
Elizabethkingia anophelis MSU001 Isolated from Anopheles stephensi: Molecular Characterization and Comparative Genome Analysis


Abstract
1. Introduction
2. Materials and Methods
2.1. Culture
2.2. MALDI-ToF MS Analyses
2.3. Antibiotic Susceptibility Testing
2.4. Genome Sequencing, Assembly, and Annotation
2.5. Bioinformatics
3. Results
3.1. Biochemical Characterization and Identification by MALDI-ToF/MS
3.2. Genomic Features of E. anophelis MSU001
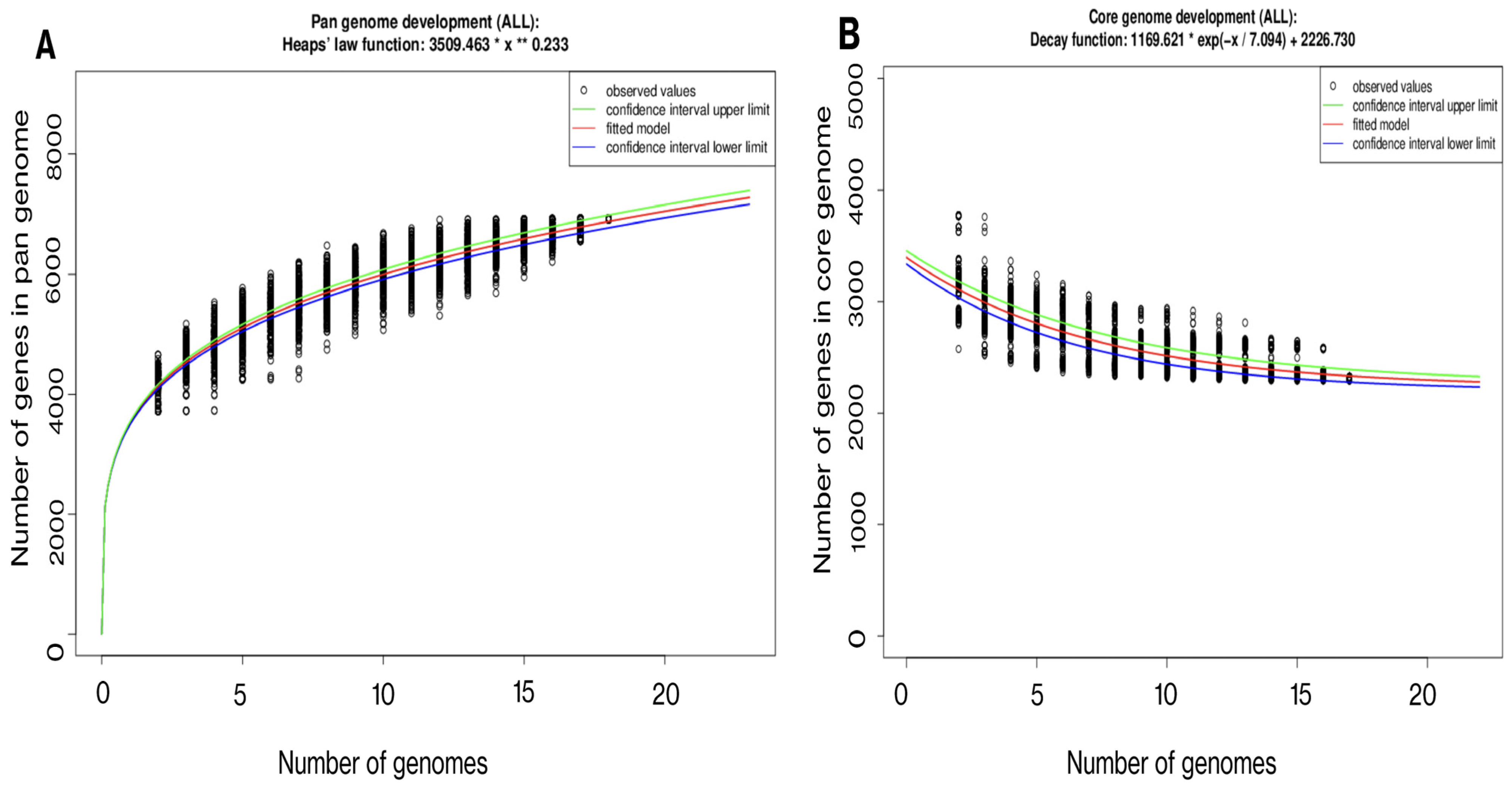
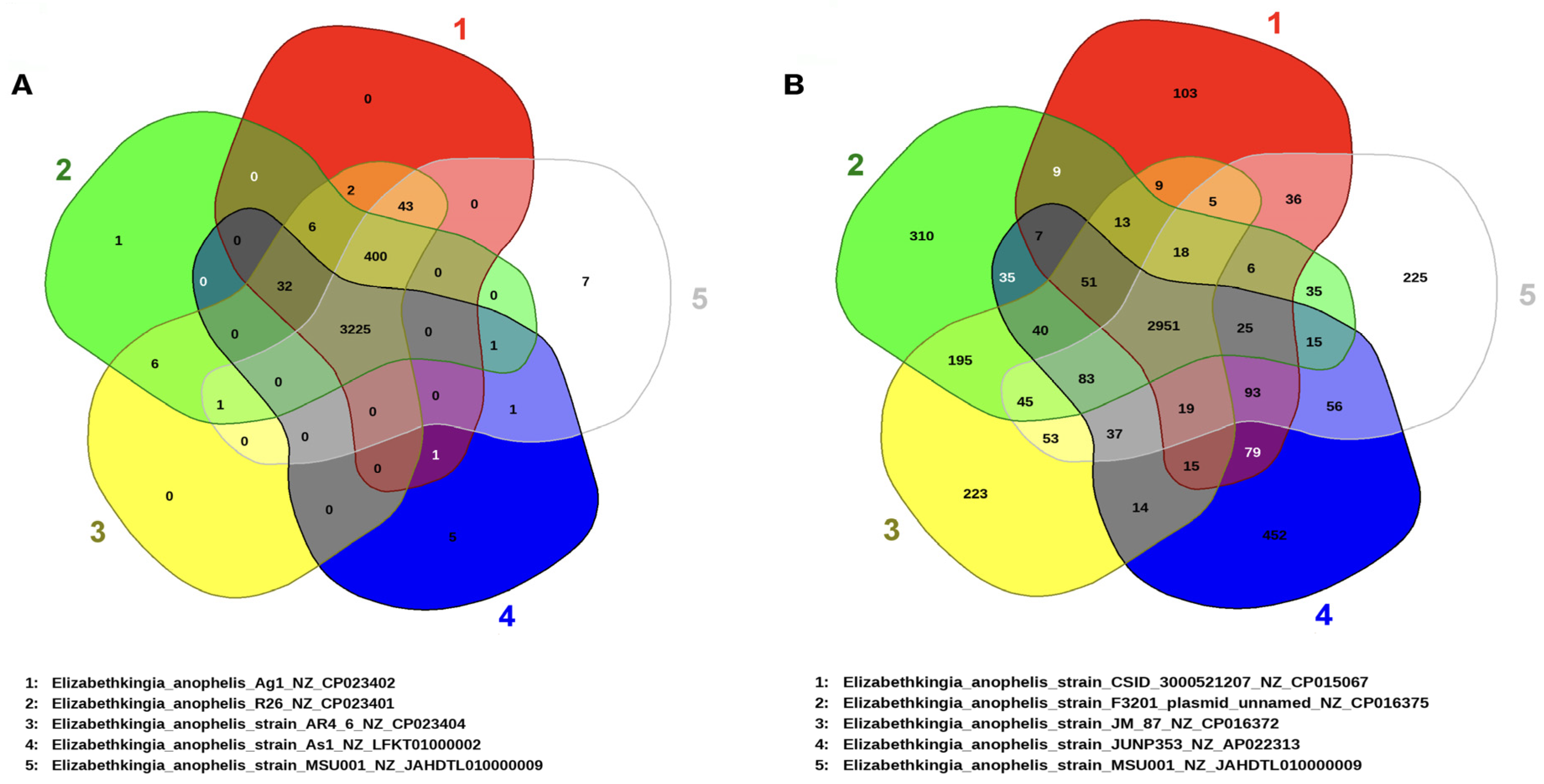
3.3. Gene Repertoire and Phylogenetic Interference of E. anophelis MSU001
3.4. Metabolites Involved in Symbiosis
3.5. Regulatory System Proteins
3.6. Carbohydrate Active Enzymes
3.7. Pathogenesis Potential Revealed by Virulence Factors and MDR Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Coyle, A.L. Elizabethkingia anophelis: Exploring the outbreak of disease in the Midwest. Nursing 2017, 47, 61–63. [Google Scholar] [CrossRef] [PubMed]
- Janda, J.M.; Lopez, D.L. Mini Review: New Pathogen Profiles: Elizabethkingia anophelis. Diagn. Microbiol. Infect. Dis. 2017, 88, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Kämpfer, P.; Matthews, H.; Glaeser, S.P.; Martin, K.; Lodders, N.; Faye, I. Elizabethkingia anophelis Sp. Nov., Isolated from the Midgut of the Mosquito Anopheles gambiae. Int. J. Syst. Evol. Microbiol. 2011, 61, 2670–2675. [Google Scholar] [CrossRef] [PubMed]
- Breurec, S.; Criscuolo, A.; Diancourt, L.; Rendueles, O.; Vandenbogaert, M.; Passet, V.; Caro, V.; Rocha, E.P.C.; Touchon, M.; Brisse, S. Genomic Epidemiology and Global Diversity of the Emerging Bacterial Pathogen Elizabethkingia anophelis. Sci. Rep. 2016, 6, 30379. [Google Scholar] [CrossRef] [PubMed]
- Ganley, J.G.; D’Ambrosio, H.K.; Shieh, M.; Derbyshire, E.R. Coculturing of Mosquito-Microbiome Bacteria Promotes Heme Degradation in Elizabethkingia anophelis. ChemBioChem 2020, 21, 1279–1284. [Google Scholar] [CrossRef]
- Mallinckrodt, L.; Huis in ’t Veld, R.; Rosema, S.; Voss, A.; Bathoorn, E. Review on Infection Control Strategies to Minimize Outbreaks of the Emerging Pathogen Elizabethkingia anophelis. Antimicrob. Resist. Infect. Control 2023, 12, 97. [Google Scholar] [CrossRef]
- Lee, Y.-L.; Hsueh, P.-R. Emerging Infections in Vulnerable Hosts: Stenotrophomonas maltophilia and Elizabethkingia anophelis. Curr. Opin. Infect. Dis. 2023, 36, 481–494. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-L.; Liu, K.-M.; Chang, H.-L.; Lin, J.-S.; Kung, F.-Y.; Ho, C.-M.; Lin, K.-H.; Chen, Y.-T. A Dominant Strain of Elizabethkingia anophelis Emerged from a Hospital Water System to Cause a Three-Year Outbreak in a Respiratory Care Center. J. Hosp. Infect. 2021, 108, 43–51. [Google Scholar] [CrossRef]
- Chen, S.; Bagdasarian, M.; Walker, E.D. Elizabethkingia anophelis: Molecular Manipulation and Interactions with Mosquito Hosts. Appl. Environ. Microbiol. 2015, 81, 2233–2243. [Google Scholar] [CrossRef]
- Akhouayri, I.G.; Habtewold, T.; Christophides, G.K. Melanotic Pathology and Vertical Transmission of the Gut Commensal Elizabethkingia meningoseptica in the Major Malaria Vector Anopheles gambiae. PLoS ONE 2013, 8, e77619. [Google Scholar] [CrossRef]
- Teo, J.; Tan, S.Y.-Y.; Liu, Y.; Tay, M.; Ding, Y.; Li, Y.; Kjelleberg, S.; Givskov, M.; Lin, R.T.P.; Yang, L. Comparative Genomic Analysis of Malaria Mosquito Vector-Associated Novel Pathogen Elizabethkingia anophelis. Genome Biol. Evol. 2014, 6, 1158–1165. [Google Scholar] [CrossRef] [PubMed]
- Steven, B.; Hyde, J.; LaReau, J.C.; Brackney, D.E. The Axenic and Gnotobiotic Mosquito: Emerging Models for Microbiome Host Interactions. Front. Microbiol. 2021, 12, 714222. [Google Scholar] [CrossRef] [PubMed]
- Onyango, M.G.; Lange, R.; Bialosuknia, S.; Payne, A.; Mathias, N.; Kuo, L.; Vigneron, A.; Nag, D.; Kramer, L.D.; Ciota, A.T. Zika Virus and Temperature Modulate Elizabethkingia anophelis in Aedes albopictus. Parasit. Vectors 2021, 14, 573. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Blom, J.; Walker, E.D. Genomic, Physiologic, and Symbiotic Characterization of Serratia marcescens Strains Isolated from the Mosquito Anopheles stephensi. Front. Microbiol. 2017, 8, 283169. [Google Scholar] [CrossRef]
- Chen, S.; Yu, T.; Terrapon, N.; Henrissat, B.; Walker, E.D. Genome Features of Asaia Sp. W12 Isolated from the Mosquito Anopheles stephensi Reveal Symbiotic Traits. Genes 2021, 12, 752. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhao, J.; Joshi, D.; Xi, Z.; Norman, B.; Walker, E.D. Persistent Infection by Wolbachia WAlbB Has No Effect on Composition of the Gut Microbiota in Adult Female Anopheles stephensi. Front. Microbiol. 2016, 7, 1485. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Johnson, B.K.; Yu, T.; Nelson, B.N.; Walker, E.D. Elizabethkingia anophelis: Physiologic and Transcriptomic Responses to Iron Stress. Front. Microbiol. 2020, 11, 804. [Google Scholar] [CrossRef] [PubMed]
- Mirza, H.C.; Tuncer, Ö.; Ölmez, S.; Şener, B.; Tuğcu, G.D.; Özçelik, U.; Gürsoy, N.C.; Otlu, B.; Büyükçam, A.; Kara, A.; et al. Clinical Strains of Chryseobacterium and Elizabethkingia Spp. Isolated from Pediatric Patients in a University Hospital: Performance of MALDI-TOF MS-Based Identification, Antimicrobial Susceptibilities, and Baseline Patient Characteristics. Microb. Drug Resist. 2018, 24, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Comba, I.Y.; Schuetz, A.N.; Misra, A.; Friedman, D.Z.P.; Stevens, R.; Patel, R.; Lancaster, Z.D.; Shah, A. Antimicrobial Susceptibility of Elizabethkingia Species: Report from a Reference Laboratory. J. Clin. Microbiol. 2022, 60, e02541-21. [Google Scholar] [CrossRef]
- Lin, J.-N.; Lai, C.-H.; Yang, C.-H.; Huang, Y.-H. Elizabethkingia Infections in Humans: From Genomics to Clinics. Microorganisms 2019, 7, 295. [Google Scholar] [CrossRef]
- Perrin, A.; Larsonneur, E.; Nicholson, A.C.; Edwards, D.J.; Gundlach, K.M.; Whitney, A.M.; Gulvik, C.A.; Bell, M.E.; Rendueles, O.; Cury, J.; et al. Evolutionary Dynamics and Genomic Features of the Elizabethkingia anophelis 2015 to 2016 Wisconsin Outbreak Strain. Nat. Commun. 2017, 8, 15483. [Google Scholar] [CrossRef] [PubMed]
- Thigpen, S.; Walblay, K.; Adil, H.; Zelencik, S.; Zelinski, C.; Nelson, K.; Cox, B.; McQuiston, J.R.; Turner, J.; Toews, K.-A.; et al. 1451. Elizabethkingia spp. Outbreak in a Ventilator-Capable Skilled Nursing Facility, Chicago 2023. Open Forum. Infect. Dis. 2023, 10, ofad500.1288. [Google Scholar] [CrossRef]
- Hu, S.; Xu, H.; Meng, X.; Bai, X.; Xu, J.; Ji, J.; Ying, C.; Chen, Y.; Shen, P.; Zhou, Y.; et al. Population Genomics of Emerging Elizabethkingia anophelis Pathogens Reveals Potential Outbreak and Rapid Global Dissemination. Emerg. Microbes Infect. 2022, 11, 2590–2599. [Google Scholar] [CrossRef] [PubMed]
- Frank, T.; Gody, J.C.; Nguyen, L.B.L.; Berthet, N.; Fleche-Mateos, A.L.; Bata, P.; Rafaï, C.; Kazanji, M.; Breurec, S. First Case of Elizabethkingia anophelis Meningitis in the Central African Republic. Lancet 2013, 381, 1876. [Google Scholar] [CrossRef] [PubMed]
- Accoti, A.; Damiani, C.; Nunzi, E.; Cappelli, A.; Iacomelli, G.; Monacchia, G.; Turco, A.; D’Alò, F.; Peirce, M.J.; Favia, G.; et al. Anopheline Mosquito Saliva Contains Bacteria That Are Transferred to a Mammalian Host through Blood Feeding. Front. Microbiol. 2023, 14, 1157613. [Google Scholar] [CrossRef] [PubMed]
- Kukutla, P.; Lindberg, B.G.; Pei, D.; Rayl, M.; Yu, W.; Steritz, M.; Faye, I.; Xu, J. Insights from the Genome Annotation of Elizabethkingia anophelis from the Malaria Vector Anopheles gambiae. PLoS ONE 2014, 9, e97715. [Google Scholar] [CrossRef] [PubMed]
- Raygoza Garay, J.A.; Hughes, G.L.; Koundal, V.; Rasgon, J.L.; Mwangi, M.M. Genome Sequence of Elizabethkingia anophelis Strain EaAs1, Isolated from the Asian Malaria Mosquito Anopheles stephensi. Genome Announc. 2016, 4, 10-1128. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liu, Y.; Chew, S.C.; Tay, M.; Salido, M.M.S.; Teo, J.; Lauro, F.M.; Givskov, M.; Yang, L. Complete Genome Sequence and Transcriptomic Analysis of the Novel Pathogen Elizabethkingia anophelis in Response to Oxidative Stress. Genome Biol. Evol. 2015, 7, 1676–1685. [Google Scholar] [CrossRef]
- Tatusova, T.; DiCuccio, M.; Badretdin, A.; Chetvernin, V.; Nawrocki, E.P.; Zaslavsky, L.; Lomsadze, A.; Pruitt, K.D.; Borodovsky, M.; Ostell, J. NCBI Prokaryotic Genome Annotation Pipeline. Nucleic Acids Res. 2016, 44, 6614–6624. [Google Scholar] [CrossRef]
- Overbeek, R.; Olson, R.; Pusch, G.D.; Olsen, G.J.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Parrello, B.; Shukla, M.; et al. The SEED and the Rapid Annotation of Microbial Genomes Using Subsystems Technology (RAST). Nucleic Acids Res. 2014, 42, D206–D214. [Google Scholar] [CrossRef]
- Alcock, B.P.; Huynh, W.; Chalil, R.; Smith, K.W.; Raphenya, A.R.; Wlodarski, M.A.; Edalatmand, A.; Petkau, A.; Syed, S.A.; Tsang, K.K.; et al. CARD 2023: Expanded Curation, Support for Machine Learning, and Resistome Prediction at the Comprehensive Antibiotic Resistance Database. Nucleic. Acids Res. 2023, 51, D690–D699. [Google Scholar] [CrossRef] [PubMed]
- Grissa, I.; Vergnaud, G.; Pourcel, C. CRISPRFinder: A Web Tool to Identify Clustered Regularly Interspaced Short Palindromic Repeats. Nucleic Acids Res. 2007, 35, W52-7. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.-H.; Ha, S.; Lim, J.; Kwon, S.; Chun, J. A Large-Scale Evaluation of Algorithms to Calculate Average Nucleotide Identity. Antonie Van Leeuwenhoek 2017, 110, 1281–1286. [Google Scholar] [CrossRef] [PubMed]
- Meier-Kolthoff, J.P.; Carbasse, J.S.; Peinado-Olarte, R.L.; Göker, M. TYGS and LPSN: A Database Tandem for Fast and Reliable Genome-Based Classification and Nomenclature of Prokaryotes. Nucleic Acids Res. 2022, 50, D801–D807. [Google Scholar] [CrossRef] [PubMed]
- Barakat, M.; Ortet, P.; Whitworth, D.E. P2RP: A Web-Based Framework for the Identification and Analysis of Regulatory Proteins in Prokaryotic Genomes. BMC Genom. 2013, 14, 269. [Google Scholar] [CrossRef] [PubMed]
- Dieckmann, M.A.; Beyvers, S.; Nkouamedjo-Fankep, R.C.; Hanel, P.H.G.; Jelonek, L.; Blom, J.; Goesmann, A. EDGAR3.0: Comparative Genomics and Phylogenomics on a Scalable Infrastructure. Nucleic Acids Res. 2021, 49, W185–W192. [Google Scholar] [CrossRef] [PubMed]
- Blom, J.; Kreis, J.; Spänig, S.; Juhre, T.; Bertelli, C.; Ernst, C.; Goesmann, A. EDGAR 2.0: An Enhanced Software Platform for Comparative Gene Content Analyses. Nucleic Acids Res. 2016, 44, W22–W28. [Google Scholar] [CrossRef] [PubMed]
- Drula, E.; Garron, M.-L.; Dogan, S.; Lombard, V.; Henrissat, B.; Terrapon, N. The Carbohydrate-Active Enzyme Database: Functions and Literature. Nucleic Acids Res. 2022, 50, D571–D577. [Google Scholar] [CrossRef]
- Baum, B.R. PHYLIP: Phylogeny Inference Package. Version 3.2. Joel Felsenstein. Q Rev. Biol. 1989, 64, 539–541. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately Maximum-Likelihood Trees for Large Alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Eriksen, H.B.; Gumpert, H.; Faurholt, C.H.; Westh, H. Determination of Elizabethkingia Diversity by MALDI-TOF Mass Spectrometry and Whole-Genome Sequencing. Emerg. Infect. Dis. 2017, 23, 320–323. [Google Scholar] [CrossRef] [PubMed]
- Champion, C.J.; Kukutla, P.; Glennon, E.K.K.; Wang, B.; Luckhart, S.; Xu, J. Anopheles gambiae: Metabolomic Profiles in Sugar-Fed, Blood-Fed, and Plasmodium falciparum-Infected Midgut. Dataset Pap. Sci. 2017, 2017, 8091749. [Google Scholar] [CrossRef]
- Brown, E.M.; Ke, X.; Hitchcock, D.; Jeanfavre, S.; Avila-Pacheco, J.; Nakata, T.; Arthur, T.D.; Fornelos, N.; Heim, C.; Franzosa, E.A.; et al. Bacteroides-Derived Sphingolipids Are Critical for Maintaining Intestinal Homeostasis and Symbiosis. Cell Host Microbe. 2019, 25, 668–680.e7. [Google Scholar] [CrossRef] [PubMed]
- Hannun, Y.A.; Obeid, L.M. Sphingolipids and Their Metabolism in Physiology and Disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 175–191. [Google Scholar] [CrossRef] [PubMed]
- Heaver, S.L.; Le, H.H.; Tang, P.; Baslé, A.; Mirretta Barone, C.; Vu, D.L.; Waters, J.L.; Marles-Wright, J.; Johnson, E.L.; Campopiano, D.J.; et al. Characterization of Inositol Lipid Metabolism in Gut-Associated Bacteroidetes. Nat. Microbiol. 2022, 7, 986–1000. [Google Scholar] [CrossRef] [PubMed]
- Hem, S.; Jarocki, V.M.; Baker, D.J.; Charles, I.G.; Drigo, B.; Aucote, S.; Donner, E.; Burnard, D.; Bauer, M.J.; Harris, P.N.A.; et al. Genomic Analysis of Elizabethkingia Species from Aquatic Environments: Evidence for Potential Clinical Transmission. Curr. Res. Microb. Sci. 2022, 3, 100083. [Google Scholar] [CrossRef] [PubMed]
- Villegas, L.E.M.; Radl, J.; Dimopoulos, G.; Short, S.M. Bacterial Communities of Aedes aegypti Mosquitoes Differ between Crop and Midgut Tissues. PLoS Negl. Trop. Dis. 2023, 17, e0011218. [Google Scholar] [CrossRef] [PubMed]
- Kadi, H.; Tanriverdi Cayci, Y.; Yener, N.; Gur Vural, D.; Bilgin, K.; Birinci, A. 16s RRNA-Based Phylogenetic Analyses of Elizabethkingia anophelis: Detection of Elizabethkingia anophelis, a Rare Infectious Agent from Blood and Determination of Antibiotic Susceptibility in Turkey. Indian J. Med. Microbiol. 2022, 40, 557–559. [Google Scholar] [CrossRef]
- McTaggart, L.R.; Stapleton, P.J.; Eshaghi, A.; Soares, D.; Brisse, S.; Patel, S.N.; Kus, J.V. Application of Whole Genome Sequencing to Query a Potential Outbreak of Elizabethkingia anophelis in Ontario, Canada. Access Microbiol. 2019, 1, e000017. [Google Scholar] [CrossRef]
- Rolando, M.; Buchrieser, C. A Comprehensive Review on the Manipulation of the Sphingolipid Pathway by Pathogenic Bacteria. Front. Cell Dev. Biol. 2019, 7, 168. [Google Scholar] [CrossRef]
- Merzendorfer, H.; Zimoch, L. Chitin Metabolism in Insects: Structure, Function and Regulation of Chitin Synthases and Chitinases. J. Exp. Biol. 2003, 206, 4393–4412. [Google Scholar] [CrossRef] [PubMed]
- Beier, S.; Bertilsson, S. Bacterial Chitin Degradation—Mechanisms and Ecophysiological Strategies. Front. Microbiol. 2013, 4, 149. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, F.H.; Gendrin, M.; Wyer, C.A.S.; Christophides, G.K. Microbiota-Induced Peritrophic Matrix Regulates Midgut Homeostasis and Prevents Systemic Infection of Malaria Vector Mosquitoes. PLoS Pathog. 2017, 13, e1006391. [Google Scholar] [CrossRef] [PubMed]
- Kuraishi, T.; Binggeli, O.; Opota, O.; Buchon, N.; Lemaitre, B. Genetic Evidence for a Protective Role of the Peritrophic Matrix against Intestinal Bacterial Infection in Drosophila melanogaster. Proc. Natl. Acad. Sci. USA 2011, 108, 15966–15971. [Google Scholar] [CrossRef] [PubMed]
- Skaar, E.P. The Battle for Iron between Bacterial Pathogens and Their Vertebrate Hosts. PLoS Pathog. 2010, 6, e1000949. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.M.; Weerapana, E.; Ciepichal, E.; Stupak, J.; Reid, C.W.; Swiezewska, E.; Imperiali, B. Polyisoprenol Specificity in the Campylobacter jejuni N-Linked Glycosylation Pathway. Biochemistry 2007, 46, 14342–14348. [Google Scholar] [CrossRef] [PubMed]
- Lukose, V.; Walvoort, M.T.; Imperiali, B. Bacterial Phosphoglycosyl Transferases: Initiators of Glycan Biosynthesis at the Membrane Interface. Glycobiology 2017, 27, 820–833. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Lv, Y.; Xu, H.; Zheng, B.; Xiao, Y. Biofilm Formation and Antibiotic Sensitivity in Elizabethkingia anophelis. Front. Cell Infect. Microbiol. 2022, 12, 953780. [Google Scholar] [CrossRef]
- Wang, M.; Gao, H.; Lin, N.; Zhang, Y.; Huang, N.; Walker, E.D.; Ming, D.; Chen, S.; Hu, S. The Antibiotic Resistance and Pathogenicity of a Multidrug-resistant Elizabethkingia anophelis Isolate. Microbiologyopen 2019, 8, e804. [Google Scholar] [CrossRef]
- Puah, S.M.; Fong, S.P.; Kee, B.P.; Puthucheary, S.D.; Chua, K.H. Molecular Identification and Biofilm-Forming Ability of Elizabethkingia Species. Microb. Pathog. 2022, 162, 105345. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain | Original Region *a | Isolation Source *b | Size (Mb) | GC% | CDS | Gene | CRISPR Count |
|---|---|---|---|---|---|---|---|
| E. anophelis | |||||||
| As1 | USA | A. gambiae | 3.59 | 35.5 | 3237 | 3315 | 0 |
| Ag1 | USA | A. gambiae | 4.09 | 35.5 | 3686 | 3788 | 0 |
| R26 | Sweden | A. gambiae | 4.06 | 35.5 | 3635 | 3741 | 0 |
| AR4-6 | China | A. sinensis | 4.09 | 35.5 | 3678 | 3785 | 0 |
| AR6-8 | China | A. sinensis | 4.09 | 35.5 | 3678 | 3785 | 0 |
| MSU001 | USA | A. stephensi | 4.05 | 35.4 | 3857 | 3753 | 1 |
| LDVH-AR107 | France | C. carpio | 3.99 | 35.7 | 3555 | 3667 | 2 |
| OSUVM 2 | USA | E. caballus | 4.1 | 35.4 | 3644 | 3754 | 0 |
| CSID_3000521207 | USA | H. sapiens | 3.85 | 35.7 | 3412 | 3513 | 0 |
| JUNP 353 | Nepal | H. sapiens | 4.32 | 35.8 | 3897 | 4049 | 0 |
| F3201 | Kuwait | H. sapiens | 4.28 | 35.46 | 3797 | 3927 | 0 |
| 296-96 | Taiwan | H. sapiens | 4.2 | 35.8 | 3779 | 3898 | 2 |
| SUE | Taiwan | H sapiens | 4.2 | 35.8 | 3771 | 3891 | 2 |
| JM-87 | USA | Z. mays | 4.18 | 35.5 | 3695 | 3837 | 0 |
| E. meningoseptica | |||||||
| NCTC10016 | UK | H. sapiens | 3.87 | 36.5 | 3397 | 3480 | 1 |
| G4120 | France | H. sapiens | 4 | 36.4 | 3519 | 3628 | 1 |
| E. miricola | |||||||
| FL160902 | China | Frog | 4.25 | 35.7 | 3760 | 3892 | 0 |
| Elizabethkingia | Predicted Regulatory Proteins | |||||||
|---|---|---|---|---|---|---|---|---|
| TOC | TF | ODP | ||||||
| RR | PP | HK | OCS | RR | TR | SF | ||
| E. anophelis | ||||||||
| Ag1 | 26 | 9 | 16 | 31 | 23 | 118 | 16 | 12 |
| As1 | 23 | 8 | 14 | 22 | 20 | 103 | 15 | 10 |
| R26 | 26 | 9 | 16 | 31 | 23 | 118 | 16 | 11 |
| AR4-6 | 26 | 9 | 16 | 31 | 23 | 118 | 16 | 12 |
| AR6-8 | 26 | 9 | 16 | 31 | 23 | 118 | 16 | 12 |
| MSU001 | 26 | 9 | 16 | 31 | 23 | 118 | 16 | 13 |
| LDVH-AR107 | 26 | 8 | 17 | 26 | 23 | 119 | 17 | 12 |
| OSUVM 2 | 29 | 9 | 21 | 32 | 26 | 128 | 18 | 8 |
| CSID_3000521207 | 27 | 8 | 17 | 27 | 23 | 113 | 16 | 10 |
| JUNP 353 | 27 | 8 | 18 | 30 | 23 | 117 | 17 | 11 |
| F3201 | 18 | 9 | 20 | 30 | 25 | 133 | 16 | 12 |
| 296-96 | 26 | 7 | 19 | 29 | 22 | 119 | 18 | 10 |
| SUE | 27 | 7 | 18 | 29 | 23 | 118 | 18 | 11 |
| JM-87 | 30 | 9 | 21 | 28 | 27 | 124 | 18 | 9 |
| E. meningoseptica | ||||||||
| NCTC10016 | 19 | 29 | 10 | 27 | 25 | 117 | 15 | 6 |
| G4120 | 28 | 10 | 18 | 16 | 15 | 121 | 16 | 6 |
| E. miricola | ||||||||
| FL160902 | 35 | 11 | 25 | 31 | 31 | 131 | 20 | 10 |
| C8J 1080 | DnaK | EF-Tu | eno | htpB | katG | mps1-1 | mps1-2 | pglC | RmlA | |
|---|---|---|---|---|---|---|---|---|---|---|
| E. anophelis | ||||||||||
| As1 | + | + | + | + | + | + | + | + | + | + |
| Ag1 | + | + | + | + | + | + | + | + | + | + |
| R26 | + | + | + | + | + | + | + | + | + | + |
| AR4-6 | + | + | + | + | + | + | + | + | + | + |
| R6-8 | + | + | + | + | + | + | + | + | + | + |
| MSU001 | + | + | + | + | + | + | + | + | + | + |
| LDVH-AR107 | + | + | + | - | - | - | + | + | - | + |
| OSUVM 2 | - | + | + | + | + | + | + | + | - | + |
| CSID_3000521207 | + | + | + | + | + | + | + | + | - | + |
| JUNP 353 | + | + | + | - | - | + | + | + | - | + |
| F3201 | + | + | + | + | + | + | + | + | - | + |
| 296-96 | + | + | + | - | - | + | + | + | - | + |
| SUE | + | + | + | - | - | + | + | + | - | + |
| JM-87 | + | + | + | - | - | + | + | + | - | + |
| E. meningoseptica | ||||||||||
| NCTC10016 | - | + | + | - | - | + | - | - | - | + |
| G4120 | - | + | + | - | - | - | - | - | - | - |
| E. miricola | ||||||||||
| FL160902 | - | - | + | - | - | - | + | + | - | - |
| Antibiotic Class | Antimicrobial | MIC (µg/mL) * | SIR |
|---|---|---|---|
| Aminoglycosides | |||
| Amikacin | ≥64 | R | |
| Gentamicin | ≥16 | R | |
| β-lactams and β-lactamase inhibitors | |||
| Meropenem | ≥16 | R | |
| Cefazolin | ≥64 | R | |
| Cefotaxime | ≥32 | R | |
| Tobramycin | ≥16 | R | |
| Aztreonam | ≥64 | R | |
| Ampicillin | ≥32 | R | |
| Ampicillin/Sulbactam | ≥32 | R | |
| Piperacillin | ≥64 | R | |
| Ceftriaxone | ≥64 | R | |
| Piperacillin/Tazobactam | ≥128 | R | |
| Sulfonamide | Trimethoprim/Sulfamethoxazole | 40 | S |
| Quinolone | Ciprofloxacin | 0.5 | S |
| Tetracycline | Tigecycline | 4 | I |
| Nitrofuran | Nitrofurantoin | 128 | R |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, S.; Pham, S.; Terrapon, N.; Blom, J.; Walker, E.D. Elizabethkingia anophelis MSU001 Isolated from Anopheles stephensi: Molecular Characterization and Comparative Genome Analysis. Microorganisms 2024, 12, 1079. https://doi.org/10.3390/microorganisms12061079
Chen S, Pham S, Terrapon N, Blom J, Walker ED. Elizabethkingia anophelis MSU001 Isolated from Anopheles stephensi: Molecular Characterization and Comparative Genome Analysis. Microorganisms. 2024; 12(6):1079. https://doi.org/10.3390/microorganisms12061079
Chicago/Turabian StyleChen, Shicheng, Steven Pham, Nicolas Terrapon, Jochen Blom, and Edward D. Walker. 2024. "Elizabethkingia anophelis MSU001 Isolated from Anopheles stephensi: Molecular Characterization and Comparative Genome Analysis" Microorganisms 12, no. 6: 1079. https://doi.org/10.3390/microorganisms12061079
APA StyleChen, S., Pham, S., Terrapon, N., Blom, J., & Walker, E. D. (2024). Elizabethkingia anophelis MSU001 Isolated from Anopheles stephensi: Molecular Characterization and Comparative Genome Analysis. Microorganisms, 12(6), 1079. https://doi.org/10.3390/microorganisms12061079