
Isolation and Characterization of a Low-Temperature, Cellulose-Degrading Microbial Consortium from Northeastern China
 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Isolation of Microorganisms
2.2. Determination of Straw Weight Loss Rate
2.3. Microscopic Analysis
2.4. Sequencing and Gene Annotation Analysis
3. Results
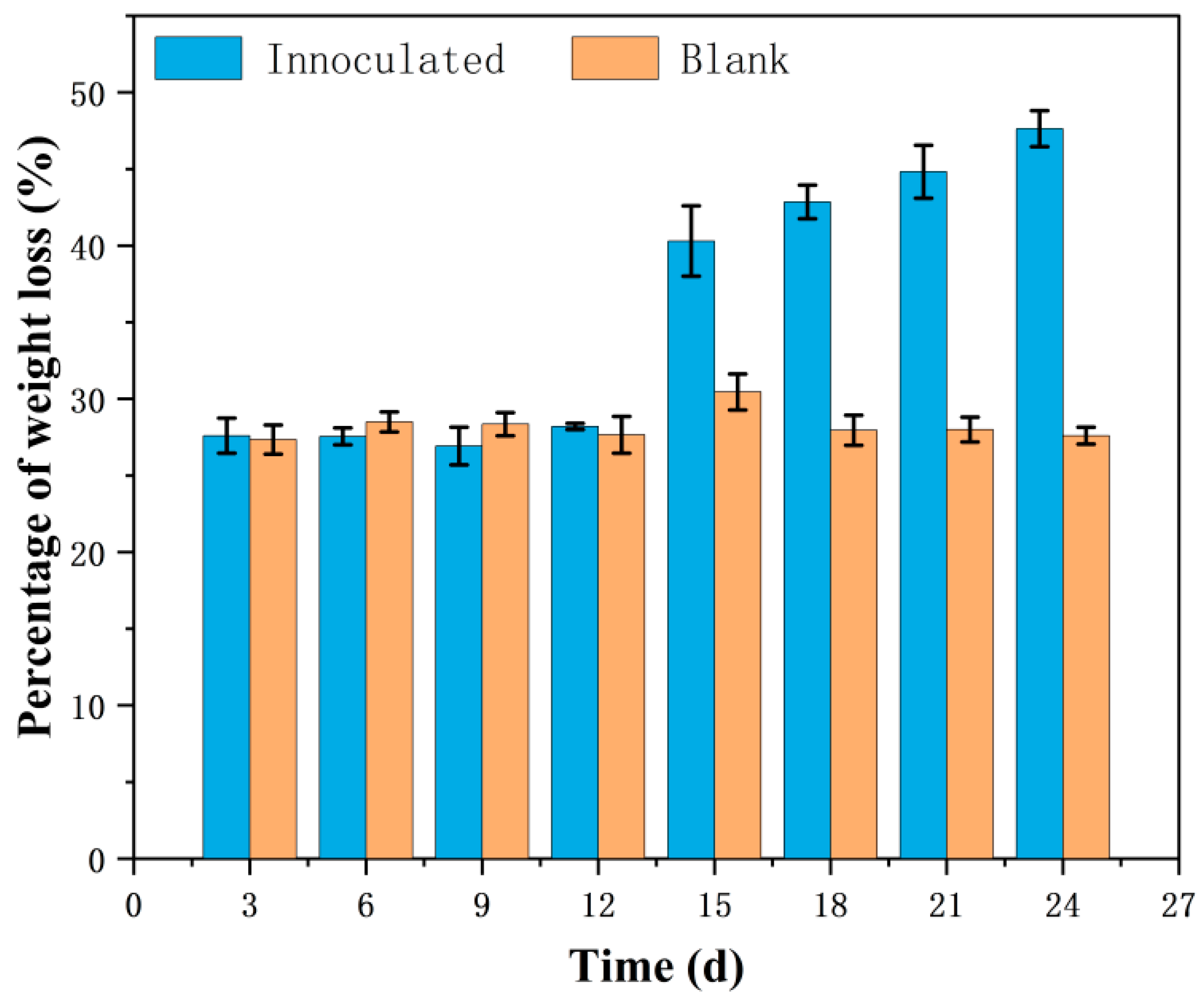
3.1. Cellulose Degradation Situation
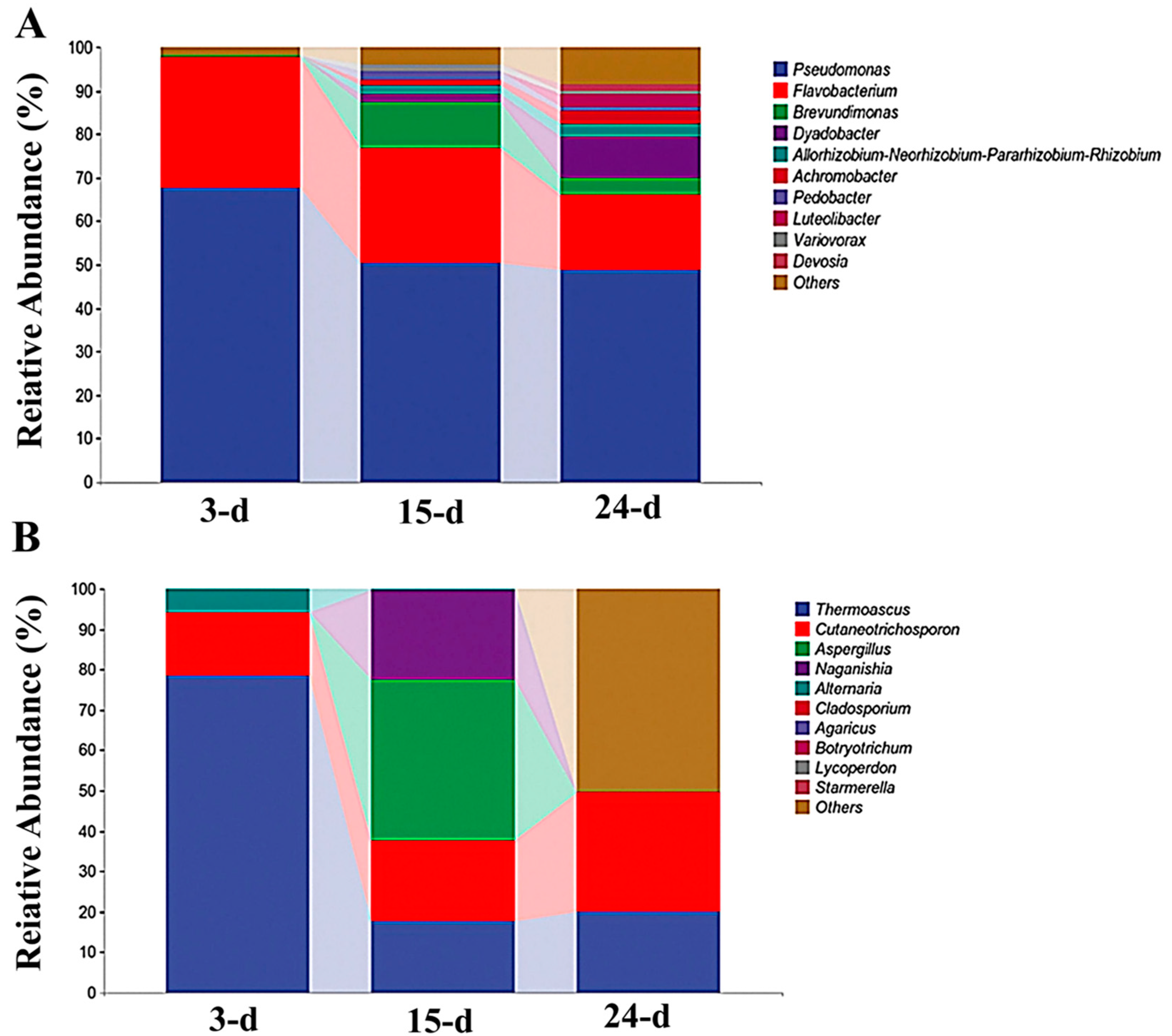
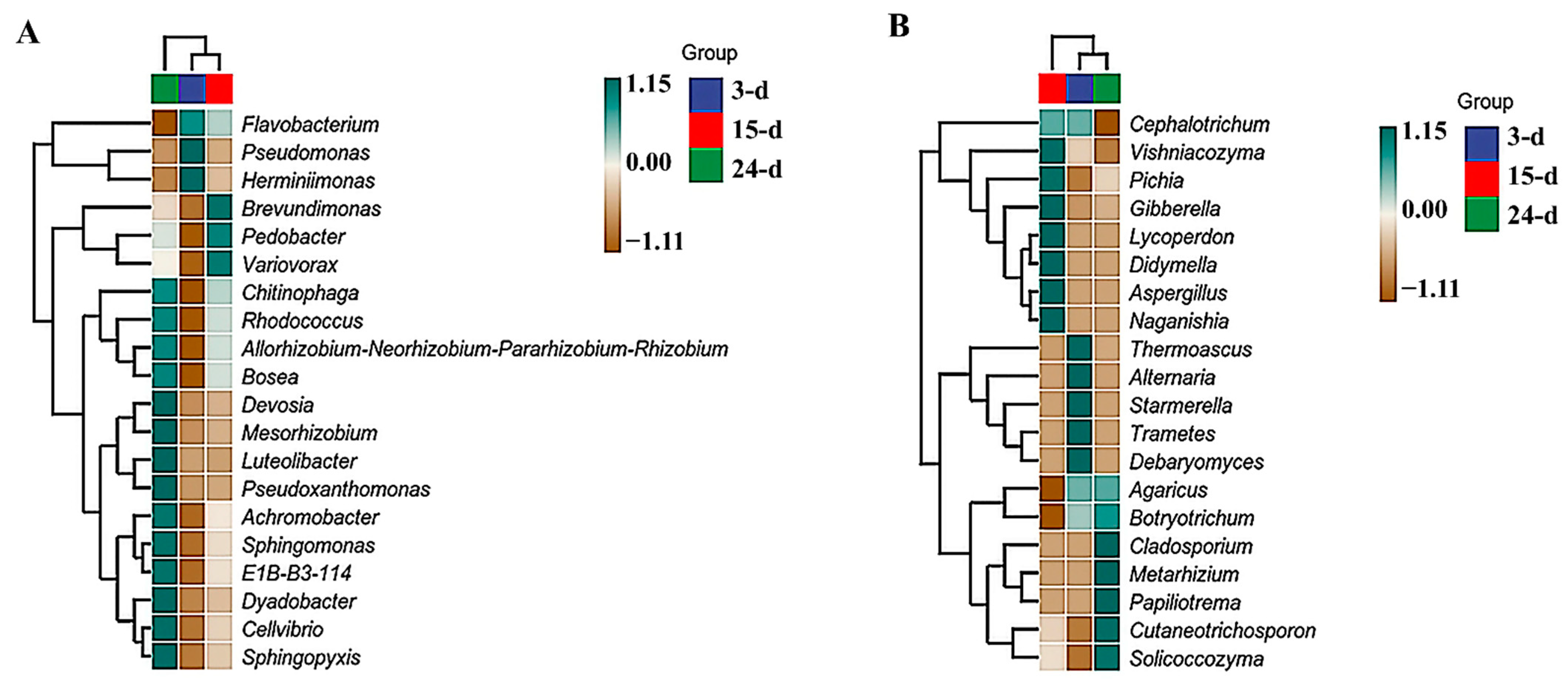
3.2. Microbial Succession
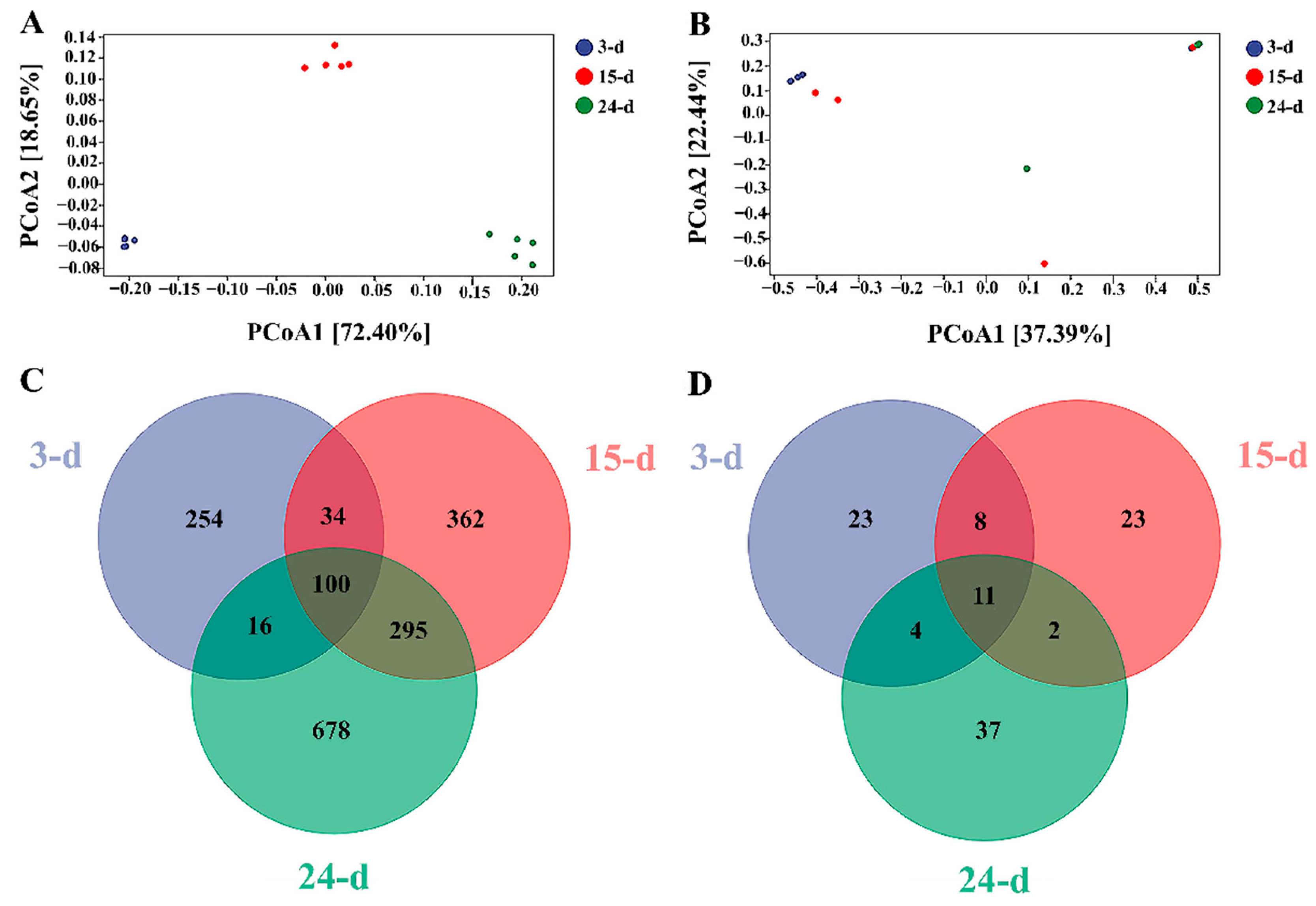
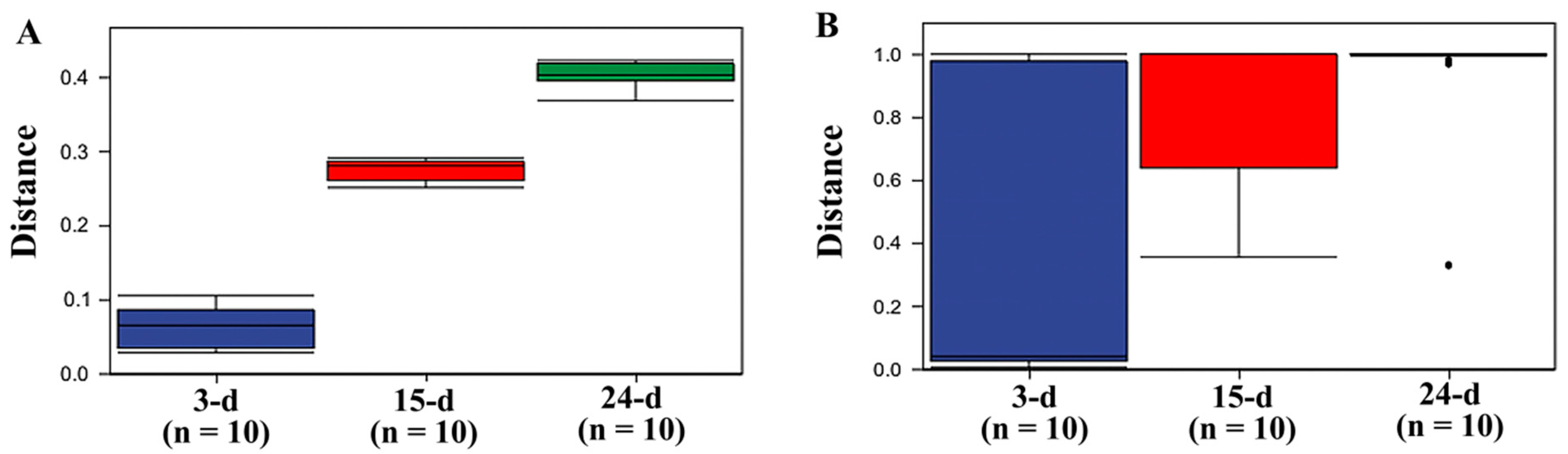
3.3. Diversity Analysis
3.4. Functional Principal Component Analysis
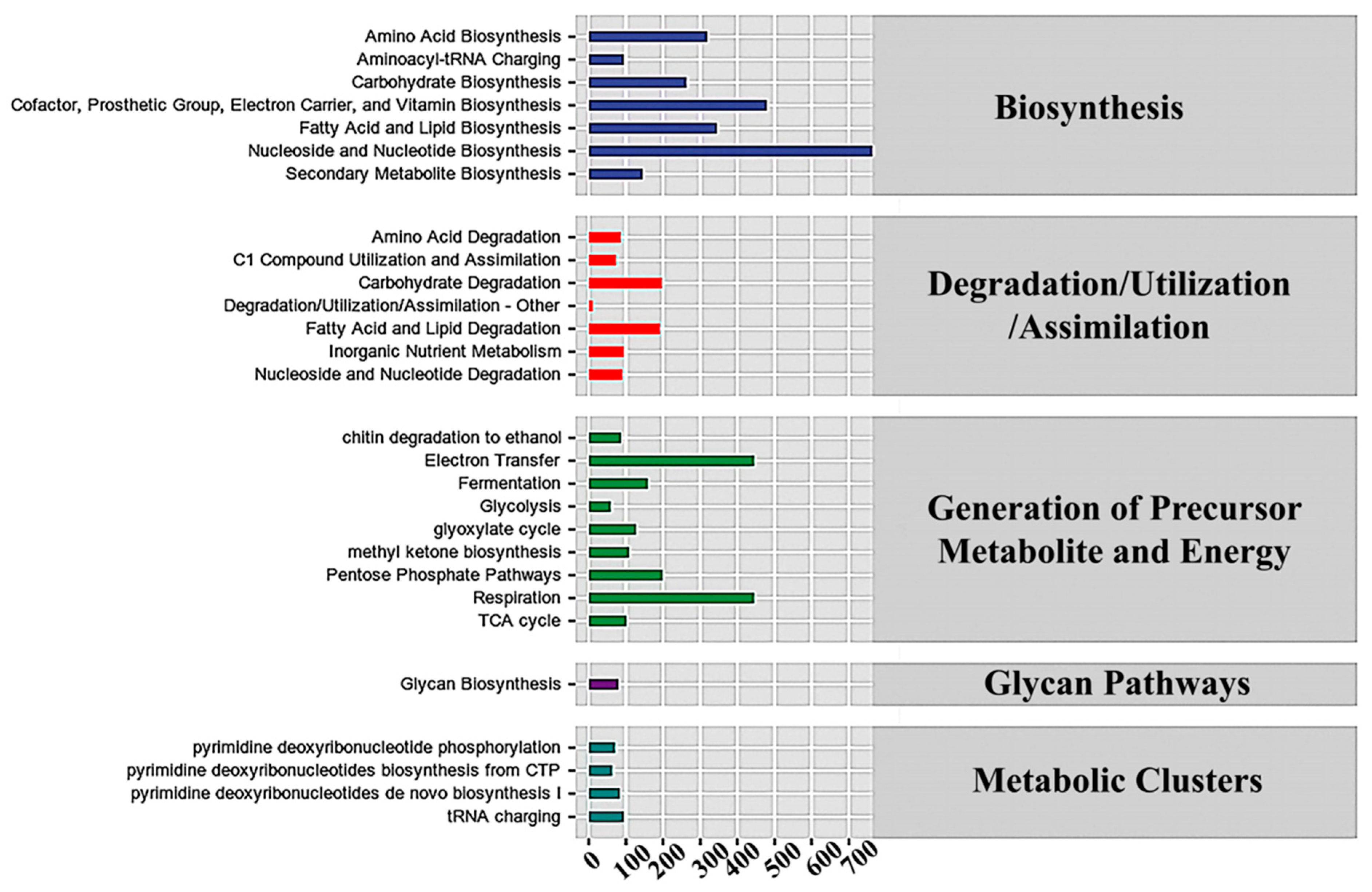
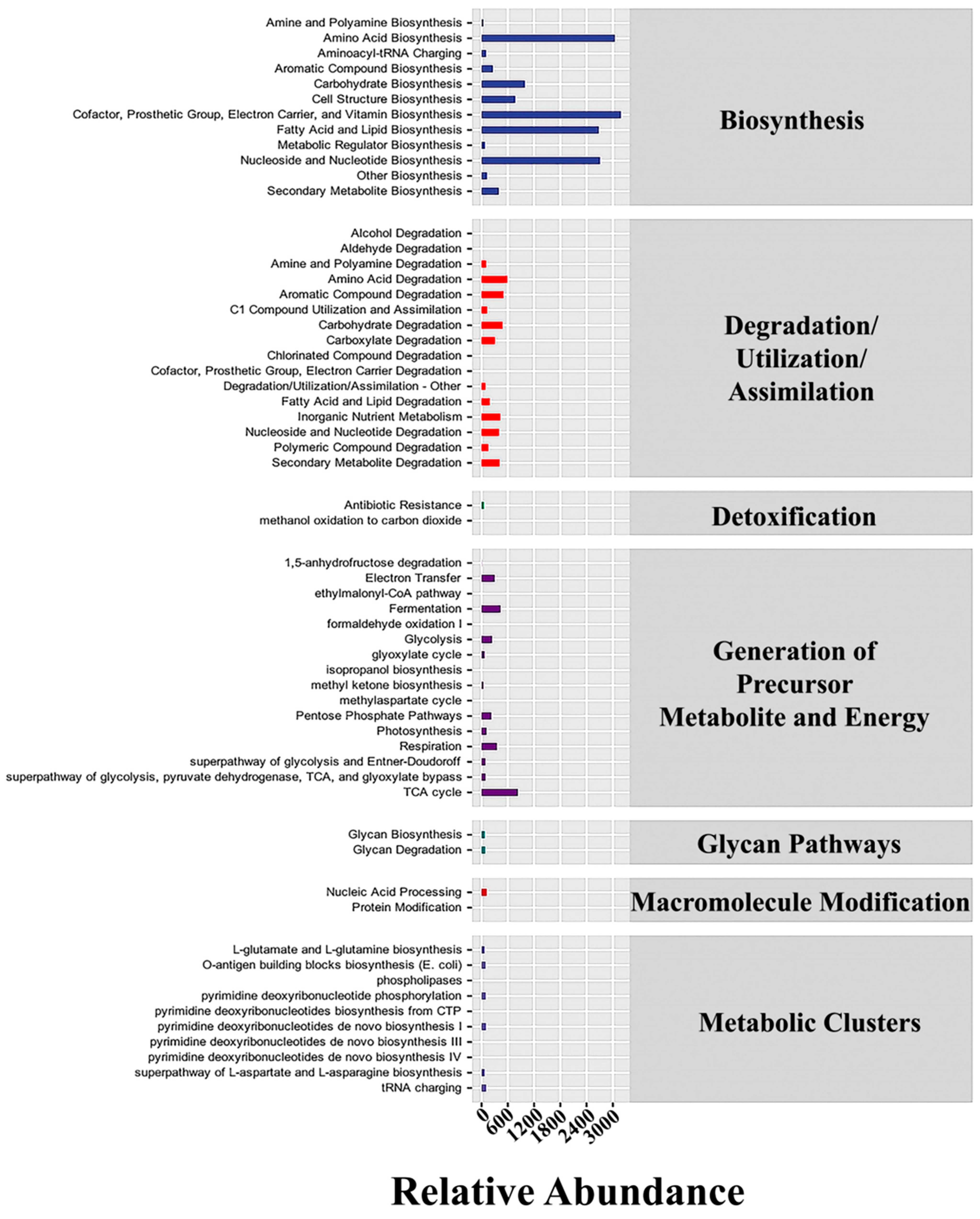
3.5. Metabolic Pathways in Bacteria and Fungi
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Tanruean, K.; Penkhrue, W.; Kumla, J.; Suwannarach, N.; Lumyong, S. Valorization of lignocellulosic wastes to produce phytase and cellulolytic enzymes from a thermophilic fungus, Thermoascus aurantiacus SL16W, under semi-solid state fermentation. J. Fungi 2021, 7, 286. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Shan, D.; Liu, X.; Sun, M. Cellulose-degrading strains: Their screeningand application to corn strawin low-temperature environments. Pol. J. Environ. Stud. 2018, 27, 2349–2355. [Google Scholar] [CrossRef]
- Qinggeer; Gao, J.-L.; Yu, X.-F.; Zhang, B.-L.; Wang, Z.-G.; Naoganchaolu, B.; Hu, S.-P.; Sun, J.-Y.; Xie, M.; Wang, Z. Screening of a microbial consortium with efficient corn stover degradation ability at low temperature. J. Integr. Agric. 2016, 15, 2369–2379. [Google Scholar] [CrossRef]
- Pedersen, M.; Johansen, K.S.; Meyer, A.S. Low Temperature Lignocellulose pretreatment: Effects and interactions of pretreatment ph are critical for maximizing enzymatic monosaccharide yields from Wheat Straw. Biotechnol. Biofuels 2011, 4, 11. [Google Scholar] [CrossRef] [PubMed]
- Santos, D.A.; Oliveira, M.M.; Curvelo, A.A.; Fonseca, L.P.; Porto, A.L. Hydrolysis of cellulose from sugarcane bagasse by cellulases from marine-derived fungi strains. Int. Biodeterior. Biodegrad. 2017, 121, 66–78. [Google Scholar] [CrossRef]
- Su, Y.; Yu, X.; Sun, Y.; Wang, G.; Chen, H.; Chen, G. Evaluation of screened lignin-degrading fungi for the biological pretreatment of corn stover. Sci. Rep. 2018, 8, 5385. [Google Scholar] [CrossRef]
- Su, Y.; Yu, X.; Sun, Y.; Wang, G.; Chen, H.; Chen, G. An efficient strategy for enhancing enzymatic saccharification with delignified fungus Myrothecium Verrucaria and solid acid. Ind. Crops Prod. 2018, 121, 396–404. [Google Scholar] [CrossRef]
- Li, Y.; Huang, H.; Wu, G.; Chang, Z. Straw degradation behaviors under different conditions of relative air humidity and ultraviolet-a irradiation. BioResources 2016, 11, 9255–9272. [Google Scholar] [CrossRef]
- Xu, X.; Xu, Z.; Shi, S.; Lin, M. Lignocellulose degradation patterns, structural changes, and enzyme secretion by inonotus obliquus on straw biomass under submerged fermentation. Bioresour. Technol. 2017, 241, 415–423. [Google Scholar] [CrossRef]
- Van Dyk, J.S.; Pletschke, B.I. A review of lignocellulose bioconversion using enzymatic hydrolysis and synergistic cooperation between enzymes—Factors affecting enzymes, conversion and Synergy. Biotechnol. Adv. 2012, 30, 1458–1480. [Google Scholar] [CrossRef]
- Lambertz, C.; Garvey, M.; Klinger, J.; Heesel, D.; Klose, H.; Fischer, R.; Commandeur, U. Challenges and advances in the heterologous expression of cellulolytic enzymes: A Review. Biotechnol. Biofuels 2014, 7, 135. [Google Scholar] [CrossRef] [PubMed]
- Behera, B.C.; Singdevsachan, S.K.; Mishra, R.R.; Dutta, S.K.; Thatoi, H.N. Diversity, mechanism and biotechnology of phosphate solubilising microorganism in mangrove—A review. Biocatal. Agric. Biotechnol. 2014, 3, 97–110. [Google Scholar] [CrossRef]
- Wei, Y.; Wu, D.; Wei, D.; Zhao, Y.; Wu, J.; Xie, X.; Zhang, R.; Wei, Z. Improved lignocellulose-degrading performance during straw composting from diverse sources with actinomycetes inoculation by regulating the key enzyme activities. Bioresour. Technol. 2019, 271, 66–74. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Ding, B.; Ali, Q.; Liu, H.; Zhao, Y.; Wang, X.; Han, Y.; Dong, H.; Divvela, P.K.; Juan, Y. Screening and isolation ofcold-adapted cellulosedegrading bacterium: A candidate for straw degradationand De novo genome sequencing analysis. Front. Microbiol. 2023, 13, 1098723. [Google Scholar] [CrossRef] [PubMed]
- Maruthamuthu, M.; Jimenez, D.J.; Stevens, P.; van Elsas, J.D. A multi-substrate approach for functional metagenomics-based screening for (hemi)cellulases in two wheat straw-degrading microbial consortia unveils novel thermoalkaliphilic enzymes. BMC Genom. 2016, 17, 86. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Jiang, W.; Xin, F.; Zhang, W.; Jiang, M. Thermophiles: Potential chassis for Lignocellulosic Biorefinery. Trends Biotechnol. 2022, 40, 643–646. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Wang, X.; Wu, Y.; Wang, X.; Zhao, J.; Liu, Y.; Chen, Z.; Jiang, Z.; Tian, W.; Zhang, J. Effects of thermophiles inoculation on the efficiency and maturity of rice straw composting. Bioresour. Technol. 2022, 354, 127195. [Google Scholar] [CrossRef] [PubMed]
- Lü, Y.; Li, N.; Gong, D.; Wang, X.; Cui, Z. The effect of temperature on the structure and function of a cellulose-degrading microbial community. Appl. Biochem. Biotechnol. 2012, 168, 219–233. [Google Scholar] [CrossRef] [PubMed]
- Boboua, S.Y.; Wang, S.; Tao, D.; Sun, Y.; Zheng, G. Effect of low temperature resistant microbial consortium LTF-27 system on biogas anaerobic digestion performance and optimization of acid production. SSRN Electron. J. 2023. [Google Scholar] [CrossRef]
- Wang, Y.; Cai, J.; Li, D. Efficient degradation of rice straw through a novel psychrotolerant Bacillus cereus at low temperature. J. Sci. Food Agric. 2022, 103, 1394–1403. [Google Scholar] [CrossRef]
- Öquist, M.G.; Sparrman, T.; Klemedtsson, L.; Drotz, S.H.; Grip, H.; Schleucher, J.; Nilsson, M. Water availability controls microbial temperature responses in Frozen Soil CO2 Production. Glob. Change Biol. 2009, 15, 2715–2722. [Google Scholar] [CrossRef]
- Stevenson, A.; Burkhardt, J.; Cockell, C.S.; Cray, J.A.; Dijksterhuis, J.; Fox-Powell, M.; Kee, T.P.; Kminek, G.; McGenity, T.J.; Timmis, K.N.; et al. Multiplication of microbes below 0.690 water activity: Implications for terrestrial and extraterrestrial life. Environ. Microbiol. 2014, 17, 257–277. [Google Scholar] [CrossRef]
- Hoehler, T.M.; Jørgensen, B.B. Microbial Life Under Extreme Energy Limitation. Nat. Rev. Microbiol. 2013, 11, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Lorv, J.S.; Rose, D.R.; Glick, B.R. Bacterial ice crystal controlling proteins. Scientifica 2014, 2014, 976895. [Google Scholar] [CrossRef] [PubMed]
- Bore, E.K.; Apostel, C.; Halicki, S.; Kuzyakov, Y.; Dippold, M.A. Microbial metabolism in soil at subzero temperatures: Adaptation mechanisms revealed by position-specific 13C labeling. Front. Microbiol. 2017, 8, 265512. [Google Scholar] [CrossRef] [PubMed]
- Cacace, G.; Mazzeo, M.F.; Sorrentino, A.; Spada, V.; Malorni, A.; Siciliano, R.A. Proteomics for the elucidation of cold adaptation mechanisms in listeria monocytogenes. J. Proteom. 2010, 73, 2021–2030. [Google Scholar] [CrossRef] [PubMed]
- De Maayer, P.; Anderson, D.; Cary, C.; Cowan, D.A. Some like it cold: Understanding the survival strategies of Psychrophiles. EMBO Rep. 2014, 15, 508–517. [Google Scholar] [CrossRef] [PubMed]
- Ayala-del-Río, H.L.; Chain, P.S.; Grzymski, J.J.; Ponder, M.A.; Ivanova, N.; Bergholz, P.W.; Di Bartolo, G.; Hauser, L.; Land, M.; Bakermans, C.; et al. The genome sequence of Psychrobacter arcticus 273-4, a psychroactive Siberian permafrost bacterium, reveals mechanisms for adaptation to low-temperature growth. Appl. Environ. Microbiol. 2010, 76, 2304–2312. [Google Scholar] [CrossRef] [PubMed]
- Casanueva, A.; Tuffin, M.; Cary, C.; Cowan, D.A. Molecular adaptations to psychrophily: The impact of ‘OMIC’ technologies. Trends Microbiol. 2010, 18, 374–381. [Google Scholar] [CrossRef] [PubMed]
- Qin, G.; Zhu, L.; Chen, X.; Wang, P.G.; Zhang, Y. Structural characterization and ecological roles of a novel exopolysaccharide from the deep-sea psychrotolerant Bacterium pseudoalteromonas sp. SM9913. Microbiology 2007, 153, 1566–1572. [Google Scholar] [CrossRef]
- Sandhya, V.; Ali, S.Z. The production of exopolysaccharide by pseudomonas putida gap-p45 under various abiotic stress conditions and its role in soil aggregation. Microbiology 2015, 84, 512–519. [Google Scholar] [CrossRef]
- Nichols CA, M.; Guezennec, J.; Bowman, J.P. Bacterial exopolysaccharides from extreme marine environments with special consideration of the Southern Ocean, sea ice, and deep-sea hydrothermal vents: A Review. Mar. Biotechnol. 2005, 7, 253–271. [Google Scholar] [CrossRef] [PubMed]
- Kandror, O.; DeLeon, A.; Goldberg, A.L. Trehalose synthesis is induced upon exposure of Escherichia coli to cold and is essential for viability at low temperatures. Proc. Natl. Acad. Sci. USA 2002, 99, 9727–9732. [Google Scholar] [CrossRef] [PubMed]
- Johns, G.C. Evolutionary convergence in adaptation of proteins to temperature: A4-lactate dehydrogenases of Pacific damselfishes (Chromis spp.). Mol. Biol. Evol. 2003, 21, 314–320. [Google Scholar] [CrossRef] [PubMed]
- Berry, E.D.; Foegeding, P.M. Cold temperature adaptation and growth of microorganisms. J. Food Prot. 1997, 60, 1583–1594. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, R.C.; Singh, R.; Eltis, L.D.; Mohn, W.W. Bacterial contributions to delignification and lignocellulose degradation in forest soils with metagenomic and quantitative stable isotope probing. ISME J. 2018, 13, 413–429. [Google Scholar] [CrossRef] [PubMed]
- Janusz, G.; Pawlik, A.; Sulej, J.; Świderska-Burek, U.; Jarosz-Wilkołazka, A.; Paszczyński, A. Lignin degradation: Microorganisms, enzymes involved, genomes analysis and Evolution. FEMS Microbiol. Rev. 2017, 41, 941–962. [Google Scholar] [CrossRef] [PubMed]
- Kato, S.; Haruta, S.; Cui, Z.J.; Ishii, M.; Igarashi, Y. Effective cellulose degradation by a mixed-culture system composed of a cellulolytic clostridium and aerobic non-cellulolytic bacteria. FEMS Microbiol. Ecol. 2004, 51, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Cheng, W.; Li, S.; Tang, X.; Wei, Z. The “quality” and “quantity” of microbial species drive the degradation of cellulose during composting. Bioresour. Technol. 2021, 320, 124425. [Google Scholar] [CrossRef]
- Wongwilaiwalin, S.; Rattanachomsri, U.; Laothanachareon, T.; Eurwilaichitr, L.; Igarashi, Y.; Champreda, V. Analysis of a thermophilic lignocellulose degrading microbial consortium and multi-species lignocellulolytic enzyme system. Enzym. Microb. Technol. 2010, 47, 283–290. [Google Scholar] [CrossRef]
- Guo, P.; Wang, X.; Zhu, W.; Yang, H.; Cheng, X.; Cui, Z. Degradation of corn stalk by the composite microbial system of MC1. J. Environ. Sci. 2008, 20, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Kaeberlein, T.; Lewis, K.; Epstein, S.S. Isolating “uncultivable” microorganisms in pure culture in a simulated natural environment. Science 2002, 296, 1127–1129. [Google Scholar] [CrossRef] [PubMed]
- McArdle, B.H.; Anderson, M.J. Fitting multivariate models to community data: A comment on distance-based redundancy analysis. Ecology 2001, 82, 290–297. [Google Scholar] [CrossRef]
- Deacon, J.W. Decomposition of filter paper cellulose by thermophilic fungi acting singly, in combination, and in sequence. Trans. Br. Mycol. Soc. 1985, 85, 663–669. [Google Scholar] [CrossRef]
- Fidio, N.; Liuzzi, F.; Mastrolitti, S.; Albergo, R.; Bari, I. Single cell oil production from undetoxified Arundo donax L. hydrolysate by Cutaneotrichosporon curvatus. J. Microbiol. Biotechnol. 2019, 29, 256–267. [Google Scholar] [CrossRef] [PubMed]
- Caporusso, A.; De Bari, I.; Valerio, V.; Albergo, R.; Liuzzi, F. Conversion of cardoon crop residues into single cell oils by Lipomyces Tetrasporus and Cutaneotrichosporon curvatus: Process optimizations to overcome the microbial inhibition of lignocellulosic hydrolysates. Ind. Crops Prod. 2021, 159, 113030. [Google Scholar] [CrossRef]
- Tamayo-Ordóñez, M.C.; Contreras-Esquivel, J.C.; Ayil-Gutiérrez, B.A.; De la Cruz-Arguijo, E.A.; Tamayo-Ordóñez, F.A.; Ríos-González, L.J.; Tamayo-Ordóñez, Y.J. Interspecific evolutionary relationships of alpha-glucuronidase in the genus aspergillus. Fungal Biol. 2021, 125, 560–575. [Google Scholar] [CrossRef]
- Romero, S.M.; Giudicessi, S.L.; Vitale, R.G. Is the fungus aspergillus a threat to cultural heritage? J. Cult. Herit. 2021, 51, 107–124. [Google Scholar] [CrossRef]
- Sohail, M.; Barzkar, N.; Michaud, P.; Tamadoni Jahromi, S.; Babich, O.; Sukhikh, S.; Das, R.; Nahavandi, R. Cellulolytic and xylanolytic enzymes from yeasts: Properties and industrial applications. Molecules 2022, 27, 3783. [Google Scholar] [CrossRef]
- Carvalho, J.K.; Panatta, A.A.; Silveira, M.A.; Tav, C.; Johann, S.; Rodrigues, M.L.; Martins, C.V. Yeasts isolated from a lotic continental environment in Brazil show potential to produce amylase, cellulase and protease. Biotechnol. Rep. 2021, 30, e00630. [Google Scholar] [CrossRef]
- Cheng, Y.; Huang, M.; Shen, X.; Jiang, C. Enhanced Cornstalk decomposition by a psychrotrophic bacterial consortium comprising cellulose, hemicellulose, and lignin degraders with biochar as a carrier for carbon neutrality. Bioresour. Technol. 2022, 344, 126259. [Google Scholar] [CrossRef]
- Han, S.; Yu, X.; Zhang, S.; Ouyang, Y.; Hu, S.; Borjigin, Q.; Bai, J.; Guo, J.; Gao, J. Construction of Bacterial Consortium for Cornstalk degradation in Soil. SSRN Electron. J. 2022. [Google Scholar] [CrossRef]
- Mukhia, S.; Kumar, A.; Kumari, P.; Kumar, R.; Kumar, S. Multilocus sequence based identification and adaptational strategies of pseudomonas sp. from the supraglacial site of Sikkim Himalaya. PLoS ONE 2022, 17, e0261178. [Google Scholar] [CrossRef]
- Xu, C.; Cui, D.; Lv, X.; Zhong, G.; Liu, J. Heterogeneous distribution of carbofuran shaped by Pseudomonas stutzeri biofilms enhances biodegradation of agrochemicals. SSRN Electron. J. 2023. [Google Scholar] [CrossRef]
- Pereira, A.R.; Assis, N.V.; Paranhos, A.G.; Lima, D.R.; Baeta, B.E.; Aquino, S.F.; de Silva, S. Effect of inoculum composition on the microbial community involved in the anaerobic digestion of sugarcane bagasse. Environ. Technol. 2023, 45, 2205–2217. [Google Scholar] [CrossRef]
- Zou, L.; Ouyang, S.; Hu, Y.; Zheng, Z.; Ouyang, J. Efficient lactic acid production from dilute acid-pretreated lignocellulosic biomass by a synthetic consortium of engineered pseudomonas putida and bacillus coagulans. Biotechnol. Biofuels 2021, 14, 227. [Google Scholar] [CrossRef]
- Tao, J.; Chen, Q.; Chen, S.; Lu, P.; Chen, Y.; Jin, J.; Li, J.; Xu, Y.; He, W.; Long, T.; et al. Metagenomic insight into the microbial degradation of organic compounds in fermented plant leaves. Environ. Res. 2022, 214, 113902. [Google Scholar] [CrossRef]
- Tsudome, M.; Tachioka, M.; Miyazaki, M.; Tsuda, M.; Takaki, Y.; Deguchi, S. Marinagarivorans cellulosilyticus sp. nov., a cellulolytic bacterium isolated from the deep-sea off Noma-Misaki, Japan. Int. J. Syst. Evol. Microbiol. 2023, 73, 005748. [Google Scholar] [CrossRef]
- Novak, J.K.; Gardner, J.G. Galactomannan utilization by Cellvibrio japonicus relies on a single essential α-galactosidase encoded by the aga27a gene. Mol. Microbiol. 2023, 119, 312–325. [Google Scholar] [CrossRef]
- Hernández-Lara, A.; Ros, M.; Cuartero, J.; Vivo, J.-M.; Lozano-Pastor, P.; Pascual, J.A. Effects of solarisation combined with compost on soil pathogens and the microbial community in a spinach cropping system. Agric. Ecosyst. Environ. 2023, 346, 108359. [Google Scholar] [CrossRef]
- Lu, Y.; Jiao, L.; Sun, G.; Wang, J.; Liu, S.; Li, R.; Zhang, Y.; Guo, Y.; Guo, J.; Jiang, X.; et al. Preservation status and microbial community of waterlogged archaeological woods over 7800 years old at the Jingtoushan site, China. Wood Sci. Technol. 2023, 57, 537–556. [Google Scholar] [CrossRef]
- Bucci, P.; Coppotelli, B.; Morelli, I.; Zaritzky, N.; Caravelli, A. Micronutrients and COD/N ratio as factors influencing granular size and SND in aerobic granular sequencing batch reactors operated at low organic loading. J. Water Process. Eng. 2022, 46, 102625. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Type | Sample Time | Coverage | Chao1 | Observed | Shannon | Simpson |
|---|---|---|---|---|---|---|
| Bacteria | 3 days | 0.999 | 164.28 | 151.34 | 2.074 | 0.685 |
| 15 days | 0.998 | 392.01 | 352.52 | 3.523 | 0.811 | |
| 24 days | 0.997 | 560.31 | 500.02 | 4.476 | 0.875 | |
| Fungi | 3 days | 0.999 | 17.06 | 16.88 | 0.512 | 0.163 |
| 15 days | 0.999 | 16.83 | 16.8 | 0.923 | 0.33 | |
| 24 days | 0.999 | 17.32 | 17.22 | 1.022 | 0.322 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ji, J.; Escobar, M.; Cui, S.; Zhang, W.; Bao, C.; Su, X.; Wang, G.; Zhang, S.; Chen, H.; Chen, G. Isolation and Characterization of a Low-Temperature, Cellulose-Degrading Microbial Consortium from Northeastern China. Microorganisms 2024, 12, 1059. https://doi.org/10.3390/microorganisms12061059
Ji J, Escobar M, Cui S, Zhang W, Bao C, Su X, Wang G, Zhang S, Chen H, Chen G. Isolation and Characterization of a Low-Temperature, Cellulose-Degrading Microbial Consortium from Northeastern China. Microorganisms. 2024; 12(6):1059. https://doi.org/10.3390/microorganisms12061059
Chicago/Turabian StyleJi, Jiaoyang, Maia Escobar, Shijia Cui, Wei Zhang, Changjie Bao, Xuhan Su, Gang Wang, Sitong Zhang, Huan Chen, and Guang Chen. 2024. "Isolation and Characterization of a Low-Temperature, Cellulose-Degrading Microbial Consortium from Northeastern China" Microorganisms 12, no. 6: 1059. https://doi.org/10.3390/microorganisms12061059
APA StyleJi, J., Escobar, M., Cui, S., Zhang, W., Bao, C., Su, X., Wang, G., Zhang, S., Chen, H., & Chen, G. (2024). Isolation and Characterization of a Low-Temperature, Cellulose-Degrading Microbial Consortium from Northeastern China. Microorganisms, 12(6), 1059. https://doi.org/10.3390/microorganisms12061059