources for 10 cultivable environmental AEFB samples designated SDF strains (Table 2). We presented high-quality whole-genome sequences from 10 SDF strains based on Illumina. These samples corresponded to four orders, four families, six genera, and nine species allocated to the phylum Bacillota, class Bacilli (Table 2). Six samples were assigned to the order Bacillales, family Bacillaceae. Among them, four strains belonged to three different Bacillus spp.: Bacillus pumilus SDF0011, Bacillus safensis SDF0016, Bacillus velezensis SDF0141, and Bacillus velezensis SDF0150. The family Bacillaceae was also represented by two other genera and species of named strains, Heyndrickxia oleronia SDF0015 and Peribacillus simplex SDF0024, referred to here as Pe. simplex SDF0024. Inside the order Caryophanales, the family Caryophanaceae, genus Lysinibacillus were represented by two strains, Lysinibacillus fusiformis SDF0005 and Lysinibacillus sphaericus SDF0037. The strain Paenibacillus popilliae SDF0028 belonged to the order Paenibacillales, family Paenibacillaceae, and genus Paenibacillus. Finally, Brevibacillus brevis SDF0063 was allocated to the order Brevibacillales, family Brevibacillaceae and is referred to here as Br. brevis SDF0063. Genome analysis of the SDF strains uncovered considerable differences in genome size, scaffold number, N50, GC content, coding sequences (CDSs), protein-coding regions, pseudo genes, rRNAs, and tRNAs, as detailed in Table 2. Briefly, the genome sizes ranged from 3,674,191 to 6,580,875 bp, with the scaffold numbers varying from 15 up to 75 and GC content (%) spanning from 34.7 to 47.3.
A maximum likelihood method was applied to reconstruct a phylogenetic tree based on the results of the OrthologSorter tool (available in https://git.facom.ufms.br/bioinfo/orthologsorter, accessed on 25 February 2025) [44] in 10 SDF strain whole-genomes (Table 2) and S. pseudintermedius as an outgroup (Figure 1), which resulted in two major clades. The Lysinibacillus spp., L. fusiformis SDF0005, and L. sphaericus SDF0037 clustered together in the most distinct branch. The genomes of H. oleronia SDF0015 and Pe. simplex SDF0024 formed a branch that also included the four Bacillus strains, B. pumilus SDF0011, B. safensis SDF0016, Bacillus velezensis SDF0141, and Bacillus velezensis SDF0150, on the second major clade. The strains P. popilliae SDF0028 and Br. brevis SDF0063 were also positioned as the most distinct SDF strains analysed.
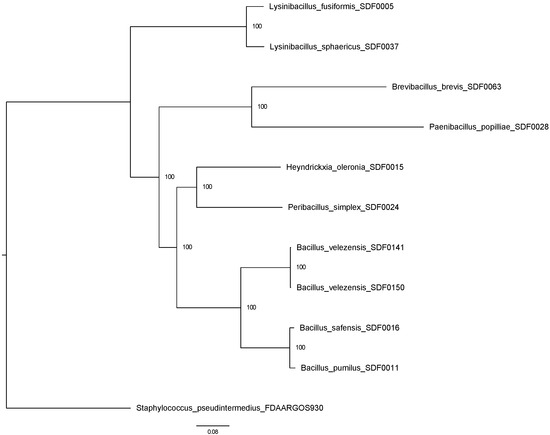
Figure 1.
Phylogenetic relationship of the ten SDF strains based on a whole-genome analysis. An unrooted maximum likelihood tree was constructed using RAxML (PROTCATJTT model) with 918 core protein families. Bootstrap values (1000 replicates) are shown at the nodes. Staphylococcus pseudintermedius was used as an outgroup. Strain classifications are indicated in the branches, and a distance scale bar is displayed at the bottom.
3.2. MEP Pathway Reconstruction
BGCs are a locally clustered group of two or more genes in a particular genome. antiSMASH is an in silico pipeline offering the detection and analysis of many BGC types [49]. These gene clusters encode biosynthetic pathways for specialised metabolite production with diverse functions, including chemical variants [53]. Previously, using the antiSMASH 6.0 bacterial standalone version [49], we identified 153 putative BGCs codifying for 20 different classes of specialised metabolites synthesis in 10 SDF strains (Table 2) deposited at CBafes [41]. Among these, 16 were related to terpene synthesis. In this work, the potential of these SDF strains for terpene biosynthesis was further addressed by taking advantage of these 16 high-quality BGC sequences.
The algorithm PathoLogic (http://bioinformatics.ai.sri.com/ptools/, accessed on 1 March 2023) predicted that all the corresponding gene sequences coding for the seven enzymes (DXS, DXR, MCT, CMK, MDS, HDS, and HDR) that catalyse the MEP pathway reactions (Table 1) would be found among the 10 SDF genomes (Table 2). The PGDB obtained is represented in a diagram aggregating a system-oriented view of the metabolic routes of the 10 SDF strains generated by the Omics Dashboard tool (Figure 2). In addition to MEP route enzymes, this tool also detected the enzyme IDI (Table 1), responsible for both IPP isomerisation to DMAPP and the subsequent IPP and DMAPP condensation that generates the first substrate in the terpenes’ production (Table 1). The information coding for the enzyme IDI was present in all SDF strains analysed, except for the L. fusiformis SDF0005 strain (Figure 2).
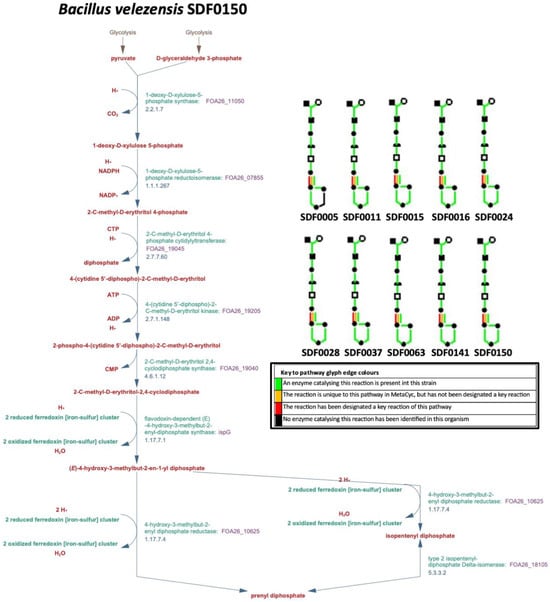
Figure 2.
Methylerythrotol-phosphate pathway reconstruction. The Pathway Tools software created a Pathway/Genome Database (PGDB) that includes the predicted metabolic pathways of the respective strains. The Omics Dashboard tool was then applied to align the metabolomic data, generating diagrams to provide an aggregated system-oriented view of the predicted metabolic route information found in the genome of Bacillus velezensis SDF0150 (larger diagram). Smaller diagrams representing these routes in Bacillus velezensis SDF0150 and the remaining nine strains are displayed in the top right corner. A key to the colours of the pathway glyph edges is indicated. The SDF strain designation is indicated: Lysinibacillus fusiformis SDF0005, Bacillus pumilus SDF0011, Heyndrickxia oleronia SDF0015, Bacillus safensis SDF0016, Peribacillus simplex SDF0024, Paenibacillus popilliae SDF0028, Lysinibacillus sphaericus SDF0037, Brevibacillus brevis SDF0063, and Bacillus velezensis SDF0141.
3.3. Detection of Polyprenyl Synthase Enzymes
Using BLASTp, the alignment and comparison of the amino acid sequences between the database we created (6146 files; see Section 2) and SDF sequences revealed the presence of the PPS family —the enzymes responsible for the conversion of IPP to GPP, FPP, and GGPP (Table 1). The strains L. fusiformis SDF0005; H. oleronia SDF0015; Pe. simplex SDF0024; B. velezensis SDF0141; and B. velezensis SDF0150 presented a >98% amino acid similarity, while H. oleronia SDF0015 exhibited a 67.86% one (Table 3). The amino acid sequences for PPS were not detected in the remaining SDF strains.
Table 3.
Occurrence of the polyprenyl synthase family across the ten SDF strains analysed.
3.4. Prediction of Biosynthetic Gene Clusters Associated with Terpene Synthesis
At least three gene sequences inside the 16 BGCs uncovered by antiSMASH [41] directed the synthesis of three TSs: (i) a sqhC gene, coding for a squalene-hopene cyclase (SHC); (ii) a gene encoding undetermined activity related to the production of the phytoene and/or squalene synthase (PSS); and (iii) a crti gene, coding for a phytoene desaturase (PDS). Table 3 outlines the TS coding sequences for the strains used in this study.
Out of these 10 SDF strains, the sequences coding for the SHC were detected in eight genomes: B. pumilus SDF0011; H. oleronia SDF0015; B. safensis SDF0016; Pe. simplex SDF0024; P. popilliae SDF0028; Br. brevis SDF0063; B. velezensis SDF0141, and B. velezensis SDF0150 (Table 4; Figure 3). The sequences coding for the enzymes of the PSS synthase family were found in seven genomes: L. fusiformis SDF0005, B. pumilus SDF0011, B. safensis SDF0016, Pe. simplex SDF0024, L. sphaericus SDF0037, B. velezensis SDF0141, and B. velezensis SDF0150 (Table 4; Figure 4). The BGC structures involved in terpene production varied among the analysed strains, as detected by antiSMASH. Depending on the species, the genes responsible for synthesising SHC and PSS were flanked by different genetic elements within the BGCs. However, an exception was noted for the strains of B. velezensis SDF0141 and B. velezensis SDF0150, which exhibited structurally similar BGCs (Figure 3 and Figure 4).
Table 4.
Gene coding for the TS enzymes identified in the ten SDF strains analysed using antiSMASH.
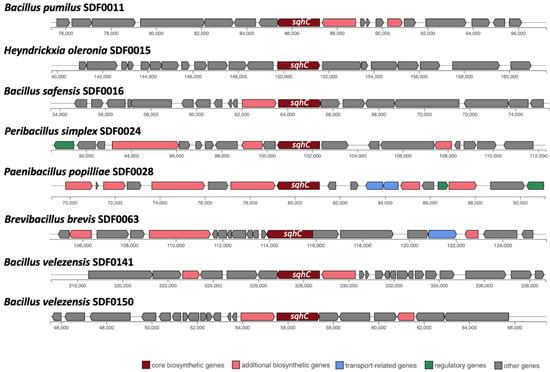
Figure 3.
Structure of biosynthetic gene clusters involved in squalene-hopene cyclase expression in the genome of the SDF strains. AntiSMASH identified sqhC, which directs the production of the squalene-hopene cyclase (SHC) as a core biosynthetic gene (brown) in eight strains. Predicted gene functions (colour-coded) are shown at the bottom.
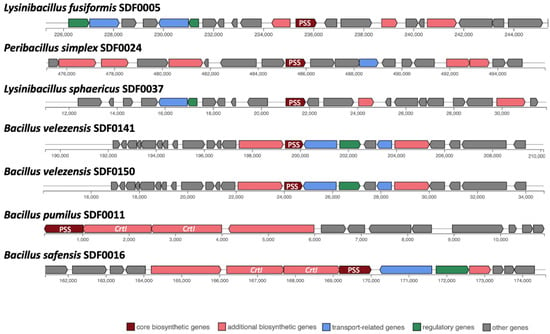
Figure 4.
Biosynthetic gene clusters involved in the phytoene/squalene synthase and phytoene desaturase in SDF strains. A gene sequence that directs the phytoene and/or squalene synthase (PSS) family enzyme production was identified as a core biosynthetic gene (brown) inside the BGCs detected in seven SDF genomes by antiSMASH. In Bacillus pumilus SDF0011 and Bacillus safensis SDF0016, the gene codifying PSS was adjacent to a copy of the gene crtI, which drives phytoene desaturase synthesis. The colour boxes (bottom) indicate the predicted gene functions in the biosynthesis of terpenes.
In contrast, the genomes of B. pumilus SDF0011 and Bacillus safensis SDF0016 contained the genes encoding the PSS and SHC enzymes positioned adjacent to two copies of the crtI gene, which codes for a PDS (Figure 4). The BGC containing these two gene copies in the genomes of these two strains are reported to take part in carotenoid production [54]. Furthermore, these latter BGCs presented a 50% similarity compared to the corresponding sequence described for strain Halobacillus halophylus DSM2266 in the MIBiG platform (reference number BGC0000645) used by antiSMASH as a reference to predict this metabolite. The similarity percentages (0–100%) indicate the number of genes within the reference that have a hit to any genes in a particular BGC related to terpene production and that were recognised by antiSMASH.
3.5. Distribution of the Enzyme Set for Terpene Production Among the 10 SDF Strains
The distribution of the predicted enzymes obtained from the in silico translation of the corresponding gene sequences coding for the seven MEP pathway enzymes (DXS, DXR, MCT, CMK, MDS, HDS, and HDR), along with the enzyme IDI, were also analysed in the SDF strain genomes. The PPS family, the three TS enzymes SHC, PSS, and PDS, were also included. To this end, Pearson’s correlation [52] was employed to cluster the 10 SDF strains (Table 2; Figure 5) based on the ensemble of enzymes engaged in the terpene’s synthesis detected. We constructed a heatmap [51] representing the presence or absence of an enzyme in a particular strain to enhance the potential visual distribution of the enzyme distributions among the strains. Two major clades were distinguished (Figure 5). The first comprised B. pumilus SDF0011 and B. safensis SDF0016, which shared an identical profile, or 11 out of the 12 enzymes detected. The second major clade was further split. Pe. simplex SDF0024, B. velezensis SDF0141, and B. velezensis SDF0150 also shared the same profile, bearing an equivalent set of 11 enzymes (Figure 5). The next subclade embraced Lysinibacillus sphaericus SDF0037 and L. fusiformis SDF0005, despite the dissimilar enzymatic profiles (Figure 5). Although H. oleronia SDF0015 is the only representative of a subclade, the correlation showed that the enzymatic set for terpene production in this strain was compatible with the subclade encompassed by P. popilliae SDF0028 and Br. brevis SDF0063, except that one out of nine enzymes was missing.
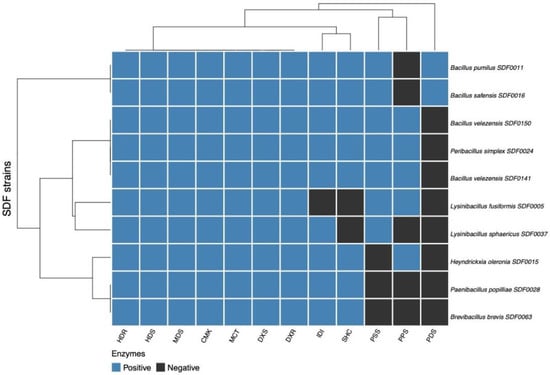
Figure 5.
Distribution of the ensemble of enzymes engaged in the terpenes’ synthesis among the ten SDF strains. A heatmap (Person’s correlation-based) shows the SDF strains clustered into two sections based on the presence (blue squares) or absence (black squares) of the respective gene coding for the enzyme in a particular species genome (right). The protein set (Table 1) includes catalysts of the MEP route detected by the Pathways tools, along with the polyprenyl synthase family (PPS) detected by BLASTp, and the TS squalene-hopene cyclase, phytoene and/or squalene synthase (PSS), and phytoene desaturase (PDS) identified by antiSMASH are described on the bottom.
3.6. SDF Strains’ Evolutionary Relationship Based on Two TS Amino Acid Sequences
As described above, the BGC sequences involved in the terpenes’ production detected by the antiSMASH tool encompassed three TS enzyme gene sequences (Table 4). A phylogenetic tree was reconstructed based on multiple alignments of the eight SHC amino acid sequences obtained by in silico translation from the corresponding gene sequence of these SDF strains (Figure 6A). The amino acid sequence of Pseudomonas sp. was included as an outgroup. The inferred evolutionary relationship among these SDF strains was clustered into two main clades. In the first, the respective SHC sequences found in the strains B. velezensis SDF0141 and B. velezensis SDF0150 were equivalent. The enzyme sequence obtained for B. pumilus SDF0011 was very close to that obtained for B. safensis SDF0016. The SHC sequences from H. oleronia SDF0015 and Pe. simplex SDF0024 revealed the highest evolutionary relationship in the second main clade generated (Figure 6A). The molecular correlation between the SHC primary chains of these two strains was closer to the corresponding sequences obtained for the strains P. popilliae SDF0028 and Br. brevis SDF0063, also positioned in the second clade (Figure 6A).
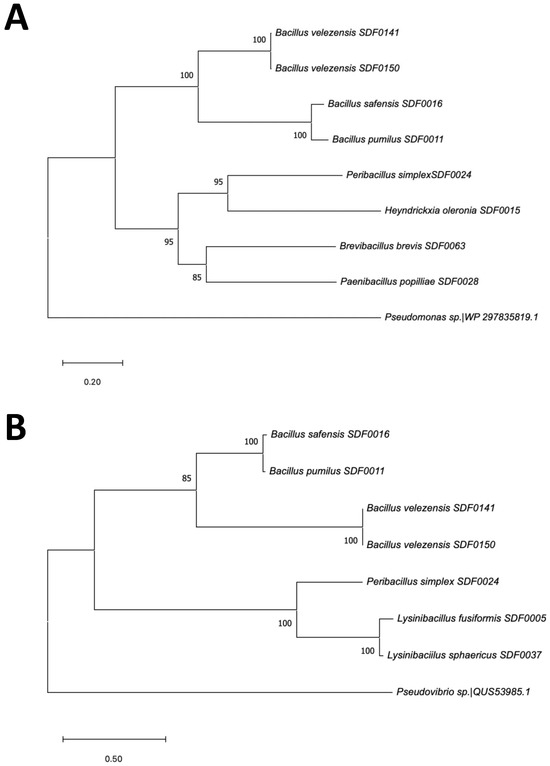
Figure 6.
Phylogenetic relationship of the SDF strains based on terpene synthase sequences. The evolutionary history of the SDF strains was determined by aligning the deduced amino acid sequences of two terpene synthases using ClustalW. The sequences were obtained by translating the respective SDF gene sequences in silico. Phylogenetic trees were reconstructed using the maximum likelihood method in MEGA version 11.0. The tree nodes show bootstrap values as percentages of 1000 replications. SDF strain designations are indicated in the branches. A distance scale bar is displayed at the bottom. The evolutionary relationships among (A) eight SDF strains based on the amino acid sequence of the SHC enzyme and the homolog amino acid sequence of Pseudomonas sp. as an outgroup and (B) seven SDF strains based on the amino acid sequence of the phytoene/squalene synthase family (PSS) and the homolog amino acid sequence of Pseudovibrio brasiliensis as an outgroup.
Likewise, the phylogenetic tree generated from seven amino acid sequences corresponding to the PSS enzyme family genomes and Pseudovibrio brasiliensis as an outgroup also divided these seven SDF strains into two main clades (Figure 6B). The first was further separated into two subclades. The sequences of this enzyme obtained for the strains B. velezensis SDF0141 and B. velezensis SDF0150 pointed out a close evolutionary relationship. The sequences of the strains B. pumilus SDF0011 and B. safensis SDF0016 positioned in the other subclade also displayed a high-level molecular relationship (Figure 6B). The second clade showed that the strains L. fusiformis SDF0005 and L. sphaericus SDF0037 presented amino acid sequences with the highest molecular relationship. Strain Pe. simplex SDF0024 was positioned apart from the other two inside this clade (Figure 6B). Because the gene sequence of PDS was found in 2 out of 10 SDF strains, it was not considered for further phylogenetic analyses.
4. Discussion
The phylum Firmicutes was recently renamed Bacillota [34]. Inside the class Bacilli, the order Bacillales, which allocated AEFB species, displayed an immense diversity, spanning several families, genera, and species [20,35]. Lately, new taxa have been established to reposition strains otherwise considered members of the order Bacillales (https://lpsn.dsmz.de/contact, accessed on 19 December 2024) [55]. This reorganisation considers the family Bacillaceae the only member of this order, includes Bacillus as the type genus of these taxa, and Bacillus subtilis remains the type species of the genus Bacillus.
Historically, the genus Bacillus represented a large assemblage of genetically and evolutionarily unrelated microorganisms. Thus, the genus has long been recognised as housing members exhibiting an extensive polyphyly and with very little in common with each other [34,55,56,57]. To more adequately represent the overall genetic diversity within this genus, it was proposed that the vast majority of Bacillus spp. needed to be reclassified into other genera, families, and orders. The revision of the genus Bacillus led to reallocating (not limited to) two Bacillus spp. to novel genera that could better accommodate them. The former Bacillus oleronius is now designated Heyndrickxia oleronia [58]. Likewise, Bacillus simplex was reallocated into a novel genus designated Peribacillus, species Peribacillus simplex [59].
Other misclassified Bacillus spp. were transferred to specific genera and reallocated to the family Caryophanaceae, which in turn were transferred to the order Caryophanales, including the genus Lysinibacillus [56]. In this context, the genera Paenibacillus and Brevibacillus were moved from the genus Bacillus, family Bacillaceae, being the genus Paenibacillus, to the family Paenibacillaceae, order Paenibacillales, and the genus Brevibacillus to the novel family Brevibacillaceae, order Brevibacillales [57].
4.1. Uncovering Enzymes from the MEP Pathway and the Polyprenyl Synthase Family in the SDF Strains
We previously identified 153 putative BGCs in the genomes of 10 SDF strains (Table 2). Among these, 20 classes of specialised metabolites were identified [41]. In the current work, we focused on 16 BGCs, which were linked to terpene synthesis, to assess the genomic potential of these environmental AEFB for the biosynthesis of these molecules. Bacteria can produce terpenes through the MEP pathway by synthesising IPP, a precursor for these isoprenes [14]. Therefore, we evaluated the putative genetic information associated with the biosynthesis of essential terpene precursors within the MEP pathway. Through the Pathways tools, our study has uncovered that all the 10 SDF strains (Table 2) examined contain a significant number of genetic determinants encoding the enzymes DXS, DXR, MCT, CMK, MDS, HDS, and HDR, which are associated with the MEP pathway (Table 1; Figure 2). These findings suggest that the genetic information coding for these pathway enzymes was conserved among these SDF strains, which can be carriers of the basic apparatus for terpene production.
Indeed, information for MEP pathway catalysts (Table 1) is an ancestral characteristic of prokaryotes. According to Zeng and Dehesh [60], there is substantial evidence of the vertical transfer of genetic information encoding MEP pathway constituents between plastids—initially present in the phylum Cyanobacteria—and plants. This evidence supports the idea that the genetic determinants of the MEP pathway are an ancestral characteristic shared among the SDF strains evaluated in this study and potentially also by AEFB. For instance, the genetic information related to enzyme production found in the B. velezensis SDF0150 strain has also been identified in the genome of Synechocystis sp., which is classified under the phylum Cyanobacteria [61]. Additionally, this metabolic pathway is well characterised in the phylum Actinomycetota, notably in species of the genus Streptomyces [62]. These compelling data underscore the necessity for further investigation to explore the potential implications of these fundamental genetic determinants in terpene biosynthesis within AEFB.
The PPS family amino acid sequences obtained by in silico translation (Table 3) were compared to a sequence database we assembled, with more than 6000 trusted files of known sequences involved in these catalysts’ synthesis (see Section 2). We performed pairwise all-against-all protein sequence alignments of all the genomes, using Blastp with an E-value cutoff of 1 × 10−5. The best hits were found to be B. velezensis SDF0141 and B. velezensis SDF0150, in which the identity scored >99% of the top-scoring hit in the database (Table 3), an ortholog of a B. velezensis strain sequence. Likewise, L. fusiformis SDF0005 and Pe. simplex SDF0024 scored a >98% identity (Table 3) if compared to the top-scoring hit in the database representative of their respective species. These findings strongly indicate the presence of the gene sequences coding for this enzyme in the genome of these SDF strains.
In contrast, H. oleronia SDF0015 showed a 67.86% identity if compared to the ortholog sequences representing B. pumilus in the database. (Table 3) This result suggested that this latter strain may lack the genetic information necessary for synthesising the PPS family enzyme. Alternatively, it possesses a structurally distinct catalyst compared to those found in AEFB, likely due to unique characteristics associated with H. oleronia. Consequently, it is reasonable to infer that the remaining SDF strains lacking at least one enzyme from the PPS family may experience limitations in terpene production, as they do not bear the necessary enzymatic machinery to synthesise intermediates for isoprene precursors.
4.2. Genomic Potential of Selected SDF Strains for Terpene Production
GPP, FPP, or GGPP are used as precursor molecules in natural terpene syntheses by different TSs. antiSMASH identified the sequence encoding the enzyme of the PSS in at least seven SDF strains (Figure 4; Table 4). Phytoene and squalene molecules are constructed from two molecules of FPP and two GGPP molecules, respectively [63,64]. The enzymes squalene synthase (SQS) and phytoene synthase (PHS) are closely related [64], and SQS is also reported for the synthesis of phytoene [63]. This connection between these enzymes may explain why antiSMASH did not discriminate between the genetic sequences for phytoene and squalene production. The SDF strains containing genes that encode enzymes from the PPS family and PSS putatively presented the necessary genetic apparatus to produce either phytoene or squalene, as illustrated in the genomes of Pe. simplex SDF0024, B. velezensis SDF0141, and B. velezensis SDF0150.
Squalene is known for its antioxidant and antitumor properties, and this terpene also enhances the human immune system, as reported by Sanchez-Quesada et al. [65]. This specialised metabolite is commonly used as an additive and supplement in the food and personal care industries [66]. Squalene synthesis has been documented for various organisms, including AEFB, which corroborates the findings in this study. Beyond its benefits, squalene serves as an intermediate compound in the biosynthetic pathway of sterols such as hopanoids [67].
Hopanoids are synthesised from squalene by the enzyme SHC [67]. The sqhC gene sequence encodes the SHC enzyme and was identified by antiSMASH in eight SDF strains (Figure 3; Table 4). These data indicate that the SDF strains qualified to produce SHC and PSS possess the required components for the final enzymatic reactions in hopanoid biosynthesis. This condition was observed in three SDF genomes, specifically Pe. simplex SDF0024, B. velezensis SDF0141, and B. velezensis SDF0150 (Table 4). Hopanoids play a crucial role by integrating into the biological membranes of the producing cells, regulating fluidity and permeability [67,68]. Consequently, these terpenes keep the bacterial cytoplasmic membrane stable, which is particularly significant given the absence of cholesterol in the membranes of these prokaryotes. While a lack of hopanoids does not hinder bacterial growth, it does affect tolerance to stressful conditions, such as high temperatures and anaerobic or acidic environments [69].
Interestingly, the antiSMASH analysis revealed that the organisation of hopanoid biosynthesis genes in the SDF strains deviated from the typical BGC pattern. While the sqhC gene encoding SHC was identified in eight strains, and genes encoding PSS enzymes were found in seven, these genes were not consistently clustered with other hopanoid biosynthesis genes (Figure 3 and Figure 4). This variability suggests that the genetic architecture of hopanoid biosynthesis in SDF strains might be more complex and diverse than previously recognised. Further investigation into the arrangement and regulation of these genes may shed light on this variation in its evolutionary and functional significance. Despite this variability, key hopanoid biosynthesis gene identifications in the genomes of these strains underscore the potential of AEFB as a source of these important membrane components.
The software antiSMASH 6.0 bacterial standalone version [49] (https://antismash.secondarymetabolites.org/#!/start, accessed on 4 July 2023) detected a 50% similarity in the BGC sequence involved in the carotenoid’s synthesis in the genomes of B. pumilus SDF0011 and B. safensis SDF0016. The reference strain employed for comparison was Halobacillus halophilus DSM2266 (reference number BGC0000645), available through the MIBiG platform. Although the SDF strains did not share the same genetic framework as the reference strain, they possessed the essential genetic information required to express the final enzymatic activities involved in lycopene synthesis. Lycopene, which closely resembles beta-carotene and is widely produced by plants [70], imparts the reddish pigmentation characteristic of tomatoes and watermelons, among other vegetables [71]. This terpene is known for its antioxidant, anti-inflammatory, and antitumor attributes, making it valuable for various applications in the pharmaceutical and food industries.
It is noteworthy that lycopene synthesis in AEFB has been reported in the context of heterologous expression in B. amyloliquefaciens and B. subtilis, as documented by Zou et al. [72] and Luo, Bao, and Zhu [73]. Additionally, studies have explored natural lycopene production in other AEFB species. For instance, Osawa et al. [74] investigated the synthesis of an oxidised lycopene in Cytobacillus firmus [59], formerly Bacillus firmus [75], while Hwang et al. [76] also detected genes for lycopene synthesis in Metabacillus flavus. The findings of our study are consistent with these previous reports and underscore the potential of AEFB as a source of lycopene. Further investigation is needed to fully elucidate these mechanisms and the evolutionary significance of lycopene production in these bacteria.
Figure 7 summarises the catalytic steps required for terpene production from the MEP pathway to the final reactions by different TSs and the SDF strain carriers for their respective enzymes. Nonetheless, it is essential to note that even though a specific strain of SDF may lack a particular enzyme required for terpene synthesis, this does not automatically imply that the cell is a non-terpene producer. Our study concentrated on the genomic potential of the SDF strain to generate terpenes. Therefore, any inaccuracies in the prior steps, such as the purification, extraction, sequencing, and annotation of genome sequences, may lead to erroneous results. Additionally, the biosynthetic pathways of terpenes entail multiple enzymatic reactions [77]. Even if the information for a specific enzyme is not detected, the SDF strains examined might still be able to synthesise the predicted terpene because of the promiscuous nature of the enzymes found within the pathway. A complete enzymatic route detection for a given terpene molecule does not assure the synthesis of the corresponding product by this SDF strain due to the complex mechanisms of gene expression. Further research could reveal the synthesis in vitro of the molecules detected in this study.
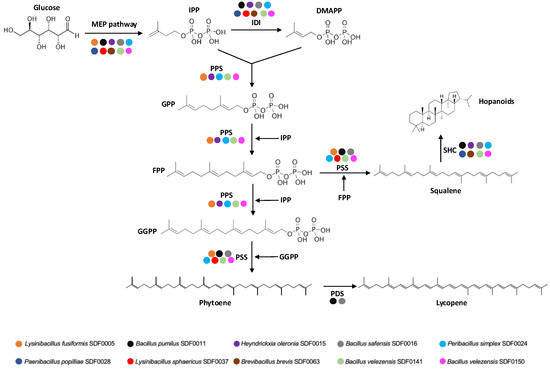
Figure 7.
Putative biosynthetic pathway of methylerythritol phosphate and terpene biosynthesis metabolic pathways in the ten SDF strains studied. The reaction steps to synthesise terpenes from isopentenyl diphosphate (IPP), the final product of the methylerythritol-phosphate (MEP) route, and the respective catalysts are indicated. The coloured dots indicate the gene sequence coding for an enzyme detected in a particular SDF strain described at the bottom. DMAPP: dimethylallyl pyrophosphate. IDI: isopentenyl diphosphate isomerase. GPP: geranyl pyrophosphate. FPP: farnesyl pyrophosphate. GGPP: geranyl-geranyl pyrophosphate. PSS: phytoene and/or squalene synthase. SHC: squalene-hopene cyclase. PDS: phytoene desaturase.
4.3. The Evolutionary Nature of Terpene Production in the SDF Stains
To explore the evolutionary nature of the enzymes involved in terpene production in the evaluated SDF strains, we aligned the amino acid sequences of the SHC (Figure 3) and PSS enzymes (Figure 4) obtained through in silico translation from the gene sequences inside the BGC identified by antiSMASH in this study. This alignment was used to generate phylogenetic trees, as shown in Figure 6A,B. However, we did not reconstruct the phylogenetic tree for the PDS enzyme, as it was detected only in two SDF strains analysed (Table 4).
The primary sequences of the SHC (Figure 6A) and PSS (Figure 6B) enzymes of B. velezensis SDF0140 and B. velezensis SDF0150 exhibited a robust molecular relationship, as both strains belong to the same species. In contrast, the strains B. pumilus SDF0011 and B. safensis SDF0016 showed a significant evolutionary relationship based on their TS amino acid sequences. These two latter species are of biotechnological and pharmaceutical significance and are closely related according to classical phenotypic characteristics and 16S rRNA gene sequences. Consequently, they are challenging to distinguish by these conventional methodologies [28,30,31,78]. Both species comprise a clonally diverse population inside the B. subtilis complex [78].
Furthermore, the phylogenetic trees generated using both the SHC (Figure 6A) and PSS (Figure 6B) enzymes revealed similar topologies among B. pumilus SDF0011, B. safensis SDF0016, B. velezensis SDF0141, and B. velezensis SDF0150. This result indicates that these four Bacillus spp. demonstrate a high level of molecular correlation in their respective TS analyses. Their classification within the same genus likely contributes to this significant conservation of catalytic properties.
Interestingly, the molecular relationship observed in Figure 6B shows that the PSS enzyme positions the two species of the genus Lysinibacillus (L. fusiformis SDF0005 and L. sphaericus SDF0037) and P. simplex SDF0024 in the same clade. This grouping contrasts with the global genome phylogeny described in Figure 1. Thus, it may suggest a possible horizontal transfer of this sequence at some point in the evolution of these taxa allocated to different orders of the class Bacilli. However, further investigation is necessary since the molecular relationships for the other SDF strains align with the expected phylogenetic relationships (Figure 6A,B).
In the heatmap (Figure 5), the enzymatic profiles for terpene production in the SDF strains highlighted a strong molecular relationship between B. pumilus SDF0011 and B. safensis SDF0016, as they share the same enzymatic set. Figure 5 also reveals that B. velezensis SDF0141 and B. velezensis SDF0150 possess an identical catalyst set for terpene production. These results reinforce the molecular relationship among these SDF strains, although the disposition of SDF strains belonged to Bacillus spp. did not display the same distribution observed in the phylogenetic tree of TS amino acid sequences (Figure 6).
The strains H. oleronia SDF0015 and Pe. simplex SDF0024 exhibited a high degree of molecular similarity in their respective amino acid sequences of the SHC enzyme (Figure 6A). The amino acid sequences of SHC from H. oleronia SDF0015 and Pe. simplex SDF0024 displayed a superior evolutionary distance compared to SHC sequences obtained from the SDF strains within the genus Bacillus, therefore positioned in different clades (Figure 6A). Bacillus, Heyndrickxia, and Peribacillus are all classified within the family Bacillaceae, and species in these genera present significant levels of polyphyly [56]. This observation further corroborated phylogenetic relationships derived from the SHC amino acid sequences (Figure 6A).
P. popilliae SDF0028 and Br. brevis SDF0063 presented the highest molecular relationship in their SHC amino acid sequences if compared to the SHC sequences of the remaining six SDF strains analysed (Figure 6A). The genera Bacillus, Peribacillus, and Heyndrickxia, part of the family Bacillaceae, belong to the order Bacillales. At the same time, the genera Paenibacillus and Brevibacillus are classified under the orders Paenibacillales and Brevibacillales, respectively [57,58]. Therefore, the minimal molecular relationship between the SHC amino acid sequences of P. popilliae SDF0028 and Br. brevis SDF0063 and the other SDF strains aligns with the anticipated evolutionary distance for these species. Additionally, the same enzymatic set for terpene production was observed between these two SDF strains (Figure 5), supporting even more the phylogenetic relationship obtained by the sequence of the SHC of P. popilliae SDF0028 and Br. brevis SDF0063 (Figure 6A). Notably, the amino acid sequences of the SHC from H. oleronia SDF0015 and Pe. simplex SDF0024 were recognised to be evolutionarily closer to those of P. popilliae SDF0028 and Br. brevis SDF0063 than the SHC sequences from SDF strains within the genus Bacillus (Figure 6A).
L. fusiformis SDF0005 and L. sphaericus SDF0037 shared highly conserved PSS amino acid sequences (Figure 6B). This commonality might be attributed to the two SDF strains from the genus Lysinibacillus. The phylogenetic tree generated using the sequences revealed a clear distinction between the SDF strains of the genus Bacillus, which were grouped in one clade, and the SDF strains of the genus Lysinibacillus, which formed a separate clade (Figure 6B). These results discriminated between the members of the families Bacillaceae and Caryophanaceae based on the amino acid sequence of this catalyst. Furthermore, a relevant molecular relationship for the terpene production enzymatic set was already observed between Lysinibacillus spp. as shown in the heatmap (Figure 5). Markedly, the sequences of L. fusiformis SDF0005 and L. sphaericus SDF0037 demonstrated a closer evolutionary relationship with the sequence of Pe. simplex SDF0024, which is allocated within the family Bacillaceae.
The phylogenetic trees derived from the molecular analyses of the TSs detected in the SDF strains examined in this study agreed with the phylogenetic relationships for the complete genomes of the SDF strains evaluated (Figure 1). The results indicated that the TS enzymes responsible for terpene production in these investigated environmental strains are evolutionarily conserved. In addition, the production of terpenes—strikingly, squalene and hopanoids—appears to be an ancestral characteristic of the AEFB evaluated in this study.
As mentioned above, the phylogenetic trees derived from the amino acid sequences of TS enzymes (Figure 6A,B) indicated a closer molecular relationship between B. velezensis SDF0141 and B. velezensis SDF0150 to B. pumilus SDF0011 and B. safensis SDF0016. In contrast, the heatmap (Figure 5) groups these two B. velezensis strains with Pe. simplex SDF0024, along with L. fusiformis SDF0005 and L. sphaericus SDF0037. Additionally, the strains P. popilliae SDF0028 and Br. brevis SDF0063 demonstrated a similar enzyme content for terpene production and clustered with the strain H. oleronia SDF0015 (Figure 5).
The cluster of catalysts implicated in terpene production shown in Figure 5 shares similarities with the phylogenetic trees generated from the TS amino acid sequences (Figure 6A,B). Nevertheless, the cluster does not fully align with the expected evolutionary relationships among these species. This analysis suggested that the ability to produce terpene molecules may vary among the SDF strains and might not be influenced by phylogenetic factors. Furthermore, the enzyme set involved in terpene production cannot be used as a molecular marker to establish evolutionary relationships among the SDF strains analysed and, by extension, for AEFB.
This study is based on in silico analyses. Thus, experimental approaches to confirm terpene production potential, alongside possible active applications, are significant. In future work, we plan to include transcriptomic analyses under conditions that induce a specialised metabolism. This approach will allow us to verify the expression of genes associated with the predicted terpene biosynthetic pathways. Techniques such as RNA sequencing (RNA-Seq) and quantitative-reverse transcription polymerase chain reaction (RT-qPCR) will allow us to assess whether these genes are transcriptionally active, and the conditions in which they are expressed. For instance, RT-qPCR permits the precise quantification of a specific gene expression by converting mRNA into cDNA, followed by the real-time monitoring of amplification using fluorescent markers. Still, transcriptomic data can be complemented with metabolomic profiling using mass spectrometry to detect and quantify terpene compounds produced by these environmental strains. This integrative approach will allow us to correlate gene expression with metabolite production, providing strong evidence for the functional activity of the predicted pathways.
5. Conclusions
AEFB are ubiquitous and characterised by producing several specialised metabolites, among which terpenes are a significant class. While the synthesis of terpenes has been demonstrated in prokaryotes, research addressing specifically the production of these compounds in AEFB is still scarce. Our analyses revealed that strains of these taxa possess the BGCs required to synthesise at least three terpenes: squalene, hopanoids, and lycopene. We successfully identified the metabolic pathways for synthesising terpene precursors in all 10 genomes of the evaluated SDF strains. The detected amino acid sequences of terpene synthases indicated functional equivalence among these catalysts. Although the identified terpene classes represent only a narrow fraction of these isoprene molecules, our findings can support future investigations to broaden the understanding of the physiological and ecological roles of terpenes in AEFB. Natural sources for terpenes are insufficient to meet the growing need, since these metabolites are extensively used across numerous industries. Therefore, synthetic biology and metabolic engineering methods can be used to create cell factories for the enhanced production of terpenes. These approaches grant the study and manipulation of BGCs under controlled parameters. Besides leading to novel functions, these strategies can help minimise the TS enzyme’s promiscuity by adding precursors that can directly synthesise a particular product and reduce the metabolic burden.
Author Contributions
Conceptualisation, F.d.A.M., W.M.C.d.S. and M.T.D.-S.; Data curation, F.d.A.M., W.M.C.d.S., M.d.M.B., B.F. and M.T.D.-S.; Formal analysis, F.d.A.M., W.M.C.d.S., T.R., N.F.d.A., B.F., D.d.A.C. and M.T.D.-S.; Funding acquisition, M.d.M.B. and N.F.d.A.; Investigation, F.d.A.M., W.M.C.d.S. and M.T.D.-S.; Methodology, F.d.A.M., W.M.C.d.S., T.R., M.d.M.B., N.F.d.A., B.F., D.d.A.C. and M.T.D.-S.; Project administration, M.T.D.-S.; Resources, F.d.A.M., W.M.C.d.S., D.d.A.C. and M.T.D.-S.; Software, F.d.A.M., W.M.C.d.S., T.R., M.d.M.B., N.F.d.A. and B.F.; Supervision, M.T.D.-S.; Validation, F.d.A.M., W.M.C.d.S., N.F.d.A. and M.T.D.-S.; Writing—original draft, M.T.D.-S.; Writing—review and editing, F.d.A.M., W.M.C.d.S. and M.T.D.-S. All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded by Brazilian agencies: NA acknowledges funding from the National Council for Scientific and Technological Development (CNPq), grant 304423/2022-04; MB acknowledges funding from the Fundação de Amparo a Pesquisa do Distrito Federal (FAP-DF) (FAP-DF), grant 0193-000.560/2009. FM and DC acknowledge CNPq fellowships. We are grateful for the support of the Research Foundation in the Federal District (FAPDF), Process No. 00193-00001612/2025-20.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are openly available in [GeneBank] at [https://www.ncbi.nlm.nih.gov/genbank/about/, accessed on 1 March 2023], reference number [VKHW00000000.1 VKHY00000000.1 VKHZ00000000.1 SADW00000000.1 VKHX00000000.1 SADY00000000.1 SADV00000000.1 SADX00000000.1 VKIB00000000.1 VKIC00000000.1].
Conflicts of Interest
The authors declare no conflicts of interest.
Correction Statement
This article has been republished with a minor correction to the Funding statement. This change does not affect the scientific content of the article.
References
- Singh, B.P.; Rateb, M.E.; Rodriguez-Couto, S.; Polizeli, M.D.L.T.D.M.; Li, W.-J. Microbial Secondary Metabolites: Recent Developments and Technological Challenges. Front. Microbiol. 2019, 10, 914. [Google Scholar] [CrossRef] [PubMed]
- Bills, G.F.; Gloer, J.B. Biologically Active Secondary Metabolites from the Fungi. Microbiol. Spectr. 2016, 4. [Google Scholar] [CrossRef] [PubMed]
- Perveen, S.; Al-Taweel, A. Terpenes and Terpenoids; IntechOpen: London, UK, 2018; pp. 1–152. ISBN 978-1-83881-529-5. [Google Scholar]
- Rudolf, J.D.; Aslup, T.; Xu, B.; Li, Z. Bacterial Terpenome. Nat. Prod. Rep. 2021, 38, 905–980. [Google Scholar] [CrossRef] [PubMed]
- Masyita, A.; Sari, R.M.; Astuti, A.D.; Yasir, B.; Rumata, N.R.; Emran, T.B.; Nainu, F.; Simal-Gandara, J. Terpenes and terpenoids as main bioactive compounds of essential oils, their roles in human health and potential application as natural food preservatives. Food Chem. 2022, 13, 100217. [Google Scholar] [CrossRef]
- Yamada, Y.; Kuzuyama, T.; Komatsu, M.; Ikeda, H. Terpene Synthases are Widely Distributed in Bacteria. Proc. Natl. Acad. Sci. USA 2015, 112, 857–862. [Google Scholar] [CrossRef]
- Pinto-Zevallos, D.M.; Hellén, H.; Hakola, H.; Nouhuys, S.V.; Halopainen, J.K. Induced defenses of Veronica spicata: Variability in herbivore-induced volatile organic compounds. Phytochem. Lett. 2013, 6, 653–656. [Google Scholar] [CrossRef]
- Zhao, D.-D.; Jiang, L.-L.; Li, H.-Y.; Yan, P.-F.; Zhang, Y.-L. Chemical Components and Pharmacological Activities of Terpene Natural Products from the Genus Paeonia. Molecules 2016, 21, 1362. [Google Scholar] [CrossRef]
- Tetali, S.D. Terpenes and isoprenoids: A wealth of compounds for global use. Planta 2019, 249, 1–8. [Google Scholar] [CrossRef]
- Quin, M.B.; Flynn, C.M.; Schimidt-Dannert, C. Traversing the Fungal Terpenome. Nat. Prod. Rep. 2014, 31, 1449–1473. [Google Scholar] [CrossRef]
- Hegazy, M.E.F.; Mohamed, T.A.; Alhammady, M.A.; Shaheen, A.M.; Reda, E.H.; Elshamy, A.I.; Aziz, M.; Paré, P.W. Molecular Architecture and Biomedical Leads of Terpenes from Red Sea Marine Invertebrates. Mar. Drugs 2015, 13, 3154–3181. [Google Scholar] [CrossRef]
- Morandini, L.; Caulier, S.; Bragard, C.; Mahillon, J. Bacillus cereus sensu lato Antimicrobial Arsenal: An Overview. Microbiol. Res. 2024, 283, 127697. [Google Scholar] [CrossRef]
- Twaij, B.M.; Hasan, M.N. Bioactive Secondary Metabolites from Plant Sources: Types, Synthesis, and Their Therapeutic Uses. Int. J. Plant Biol. 2022, 13, 4–14. [Google Scholar] [CrossRef]
- Liang, Z.; Zhi, H.; Fang, Z.; Zhang, P. Genetic engineering of yeast, filamentous fungi and bacteria for terpene production and applications in food industry. Food Res. Int. 2021, 147, 110487. [Google Scholar] [CrossRef] [PubMed]
- Kasahara, H.; Hanada, A.; Kuzuyama, T.; Takagi, M.; Kamiya, Y. Contribution of the mevalonate and methylerythritol phosphate pathways to the biosynthesis of gibberellins in Arabidopsis. J. Biol. Chem. 2002, 277, 45188–45194. [Google Scholar] [CrossRef] [PubMed]
- Pardo, E.; Rico, J.; Gil, J.V.; Orejas, M. De novo production of six key grape aroma monoterpenes by a geraniol synthase-engineered S. cerevisiae wine strain. Microb. Cell Factories 2015, 14, 136. [Google Scholar] [CrossRef]
- Lv, H.-W.; Tang, J.-G.; Wei, B.; Zhu, M.-D.; Zhang, H.-W.; Zhou, Z.-B.; Fan, B.-Y.; Wang, H.; Li, X.-N. Bioinformatics assisted construction of the link between biosynthetic gene clusters and secondary metabolites in fungi. Biotechnol. Adv. 2025, 81, 108547. [Google Scholar] [CrossRef] [PubMed]
- Mudbhari, S.; Lofgren, L.; Appidi, M.A.; Vilgalys, R.; Hettich, R.L.; Abraham, P.E. Decoding the chemical language of Suillus fungi: Genome mining and untargeted metabolomics uncover terpene chemical diversity. Msystems 2024, 9, e0122523. [Google Scholar] [CrossRef]
- Lee, S.-Y.; Kim, M.; Kim, S.-H.; Hong, C.-Y.; Ryu, S.-H.; Choi, I.-G. Transcriptomic analysis of the white rot fungus Polyporus brumalis provides insight into sesquiterpene biosynthesis. Microbiol. Res. 2016, 182, 141–149. [Google Scholar] [CrossRef]
- Harirchi, S.; Sar, T.; Ramezani, M.; Aliyu, H.; Etemadifar, Z.; Nojoumi, S.A.; Yazdian, F.; Awasthi, M.K.; Taherzadeh, M.J. Bacillales: From Taxonomy to Biotechnological and Industrial Perspectives. Microorganisms 2022, 10, 2355. [Google Scholar] [CrossRef]
- Put, H.; Gerstmans, H.; Capelle, H.V.; Fauvart, M.; Michiels, J.; Masschelein, J. Bacillus subtilis as a host for natural product discovery and engineering of biosynthetic gene clusters. Nat. Prod. Rep. 2024, 41, 1113–1151. [Google Scholar] [CrossRef]
- Zheng, D.; Ding, N.; Jiang, Y.; Zhang, J.; Ma, J.; Chen, X.; Liu, J.; Han, L.; Huang, X. Albaflavenoid, a New Tricyclic Sesquiterpenoid from Streptomyces violascens. J. Antibiot. 2016, 69, 773–775. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Sun, J.; Yang, Q.; Yang, J. Metabolic Engineering Escherichia coli for the Production of Lycopene. Molecules 2020, 25, 3136. [Google Scholar] [CrossRef]
- Tyc, O.; Song, C.; Dickschat, J.S.; Vos, M.; Garbeva, P. The Ecological Role of Volatile and Soluble Secondary Metabolites Produced by Soil Bacteria. Trends Microbiol. 2017, 25, 280–292. [Google Scholar] [CrossRef] [PubMed]
- Fritze, D. Taxonomy of the Genus Bacillus and Related Genera: The Aerobic Endospore-Forming Bacteria. Phytopathology 2004, 94, 1245–1248. [Google Scholar] [CrossRef]
- Mandic-Mulec, I.; Prosser, J.I. Diversity of Endospore-forming Bacteria in Soil: Characterisation and Driving Mechanisms. In Endospore-Forming Soil Bacteria, 1st ed.; Logan, N.A., Vos, P., Eds.; Springer: Berlin/Heidelberg, Germany, 2011; Volume 27, pp. 31–59. ISBN 978-3-642-19577-8. [Google Scholar]
- Logan, N.A. Bacillus and relatives in foodborne illness. J. Appl. Microbiol. 2012, 112, 417–429. [Google Scholar] [CrossRef] [PubMed]
- Orem, J.C.; Silva, W.M.C.; Raiol, T.; Magalhães, M.I.; Martins, P.H.; Cavalcante, D.A.; Kruger, R.H.; Brigido, M.M.; De-Souza, M.T. Phylogenetic diversity of aerobic spore-forming Bacillales isolated from Brazilian soils. Int. Microbiol. 2019, 22, 511–520. [Google Scholar] [CrossRef]
- Cavalcante, D.A.; De-Souza, M.T.; Orem, J.C.; Magalhães, M.I.A.; Martins, P.H.; Boone, T.J.; Castillo, J.A.; Driks, A. Ultrastructural analysis of spores from diverse Bacillales species isolated from Brazilian soil. Environ. Microbiol. Rep. 2019, 11, 155–164. [Google Scholar] [CrossRef]
- Martins, P.H.R.; Silva, L.P.; Orem, J.C.; Magalhães, M.I.A.; Cavalcante, D.A.; De-Souza, M.T. Protein profiling as a tool for identifying environmental aerobic endospore-forming bacteria. Open J. Bacteriol. 2020, 4, 1–7. [Google Scholar]
- Martins, P.H.R.; Rabinovitch, L.; Orem, J.C.; Silva, W.M.C.; Mesquita, F.A.; Magalhães, M.I.A.; Cavalcante, D.A.; Vivoni, A.M.; Oliveira, E.J.; Lima, V.C.P.; et al. Biochemical, physiological, and molecular characterisation of a large collection of aerobic endospore-forming bacteria isolated from Brazilian soils. Neotrop. Biol. Conserv. 2023, 18, 53–72. [Google Scholar] [CrossRef]
- Driks, A.; Eichenberger, P. The Bacterial Spore: From Molecules to Systems; ASM Press: Washington, DC, USA, 2016; pp. 1–397. ISBN 9781555816759. [Google Scholar]
- Christie, G.; Setlow, P. Bacillus spore germination: Knowns, unknowns and what we need to learn. Cell. Signal. 2020, 74, 109729. [Google Scholar] [CrossRef]
- Oren, A.; Garrity, G.M. Valid publication of the names of forty-two phyla of prokaryotes. Int. J. Syst. Evol. Microbiol. 2021, 71, 005056. [Google Scholar] [CrossRef] [PubMed]
- Galperin, M.Y.; Yutin, N.; Wolf, Y.I.; Alvarez, R.V.; Koonin, E.V. Conservation and Evolution of the Sporulation Gene Set in Diverse Members of the Firmicutes. J. Bacteriol. 2022, 204, e00079-22. [Google Scholar] [CrossRef]
- Sumi, C.D.; Yang, B.W.; Yeo, I.-C.; Hahm, Y.T. Antimicrobial peptides of the genus Bacillus: A new era for antibiotics. Can. J. Microbiol. 2015, 61, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Heilbronner, S.; Krismer, B.; Brötz-Oesterhelt, H.; Peschel, A. The microbiome-shaping roles of bacteriocins. Nat. Rev. Microbiol. 2021, 19, 726–739. [Google Scholar] [CrossRef] [PubMed]
- Salazar, B.; Ortiz, A.; Keswani, C.; Minkina, T.; Mandzhieva, S.; Singh, S.P.; Rekadwad, B.; Borriss, R.; Jain, A.; Singh, H.B.; et al. Bacillus spp. as Bio-factories for Antifungal Secondary Metabolites: Innovation Beyond Whole Organism Formulations. Microb. Ecol. 2023, 86, 1–24. [Google Scholar] [CrossRef]
- Mondol, M.A.M.; Shin, H.J.; Islam, M.T. Diversity of Secondary Metabolites from Marine Bacillus Species: Chemistry and Biological Activity. Mar. Drugs 2013, 11, 2846–2872. [Google Scholar] [CrossRef]
- Falqueto, S.A.; Pitaluga, B.F.; Sousa, J.R.; Targanski, S.K.; Campos, M.G.; Mendes, T.A.O.; Silva, G.F.; Silva, D.H.S.; Soares, M.A. Bacillus spp. metabolites are effective in eradicating Aedes aegypti (Diptera: Culicidae) larvae with low toxicity to non-target species. J. Invertebr. Pathol. 2021, 179, 107525. [Google Scholar] [CrossRef]
- Mesquita, F.A.; Silva, W.M.C.; De-Souza, M.T. In silico Analysis of the Genomic Potential for the Production of Specialized Metabolites of Ten Strains of the Bacillales Order Isolated from the Soil of the Federal District, Brazil. In Advances in Bioinformatics and Computational Biology, 1st ed.; Scherer, N.M., Melo-Minardi, R.C., Eds.; Springer: Berlin/Heidelberg, Germany, 2022; Volume 13523, pp. 158–163. ISBN 978-3-031-21175-1. [Google Scholar]
- Coil, D.; Jospin, G.; Darling, A.M. A5-miseq: An updated pipeline to assemble microbial genomes from Illumina Miseq data. Bioinformatics 2015, 31, 587–589. [Google Scholar] [CrossRef]
- Tatusova, T.; DiCuccio, M.; Badretdin, A.; Chetvernin, V.; Nawrocki, E.P.; Zaslavsky, L.; Lomsadze, A.; Pruitt, K.D.; Borodovsky, M.; Ostell, J. NCBI prokaryotic genome annotation pipeline. Nucleic Acid Res. 2016, 44, 6614–6624. [Google Scholar] [CrossRef]
- Setubal, J.C.; Almeida, N.F.; Wattam, A.R. Comparative Genomics for Prokaryotes. Methods Mol. Biol. 2018, 1704, 55–78. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Stoeckert, J., Jr.; Roos, D.S. OrthoMCL: Identification of ortholog groups for eukaryotic genomes. Genome Res. 2003, 13, 2178–2189. [Google Scholar] [CrossRef] [PubMed]
- Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 2000, 17, 540–552. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Blin, K.; Shaw, S.; Kloosterman, A.M.; Charlop-Powers, Z.; van Wezel, G.P.; Medema, M.H.; Weber, T. antiSMASH 6.0: Improving cluster detection and comparison capabilities. Nucleic Acids Res. 2021, 49, W29–W35. [Google Scholar] [CrossRef]
- Bogdanove, A.J.; Koebnik, R.; Lu, H.; Furutani, A.; Angiuoli, S.V.; Patil, P.B.; Sluys, M.A.V.; Ryan, R.P.; Meyer, D.F.; Han, S.-W.; et al. Two New Complete Genome Sequences Offer Insight into Host and Tissue Specificity of Plant Pathogenic Xanthomonas spp. J. Bacteriol. 2011, 193, 5450–5464. [Google Scholar] [CrossRef]
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef]
- Hummel, M.; Edelmann, D.; Kopp-Schneider, A. Clustering of samples and variables with mixed-type data. PLoS ONE 2017, 12, e0188274. [Google Scholar] [CrossRef]
- Medema, M.H.; Kottmann, R.; Yilmaz, P. Minimum Information about a Biosynthetic Gene cluster. Nat. Chem. Biol. 2015, 11, 625–631. [Google Scholar] [CrossRef]
- Köcher, S.; Breitenbach, J.; Müller, V.; Sandmann, G. Structure, function and biosynthesis of carotenoids in the moderately halophilic bacterium Halobacillus halophilus. Arch. Microbiol. 2009, 191, 95–104. [Google Scholar] [CrossRef]
- Xu, X.; Kóvacs, A.T. How to identify and quantify the members of the Bacillus genus? Environ. Microbiol. 2024, 26, e16593. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.S.; Patel, S. Robust Demarcation of the Family Caryophanaceae (Planococcaceae) and Its Different Genera Including Three Novel Genera Based on Phylogenomics and Highly Specific Molecular Signatures. Front. Microbiol. 2020, 10, 2821. [Google Scholar] [CrossRef] [PubMed]
- Chuvochina, M.; Mussig, A.J.; Chaumeil, P.-A.; Skarkshewski, A.; Rinke, C.; Parks, D.H.; Hugenholtz, P. Proposal of names for 329 higher taxa defined in the Genome Taxonomy Database under two prokaryotic codes. FEMS Microbiol. Lett. 2023, 370, fnad071. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.S.; Patel, S.; Saini, N.; Chen, S. Robust demarcation of 17 distinct Bacillus species clades, proposed as novel Bacillaceae genera, by phylogenomics and comparative genomic analyses: Description of Robertmurraya kyonggiensis sp. nov. and proposal for an emended genus Bacillus limiting it only to the members of the Subtilis and Cereus clades of species. Int. J. Syst. Evol. Microbiol. 2020, 70, 5753–5798. [Google Scholar] [CrossRef]
- Patel, S.; Gupta, R.S. A phylogenomic and comparative genomic framework for resolving the polyphyly of the genus Bacillus: Proposal for six new genera of Bacillus species, Peribacillus gen. nov., Cytobacillus gen. nov., Mesobacillus gen. nov., Neobacillus gen. nov., Metabacillus gen. nov. and Alkalihalobacillus gen. nov. Int. J. Syst. Evol. Microbiol. 2020, 70, 406–438. [Google Scholar] [CrossRef]
- Zeng, L.; Dehesh, K. The eukaryotic MEP-pathway genes are evolutionarily conserved and originated from Chlaymidia and cyanobacteria. BMC Genom. 2022, 22, 137. [Google Scholar] [CrossRef]
- Kaneko, T.; Tabata, S. Complete Genome Structure of the Unicellular Cyanobacterium Synechocystis sp. PCC6803. Plant Cell Physiol. 1997, 11, 1171–1176. [Google Scholar] [CrossRef]
- Dairi, T. Studies on biosynthetic genes and enzymes of isoprenoids produced by actimomycetes. J. Antibiot. 2005, 58, 227–243. [Google Scholar] [CrossRef]
- Nakashima, T.; Inoue, T.; Oka, A.; Nishino, T.; Osumi, T.; Hata, S. Cloning, expression, and characterisation of cDNAs encoding Arabidopsis thaliana squalene synthase. Proc. Natl. Acad. Sci. USA 1995, 98, 2328–2332. [Google Scholar] [CrossRef]
- Tansey, T.R.; Shechter, I. Squalene synthase: Structure and regulation. Prog. Nucleic Acid Res. Mol. Biol. 2000, 65, 157–195. [Google Scholar] [CrossRef]
- Sánchez-Quesada, C.; López-Biedma, A.; Toledo, E.; Gaforio, J.J. Squalene Stimulates a Key Innate Cell to Foster Wound Healing and Tissue Repair. Evid. -Based Complement. Altern. Med. 2018, 2018, 9473094. [Google Scholar] [CrossRef]
- Song, Y.; Guan, Z.; Merkerk, R.V.; Pramastya, H.; Abdallah, I.I.; Setroikromo, R.; Quax, W.J. Production of Squalene in Bacillus subtilis by Squalene Synthase Screening and Metabolic Engineering. J. Agric. Food Chem. 2020, 68, 4447–4455. [Google Scholar] [CrossRef] [PubMed]
- Siedenburg, G.; Jendrossek, D. Squalene-Hopene Cyclases. Appl. Environ. Microbiol. 2011, 77, 3905–3915. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, S.; Begum, F.; Rabaan, A.A.; Aljeldah, M.; Al Shammari, B.R.; Alawfi, A.; Alshengeti, A.; Sulaiman, T.; Khan, A. Classification and Multifaceted Potential of Secondary Metabolites Produced by Bacillus subtilis Group: A Comprehensive Review. Molecules 2023, 28, 927. [Google Scholar] [CrossRef]
- Belin, B.J.; Busset, N.; Giraud, E.; Molinaro, A.; Silipo, A.; Newman, D.K. Hopanoid lipids: From membranes to plant-bacteria interactions. Nat. Rev. Microbiol. 2018, 16, 304–315. [Google Scholar] [CrossRef]
- Sabio, E.; Lozano, M.; Espinosa, V.M.; Mendes, R.L.; Pereira, A.P.; Palavra, A.F.; Coelho, J.A. Lycopene and β-Carotene Extraction from Tomato Processing Waste Using Supercritical CO2. Ind. Eng. Chem. Res. 2003, 42, 6641–6646. [Google Scholar] [CrossRef]
- Li, L.; Liu, Z.; Jiang, H.; Mao, X. Biotechnological production pf lycopene by microorganisms. Appl. Microbiol. Biotechnol. 2020, 104, 10307–10324. [Google Scholar] [CrossRef]
- Zou, D.; Ye, C.; Min, Y.; Li, L. Production of a novel lycopene-rich soybean food by fermentation with Bacillus amyloliquefaciens. Food Sci. Technol. 2022, 153, 112551. [Google Scholar] [CrossRef]
- Luo, H.; Bao, Y.; Zhu, P. Development of a novel functional yogurt rich in lycopene by Bacillus subtilis. Food Chem. 2023, 407, 135142. [Google Scholar] [CrossRef]
- Osawa, A.; Iki, K.; Sandmann, G.; Shindo, K. Isolation and identification of 4,4′-diapolycopene-4,4′-dioc acid produced by Bacillus firmus GB1 and its singlet oxygen quenching activity. J. Oleo Sci. 2013, 62, 955–960. [Google Scholar] [CrossRef]
- Werner, W. Botanische Beschreinbung Häufiger am Buttersäureabbau Beteiligter Sporenbildender Bakteriensspezies; Fischer: Frankfurt, German, 1933. [Google Scholar]
- Hwang, C.Y.; Cho, E.-S.; Yoon, D.J.; Cha, I.-T.; Jung, D.-H.; Nam, Y.-D.; Park, S.-L.; Lim, S.-I.; Seo, M.-J. Genomic and Physiological Characterisation of Metabacillus flavus sp. nov., a Novel Carotenoid-Producing Bacilli Isolated from Korean Marine Mud. Microorganisms 2022, 10, 979. [Google Scholar] [CrossRef] [PubMed]
- Vattekkatte, A.; Garms, S.; Brandt, W.; Boland, W. Enhanced structural diversity in terpenoid biosynthesis: Enzymes, substrates and cofactors. Org. Biomol. Chem. 2018, 16, 348–362. [Google Scholar] [CrossRef] [PubMed]
- Branquinho, R.; Meirinhos-Soares, L.; Carriço, J.A.; Pintado, M.; Peixe, L.V. Phylogenetic and clonality analysis of Bacillus pumilus isolates uncovered a highly heterogeneous population of different closely related species and clones. FEMS Microbiol. Ecol. 2014, 90, 689–698. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).