
Neutrophils versus Protozoan Parasites: Plasmodium, Trichomonas, Leishmania, Trypanosoma, and Entameoba

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
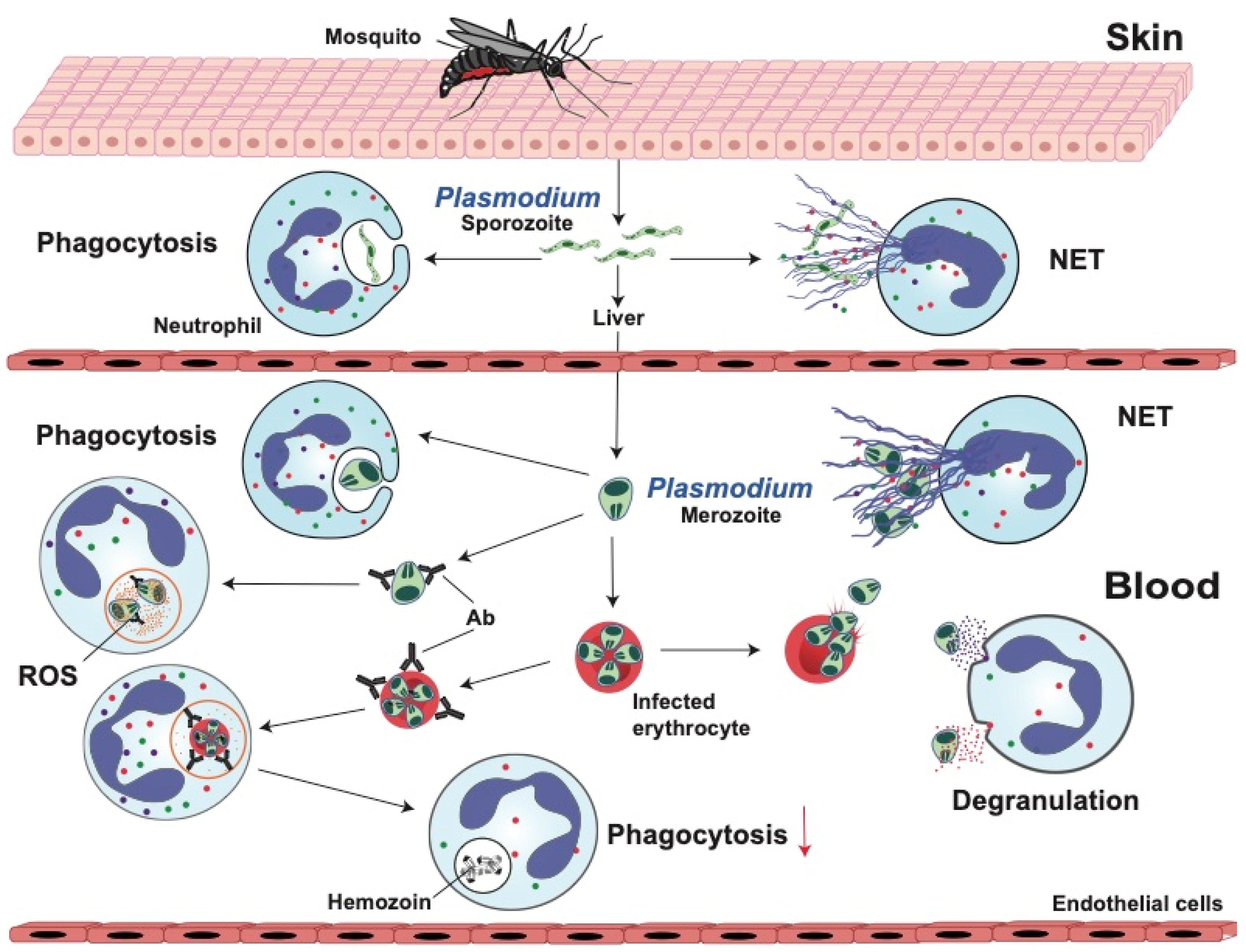
2. Malaria
2.1. Neutrophil Response Anti-Plasmodium
2.1.1. Phagocytosis
2.1.2. Reactive Oxygen Species (ROS)
2.1.3. Degranulation
2.1.4. Neutrophil Extracellular Traps (NETs)
2.2. Anti-Neutrophil Responses in Malaria
2.3. Invasive Bacterial Disease
3. Trichomoniasis
3.1. Neutrophil Response Anti T. vaginalis Parasites
3.1.1. Neutrophil Migration towards T. vaginalis Parasites
3.1.2. Neutrophils Kill T. vaginalis by Trogocytosis
3.2. Neutrophil Evasion Tactics of T. vaginalis
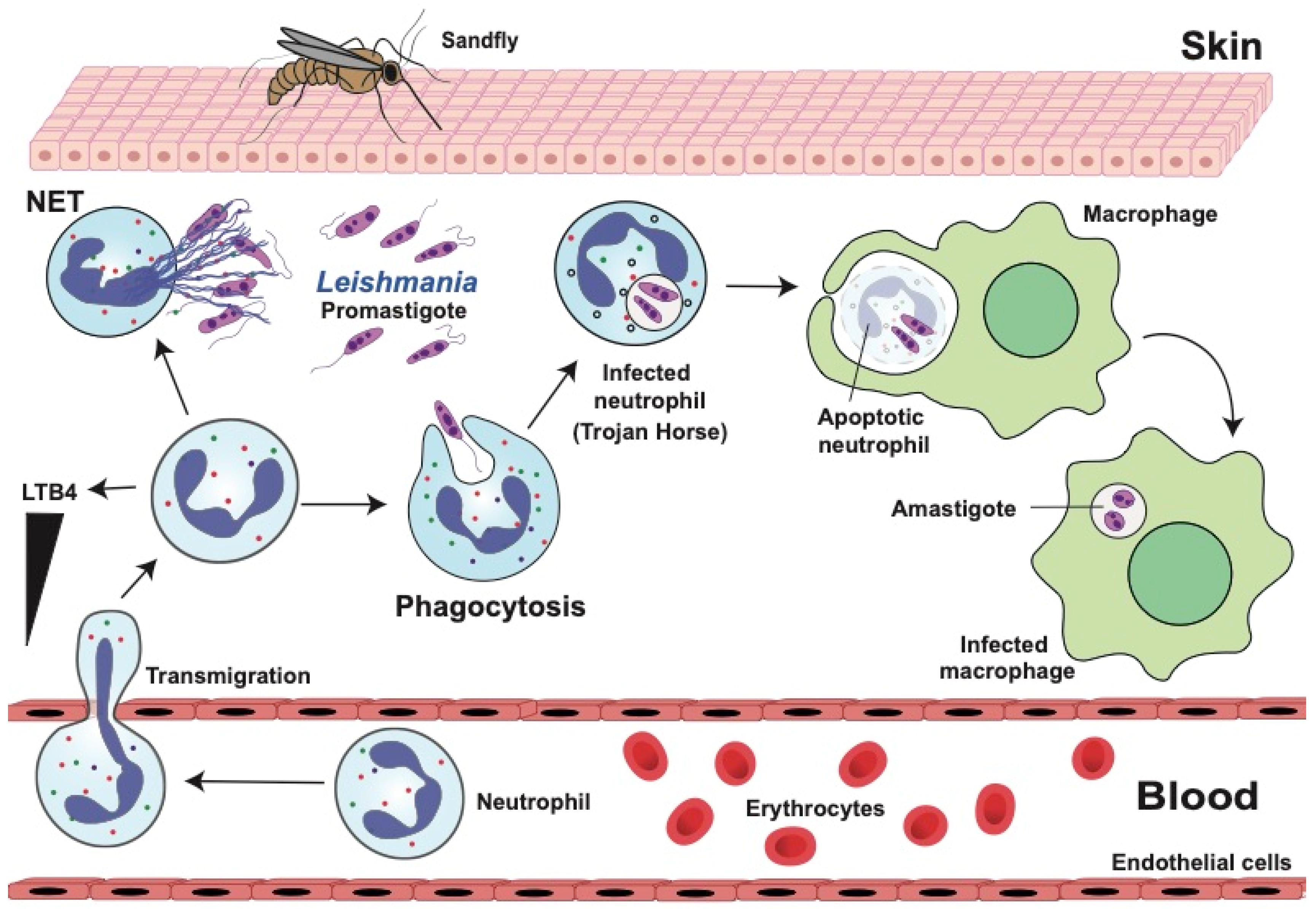
4. Leishmaniases
4.1. Dual Role of Neutrophils in Leishmaniasis
4.2. Neutrophil Response Anti-Leishmania
4.2.1. Phagocytosis and Degranulation
4.2.2. Neutrophil Extracellular Traps (NETs)
5. Chagas Disease
5.1. Innate Immune Response against T. cruzi
5.2. Dual Role of Neutrophils in Chagas Disease
5.3. Neutrophil Functions against T. cruzi Parasites
5.3.1. Phagocytosis
5.3.2. Reactive Oxygen Species (ROS)
5.3.3. Neutrophil Extracellular Traps (NETs)
6. Amoebiasis
6.1. Role of Neutrophils in Amoebiasis
6.2. Neutrophil Functions against E. histolytica
6.2.1. Reactive Oxygen Species (ROS)
6.2.2. Neutrophil Extracellular Traps (NETs)
7. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Flajnik, M.F.; Singh, N.J.; Holland, S.M. Paul’s Fundamental Immunology, 8th ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2023; p. 1748. ISBN -13: 978-1-975142-51-3. [Google Scholar]
- Aroca-Crevillén, A.; Vicanolo, T.; Ovadia, S.; Hidalgo, A. Neutrophils in physiology and pathology. Annu. Rev. Pathol. 2024, 19, 227–259. [Google Scholar] [CrossRef]
- Fine, N.; Tasevski, N.; McCulloch, C.A.; Tenenbaum, H.C.; Glogauer, M. The neutrophil: Constant defender and first responder. Front. Immunol. 2020, 11, 571085. [Google Scholar] [CrossRef]
- Petri, B.; Sanz, M.J. Neutrophil chemotaxis. Cell Tissue Res. 2018, 371, 425–436. [Google Scholar] [CrossRef]
- Rajarathnam, K.; Schnoor, M.; Richardson, R.M.; Rajagopal, S. How do chemokines navigate neutrophils to the target site: Dissecting the structural mechanisms and signaling pathways. Cell Signal. 2019, 54, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Cambier, S.; Gouwy, M.; Proost, P. The chemokines CXCL8 and CXCL12: Molecular and functional properties, role in disease and efforts towards pharmacological intervention. Cell. Mol. Immunol. 2023, 20, 217–251. [Google Scholar] [CrossRef] [PubMed]
- Nauseef, W.M.; Borregaard, N. Neutrophils at work. Nat. Immunol. 2014, 15, 602–611. [Google Scholar] [CrossRef]
- Lacy, P.; Eitzen, G. Control of granule exocytosis in neutrophils. Front. Biosci. 2008, 13, 5559–5570. [Google Scholar] [CrossRef]
- El-Benna, J.; Hurtado-Nedelec, M.; Marzaioli, V.; Marie, J.C.; Gougerot-Pocidalo, M.A.; Dang, P.M. Priming of the neutrophil respiratory burst: Role in host defense and inflammation. Immunol. Rev. 2016, 273, 180–193. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, G.T.; Green, E.R.; Mecsas, J. Neutrophils to the ROScue: Mechanisms of NADPH oxidase activation and bacterial resistance. Front. Cell. Infect. Microbiol. 2017, 7, 373. [Google Scholar] [CrossRef]
- Uribe-Querol, E.; Rosales, C. Phagocytosis: Our current understading of a universal biological process. Front. Immunol. 2020, 11, 1066. [Google Scholar] [CrossRef]
- Hidalgo, A.; Libby, P.; Soehnlein, O.; Aramburu, I.V.; Papayannopoulos, V.; Silvestre-Roig, C. Neutrophil extracellular traps: From physiology to pathology. Cardiovasc. Res. 2022, 118, 2737–2753. [Google Scholar] [CrossRef] [PubMed]
- Uribe-Querol, E.; Rosales, C. The multiple roles of trogocytosis in immunity, the Nervous System, and development. BioMed Res. Int. 2021, 2021, 601565. [Google Scholar] [CrossRef] [PubMed]
- Furze, R.C.; Rankin, S.M. Neutrophil mobilization and clearance in the bone marrow. Immunology 2008, 125, 281–288. [Google Scholar] [CrossRef]
- Greenlee-Wacker, M.C. Clearance of apoptotic neutrophils and resolution of inflammation. Immunol. Rev. 2016, 273, 357–370. [Google Scholar] [CrossRef] [PubMed]
- Borregaard, N. Neutrophils, from marrow to microbes. Immunity 2010, 33, 657–670. [Google Scholar] [CrossRef] [PubMed]
- Faurschou, M.; Borregaard, N. Neutrophil granules and secretory vesicles in inflammation. Microbes Infect. 2003, 5, 1317–1327. [Google Scholar] [CrossRef]
- Rosales, C. Neutrophils at the crossroads of innate and adaptive immunity. J. Leukoc. Biol. 2020, 108, 377–396. [Google Scholar] [CrossRef]
- Rosales, C.; Uribe-Querol, E. Phagocytosis: A fundamental process in immunity. BioMed Res. Int. 2017, 2017, 9042851. [Google Scholar] [CrossRef]
- Levin, R.; Grinstein, S.; Canton, J. The life cycle of phagosomes: Formation, maturation, and resolution. Immunol. Rev. 2016, 273, 156–179. [Google Scholar] [CrossRef] [PubMed]
- Matlung, H.L.; Babes, L.; Zhao, X.W.; van Houdt, M.; Treffers, L.W.; van Rees, D.J.; Franke, K.; Schornagel, K.; Verkuijlen, P.; Janssen, H.; et al. Neutrophils kill antibody-opsonized cancer cells by trogoptosis. Cell Rep. 2018, 23, 3946–3959. [Google Scholar] [CrossRef]
- Olivera-Valle, I.; Latorre, M.C.; Calvo, M.; Gaspar, B.; Gómez-Oro, C.; Collazos, A.; Breton, A.; Caballero-Campo, P.; Ardoy, M.; Asensio, F.; et al. Vaginal neutrophils eliminate sperm by trogocytosis. Hum. Reprod. 2020, 35, 2567–2578. [Google Scholar] [CrossRef] [PubMed]
- Ng, L.G.; Ostuni, R.; Hidalgo, A. Heterogeneity of neutrophils. Nat. Rev. Immunol. 2019, 19, 255–265. [Google Scholar] [CrossRef]
- Rosales, C. Neutrophil: A cell with many roles in inflammation or several cell types? Front. Physiol.-Integr. Physiol. 2018, 9, 113. [Google Scholar] [CrossRef]
- Naussef, W.M. How human neutrophils kill and degrade microbes. An integrated view. Immunol. Rev. 2007, 219, 88–102. [Google Scholar] [CrossRef] [PubMed]
- Herricks, J.R.; Hotez, P.J.; Wanga, V.; Coffeng, L.E.; Haagsma, J.A.; Basáñez, M.G.; Buckle, G.; Budke, C.M.; Carabin, H.; Fèvre, E.M.; et al. The global burden of disease study 2013: What does it mean for the NTDs? PLoS Neglected Trop. Dis. 2017, 11, e0005424. [Google Scholar] [CrossRef]
- Lozano, R.; Naghavi, M.; Foreman, K.; Lim, S.; Shibuya, K.; Aboyans, V.; Abraham, J.; Adair, T.; Aggarwal, R.; Ahn, S.Y.; et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: A systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012, 380, 2095–2128. [Google Scholar] [CrossRef]
- Bhakta, S.B.; Moran, J.A.; Mercer, F. Neutrophil interactions with the sexually transmitted parasite Trichomonas vaginalis: Implications for immunity and pathogenesis. Open Biol. 2020, 10, 200192. [Google Scholar] [CrossRef]
- Carlsen, E.D.; Liang, Y.; Shelite, T.R.; Walker, D.H.; Melby, P.C.; Soong, L. Permissive and protective roles for neutrophils in leishmaniasis. Clin. Exp. Immunol. 2015, 182, 109–118. [Google Scholar] [CrossRef] [PubMed]
- de Andrade, M.F.; de Almeida, V.D.; de Souza, L.M.S.; Paiva, D.C.C.; Andrade, C.M.; de Medeiros Fernandes, T.A.A. Involvement of neutrophils in Chagas disease pathology. Parasite Immunol. 2018, 40, e12593. [Google Scholar] [CrossRef]
- Mohammed, A.O.; Elghazali, G.; Mohammed, H.B.; Elbashir, M.I.; Xu, S.; Berzins, K.; Venge, P. Human neutrophil lipocalin: A specific marker for neutrophil activation in severe Plasmodium falciparum malaria. Acta Trop. 2003, 87, 279–285. [Google Scholar] [CrossRef]
- Rosales, C. Neutrophils vs. amoebas: Immunity against the protozoan parasite Entamoeba histolytica. J. Leukoc. Biol. 2021, 110, 1241–1252. [Google Scholar] [CrossRef] [PubMed]
- Schofield, L.; Grau, G.E. Immunological processes in malaria pathogenesis. Nat. Rev. Immunol. 2005, 5, 722–735. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. World Malaria Report 2021; Global Malaria Programme; WHO: Geneva, Switzerland, 2021; p. 322. ISBN 9789240040496.
- Balaji, S.N.; Deshmukh, R.; Trivedi, V. Severe malaria: Biology, clinical manifestation, pathogenesis and consequences. J. Vector Borne Dis. 2020, 57, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Laishram, D.D.; Sutton, P.L.; Nanda, N.; Sharma, V.L.; Sobti, R.C.; Carlton, J.M.; Joshi, H. The complexities of malaria disease manifestations with a focus on asymptomatic malaria. Malar. J. 2012, 11, 29. [Google Scholar] [CrossRef] [PubMed]
- Wassmer, S.C.; Grau, G.E. Severe malaria: What’s new on the pathogenesis front? Int. J. Parasitol. 2017, 47, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Aly, A.S.; Vaughan, A.M.; Kappe, S.H. Malaria parasite development in the mosquito and infection of the mammalian host. Annu. Rev. Microbiol. 2009, 63, 195–221. [Google Scholar] [CrossRef] [PubMed]
- Tannous, S.; Ghanem, E. A bite to fight: Front-line innate immune defenses against malaria parasites. Pathog. Glob. Health 2018, 112, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Amulic, B.; Moxon, C.A.; Cunnington, A.J. A more granular view of neutrophils in malaria. Trends Parasitol. 2020, 36, 501–503. [Google Scholar] [CrossRef] [PubMed]
- Babatunde, K.A.; Adenuga, O.F. Neutrophils in malaria: A double-edged sword role. Front. Immunol. 2022, 13, 922377. [Google Scholar] [CrossRef]
- Olliaro, P.; Djimdé, A.; Dorsey, G.; Karema, C.; Mårtensson, A.; Ndiaye, J.L.; Sirima, S.B.; Vaillant, M.; Zwang, J. Hematologic parameters in pediatric uncomplicated Plasmodium falciparum malaria in sub-Saharan Africa. Am. J. Trop. Med. Hyg. 2011, 85, 619–625. [Google Scholar] [CrossRef]
- Berens-Riha, N.; Kroidl, I.; Schunk, M.; Alberer, M.; Beissner, M.; Pritsch, M.; Kroidl, A.; Fröschl, G.; Hanus, I.; Bretzel, G.; et al. Evidence for significant influence of host immunity on changes in differential blood count during malaria. Malar. J. 2014, 13, 155. [Google Scholar] [CrossRef] [PubMed]
- Trubowitz, S.; Masek, B. Plasmodium falciparum: Phagocytosis by polymorphonuclear leukocytes. Science 1968, 162, 273–274. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Chakrabarti, C. Schizonts, merozoites, and phagocytosis in falciparum malaria. Ann. Clin. Lab. Sci. 1985, 15, 465–469. [Google Scholar] [PubMed]
- Healer, J.; Graszynski, A.; Riley, E. Phagocytosis does not play a major role in naturally acquired transmission-blocking immunity to Plasmodium falciparum malaria. Infect. Immun. 1999, 67, 2334–2339. [Google Scholar] [CrossRef] [PubMed]
- Celada, A.; Cruchaud, A.; Perrin, L.H. Phagocytosis of Plasmodium falciparum-parasitized erythrocytes by human polymorphonuclear leukocytes. J. Parasitol. 1983, 69, 49–53. [Google Scholar] [CrossRef] [PubMed]
- Celada, A.; Cruchaud, A.; Perrin, L.H. Independence of complement on in vitro immune phagocytosis of Plasmodium falciparum parasitised erythrocytes by human monocytes and polymorphonuclear leukocytes. Int. Arch. Allergy Appl. Immunol. 1984, 73, 363–366. [Google Scholar] [CrossRef] [PubMed]
- Boyle, M.J.; Reiling, L.; Feng, G.; Langer, C.; Osier, F.H.; Aspeling-Jones, H.; Cheng, Y.S.; Stubbs, J.; Tetteh, K.K.; Conway, D.J.; et al. Human antibodies fix complement to inhibit Plasmodium falciparum invasion of erythrocytes and are associated with protection against malaria. Immunity 2015, 42, 580–590. [Google Scholar] [CrossRef] [PubMed]
- Chua, C.L.; Robinson, L.J.; Baiwog, F.; Stanisic, D.I.; Hamilton, J.A.; Brown, G.V.; Rogerson, S.J.; Boeuf, P. High numbers of circulating pigmented polymorphonuclear neutrophils as a prognostic marker for decreased birth weight during malaria in pregnancy. Int. J. Parasitol. 2015, 45, 107–111. [Google Scholar] [CrossRef] [PubMed]
- Amodu, O.K.; Adeyemo, A.A.; Olumese, P.E.; Gbadegesin, R.A. Intraleucocytic malaria pigment and clinical severity of malaria in children. Trans. R. Soc. Trop. Med. Hyg. 1998, 92, 54–56. [Google Scholar] [CrossRef] [PubMed]
- Lufele, E.; Manning, L.; Lorry, L.; Warrel, J.; Aipit, S.; Robinson, L.J.; Laman, M. The association of intraleucocytic malaria pigment and disease severity in Papua New Guinean children with severe P. falciparum malaria. Trans. R. Soc. Trop. Med. Hyg. 2023, 117, 797–803. [Google Scholar] [CrossRef]
- Srinamon, K.; Watson, J.A.; Silamut, K.; Intharabut, B.; Phu, N.H.; Diep, P.T.; Lyke, K.E.; Fanello, C.; von Seidlein, L.; Chotivanich, K.; et al. The prognostic and diagnostic value of intraleukocytic malaria pigment in patients with severe falciparum malaria. Nat. Commun. 2022, 13, 6882. [Google Scholar] [CrossRef] [PubMed]
- Dasari, P.; Reiss, K.; Lingelbach, K.; Baumeister, S.; Lucius, R.; Udomsangpetch, R.; Bhakdi, S.C.; Bhakdi, S. Digestive vacuoles of Plasmodium falciparum are selectively phagocytosed by and impair killing function of polymorphonuclear leukocytes. Blood 2011, 118, 4946–4956. [Google Scholar] [CrossRef] [PubMed]
- Tyberghein, A.; Deroost, K.; Schwarzer, E.; Arese, P.; Van den Steen, P.E. Immunopathological effects of malaria pigment or hemozoin and other crystals. BioFactors 2014, 40, 59–78. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.; Smalley, M.E. Inhibition of the in vitro growth of Plasmodium falciparum by human polymorphonuclear neutrophil leucocytes. Clin. Exp. Immunol. 1981, 46, 106–109. [Google Scholar] [PubMed]
- Greve, B.; Lehman, L.G.; Lell, B.; Luckner, D.; Schmidt-Ott, R.; Kremsner, P.G. High oxygen radical production is associated with fast parasite clearance in children with Plasmodium falciparum malaria. J. Infect. Dis. 1999, 179, 1584–1586. [Google Scholar] [CrossRef]
- Joos, C.; Marrama, L.; Polson, H.E.; Corre, S.; Diatta, A.M.; Diouf, B.; Trape, J.F.; Tall, A.; Longacre, S.; Perraut, R. Clinical protection from falciparum malaria correlates with neutrophil respiratory bursts induced by merozoites opsonized with human serum antibodies. PLoS ONE 2010, 5, e9871. [Google Scholar] [CrossRef] [PubMed]
- Kharazmi, A.; Jepsen, S.; Andersen, B.J. Generation of reactive oxygen radicals by human phagocytic cells activated by Plasmodium falciparum. Scand. J. Immunol. 1987, 25, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Salmon, D.; Vilde, J.L.; Andrieu, B.; Simonovic, R.; Lebras, J. Role of immune serum and complement in stimulation of the metabolic burst of human neutrophils by Plasmodium falciparum. Infect. Immun. 1986, 51, 801–806. [Google Scholar] [CrossRef] [PubMed]
- Georgiadou, A.; Lee, H.J.; Walther, M.; van Beek, A.E.; Fitriani, F.; Wouters, D.; Kuijpers, T.W.; Nwakanma, D.; D’Alessandro, U.; Riley, E.M.; et al. Modelling pathogen load dynamics to elucidate mechanistic determinants of host-Plasmodium falciparum interactions. Nat. Microbiol. 2019, 4, 1592–1602. [Google Scholar] [CrossRef]
- Rodrigues, D.A.S.; Prestes, E.B.; Gama, A.M.S.; Silva, L.S.; Pinheiro, A.A.S.; Ribeiro, J.M.C.; Campos, R.M.P.; Pimentel-Coelho, P.M.; De Souza, H.S.; Dicko, A.; et al. CXCR4 and MIF are required for neutrophil extracellular trap release triggered by Plasmodium-infected erythrocytes. PLoS Pathog. 2020, 16, e1008230. [Google Scholar] [CrossRef]
- Gallego-Delgado, J.; Ty, M.; Orengo, J.M.; van de Hoef, D.; Rodriguez, A. A surprising role for uric acid: The inflammatory malaria response. Curr. Rheumatol. Rep. 2014, 16, 401. [Google Scholar] [CrossRef] [PubMed]
- Schorn, C.; Janko, C.; Krenn, V.; Zhao, Y.; Munoz, L.E.; Schett, G.; Herrmann, M. Bonding the foe—NETting neutrophils immobilize the pro-inflammatory monosodium urate crystals. Front. Immunol. 2012, 3, 376. [Google Scholar] [CrossRef] [PubMed]
- Knackstedt, S.L.; Georgiadou, A.; Apel, F.; Abu-Abed, U.; Moxon, C.A.; Cunnington, A.J.; Raupach, B.; Cunningham, D.; Langhorne, J.; Krüger, R.; et al. Neutrophil extracellular traps drive inflammatory pathogenesis in malaria. Sci. Immunol. 2019, 4, eaaw0336. [Google Scholar] [CrossRef] [PubMed]
- Sercundes, M.K.; Ortolan, L.S.; Debone, D.; Soeiro-Pereira, P.V.; Gomes, E.; Aitken, E.H.; Condino-Neto, A.; Russo, M.; D’ Império Lima, M.R.; Alvarez, J.M.; et al. Targeting neutrophils to prevent malaria-associated acute lung injury/acute respiratory distress syndrome in mice. PLoS Pathog. 2016, 12, e1006054. [Google Scholar] [CrossRef] [PubMed]
- Waisberg, M.; Molina-Cruz, A.; Mizurini, D.M.; Gera, N.; Sousa, B.C.; Ma, D.; Leal, A.C.; Gomes, T.; Kotsyfakis, M.; Ribeiro, J.M.; et al. Plasmodium falciparum infection induces expression of a mosquito salivary protein (Agaphelin) that targets neutrophil function and inhibits thrombosis without impairing hemostasis. PLoS Pathog. 2014, 10, e1004338. [Google Scholar] [CrossRef] [PubMed]
- Assumpção, T.C.F.; Ma, D.; Schwarz, A.; Reiter, K.; Santana, J.M.; Andersen, J.F.; Ribeiro, J.M.C.; Nardone, G.; Yu, L.L.; Francischetti, I.M.B. Salivary antigen-5/CAP family members are Cu2+-dependent antioxidant enzymes that scavenge O₂₋. and inhibit collagen-induced platelet aggregation and neutrophil oxidative burst. J. Biol. Chem. 2013, 288, 14341–14361. [Google Scholar] [CrossRef] [PubMed]
- Aitken, E.H.; Alemu, A.; Rogerson, S.J. Neutrophils and malaria. Front. Immunol. 2018, 9, 3005. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, C.; Demarta-Gatsi, C.; Porcherie, A.; Brega, S.; Thiberge, S.; Ronce, K.; Smith, L.; Peronet, R.; Amino, R.; Ménard, R.; et al. Plasmodium berghei histamine-releasing factor favours liver-stage development via inhibition of IL-6 production and associates with a severe outcome of disease. Cell. Microbiol. 2015, 17, 542–558. [Google Scholar] [CrossRef]
- Waisberg, M.; Cerqueira, G.C.; Yager, S.B.; Francischetti, I.M.; Lu, J.; Gera, N.; Srinivasan, P.; Miura, K.; Rada, B.; Lukszo, J.; et al. Plasmodium falciparum merozoite surface protein 1 blocks the proinflammatory protein S100P. Proc. Natl. Acad. Sci. USA 2012, 109, 5429–5434. [Google Scholar] [CrossRef]
- Church, J.; Maitland, K. Invasive bacterial co-infection in African children with Plasmodium falciparum malaria: A systematic review. BMC Med. 2014, 12, 31. [Google Scholar] [CrossRef]
- Angus, D.C.; van der Poll, T. Severe sepsis and septic shock. N. Engl. J. Med. 2013, 369, 840–851. [Google Scholar] [CrossRef]
- Singer, M.; Deutschman, C.S.; Seymour, C.W.; Shankar-Hari, M.; Annane, D.; Bauer, M.; Bellomo, R.; Bernard, G.R.; Chiche, J.D.; Coopersmith, C.M.; et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 2016, 315, 801–810. [Google Scholar] [CrossRef]
- Klevens, R.M.; Morrison, M.A.; Nadle, J.; Petit, S.; Gershman, K.; Ray, S.; Harrison, L.H.; Lynfield, R.; Dumyati, G.; Townes, J.M.; et al. Invasive methicillin-resistant Staphylococcus aureus infections in the United States. JAMA 2007, 298, 1763–1771. [Google Scholar] [CrossRef]
- Tong, S.Y.; Davis, J.S.; Eichenberger, E.; Holland, T.L.; Fowler, V.G., Jr. Staphylococcus aureus infections: Epidemiology, pathophysiology, clinical manifestations, and management. Clin. Microbiol. Rev. 2015, 28, 603–661. [Google Scholar] [CrossRef]
- Reddy, E.A.; Shaw, A.V.; Crump, J.A. Community-acquired bloodstream infections in Africa: A systematic review and meta-analysis. Lancet. Infect. Dis. 2010, 10, 417–432. [Google Scholar] [CrossRef]
- Takem, E.N.; Roca, A.; Cunnington, A. The association between malaria and non-typhoid Salmonella bacteraemia in children in sub-Saharan Africa: A literature review. Malar. J. 2014, 13, 400. [Google Scholar] [CrossRef]
- Mtove, G.; Amos, B.; Nadjm, B.; Hendriksen, I.C.; Dondorp, A.M.; Mwambuli, A.; Kim, D.R.; Ochiai, R.L.; Clemens, J.D.; von Seidlein, L.; et al. Decreasing incidence of severe malaria and community-acquired bacteraemia among hospitalized children in Muheza, north-eastern Tanzania, 2006–2010. Malar. J. 2011, 10, 320. [Google Scholar] [CrossRef]
- Muthumbi, E.; Morpeth, S.C.; Ooko, M.; Mwanzu, A.; Mwarumba, S.; Mturi, N.; Etyang, A.O.; Berkley, J.A.; Williams, T.N.; Kariuki, S.; et al. Invasive Salmonellosis in Kilifi, Kenya. Clin. Infect. Dis. 2015, 61, S290–S301. [Google Scholar] [CrossRef]
- Cunnington, A.J.; de Souza, J.B.; Walther, M.; Riley, E.M. Malaria impairs resistance to Salmonella through heme- and heme oxygenase-dependent dysfunctional granulocyte mobilization. Nat. Med. 2011, 18, 120–127. [Google Scholar] [CrossRef]
- Bannister, L.H.; Hopkins, J.M.; Fowler, R.E.; Krishna, S.; Mitchell, G.H. A brief illustrated guide to the ultrastructure of Plasmodium falciparum asexual blood stages. Parasitol. Today 2000, 16, 427–433. [Google Scholar] [CrossRef]
- Price, R.N.; Simpson, J.A.; Nosten, F.; Luxemburger, C.; Hkirjaroen, L.; ter Kuile, F.; Chongsuphajaisiddhi, T.; White, N.J. Factors contributing to anemia after uncomplicated falciparum malaria. Am. J. Trop. Med. Hyg. 2001, 65, 614–622. [Google Scholar] [CrossRef]
- Cunnington, A.J.; Njie, M.; Correa, S.; Takem, E.N.; Riley, E.M.; Walther, M. Prolonged neutrophil dysfunction after Plasmodium falciparum malaria is related to hemolysis and heme oxygenase-1 induction. J. Immunol. 2012, 189, 5336–5346. [Google Scholar] [CrossRef]
- Mooney, J.P.; Barry, A.; Gonçalves, B.P.; Tiono, A.B.; Awandu, S.S.; Grignard, L.; Drakeley, C.J.; Bottomley, C.; Bousema, T.; Riley, E.M. Haemolysis and haem oxygenase-1 induction during persistent “asymptomatic” malaria infection in Burkinabé children. Malar. J. 2018, 17, 253. [Google Scholar] [CrossRef]
- Mooney, J.P.; Galloway, L.J.; Riley, E.M. Malaria, anemia, and invasive bacterial disease: A neutrophil problem? J. Leukoc. Biol. 2019, 105, 645–655. [Google Scholar] [CrossRef]
- Martins, R.; Maier, J.; Gorki, A.D.; Huber, K.V.; Sharif, O.; Starkl, P.; Saluzzo, S.; Quattrone, F.; Gawish, R.; Lakovits, K.; et al. Heme drives hemolysis-induced susceptibility to infection via disruption of phagocyte functions. Nat. Immunol. 2016, 17, 1361–1372. [Google Scholar] [CrossRef]
- Mooney, J.P.; Butler, B.P.; Lokken, K.L.; Xavier, M.N.; Chau, J.Y.; Schaltenberg, N.; Dandekar, S.; George, M.D.; Santos, R.L.; Luckhart, S.; et al. The mucosal inflammatory response to non-typhoidal Salmonella in the intestine is blunted by IL-10 during concurrent malaria parasite infection. Mucosal Immunol. 2014, 7, 1302–1311. [Google Scholar] [CrossRef]
- Lokken, K.L.; Mooney, J.P.; Butler, B.P.; Xavier, M.N.; Chau, J.Y.; Schaltenberg, N.; Begum, R.H.; Müller, W.; Luckhart, S.; Tsolis, R.M. Malaria parasite infection compromises control of concurrent systemic non-typhoidal Salmonella infection via IL-10-mediated alteration of myeloid cell function. PLoS Pathog. 2014, 10, e1004049. [Google Scholar] [CrossRef]
- Konrad, F.M.; Knausberg, U.; Höne, R.; Ngamsri, K.C.; Reutershan, J. Tissue heme oxygenase-1 exerts anti-inflammatory effects on LPS-induced pulmonary inflammation. Mucosal Immunol. 2016, 9, 98–111. [Google Scholar] [CrossRef]
- Ekregbesi, P.; Shankar-Hari, M.; Bottomley, C.; Riley, E.M.; Mooney, J.P. Relationship between anaemia, haemolysis, inflammation and haem oxygenase-1 at admission with sepsis: A pilot study. Sci. Rep. 2018, 8, 11198. [Google Scholar] [CrossRef]
- Kasten, K.R.; Muenzer, J.T.; Caldwell, C.C. Neutrophils are significant producers of IL-10 during sepsis. Biochem. Biophys. Res. Commun. 2010, 393, 28–31. [Google Scholar] [CrossRef]
- Peyron, F.; Burdin, N.; Ringwald, P.; Vuillez, J.P.; Rousset, F.; Banchereau, J. High levels of circulating IL-10 in human malaria. Clin. Exp. Immunol. 1994, 95, 300–303. [Google Scholar] [CrossRef]
- Wenisch, C.; Parschalk, B.; Narzt, E.; Looareesuwan, S.; Graninger, W. Elevated serum levels of IL-10 and IFN-gamma in patients with acute Plasmodium falciparum malaria. Clin. Immunol. Immunopathol. 1995, 74, 115–117. [Google Scholar] [CrossRef]
- Bazzoni, F.; Tamassia, N.; Rossato, M.; Cassatella, M.A. Understanding the molecular mechanisms of the multifaceted IL-10-mediated anti-inflammatory response: Lessons from neutrophils. Eur. J. Immunol. 2010, 40, 2360–2368. [Google Scholar] [CrossRef]
- Kulkarni, U.; Karsten, C.M.; Kohler, T.; Hammerschmidt, S.; Bommert, K.; Tiburzy, B.; Meng, L.; Thieme, L.; Recke, A.; Ludwig, R.J.; et al. IL-10 mediates plasmacytosis-associated immunodeficiency by inhibiting complement-mediated neutrophil migration. J. Allergy Clin. Immunol. 2016, 137, 1487–1497.e6. [Google Scholar] [CrossRef]
- Hirt, R.P.; Sherrard, J. Trichomonas vaginalis origins, molecular pathobiology and clinical considerations. Curr. Opin. Infect. Dis. 2015, 28, 72–79. [Google Scholar] [CrossRef]
- Mercer, F.; Johnson, P.J. Trichomonas vaginalis: Pathogenesis, symbiont interactions, and host cell immune responses. Trends Parasitol. 2018, 34, 683–693. [Google Scholar] [CrossRef]
- Patel, E.U.; Gaydos, C.A.; Packman, Z.R.; Quinn, T.C.; Tobian, A.A.R. Prevalence and correlates of Trichomonas vaginalis infection among men and women in the United States. Clin. Infect. Dis. 2018, 67, 211–217. [Google Scholar] [CrossRef]
- World Health Organization. Trichomoniasis. Available online: https://www.who.int/news-room/fact-sheets/detail/trichomoniasis (accessed on 29 March 2024).
- Centers for Disease Control and Prevention. Trichomoniasis—CDC Fact Sheet. Available online: https://www.cdc.gov/std/trichomonas/stdfact-trichomoniasis.htm (accessed on 29 March 2024).
- Centers for Disease Control and Prevention. Sexually Transmitted Infections Prevalence, Incidence, and Cost Estimates in the United States. Available online: https://www.cdc.gov/std/statistics/prevalence-2020-at-a-glance.htm (accessed on 12 February 2024).
- Mtshali, A.; Ngcapu, S.; Govender, K.; Sturm, A.W.; Moodley, P.; Joubert, B.C. In vitro effect of 5-nitroimidazole drugs against Trichomonas vaginalis clinical isolates. Microbiol. Spectr. 2022, 10, e0091222. [Google Scholar] [CrossRef]
- Lustig, G.; Ryan, C.M.; Secor, W.E.; Johnson, P.J. Trichomonas vaginalis contact-dependent cytolysis of epithelial cells. Infect. Immun. 2013, 81, 1411–1419. [Google Scholar] [CrossRef]
- Cobo, E.R.; Eckmann, L.; Corbeil, L.B. Murine models of vaginal trichomonad infections. Am. J. Trop. Med. Hyg. 2011, 85, 667–673. [Google Scholar] [CrossRef]
- Rein, M.F.; Sullivan, J.A.; Mandell, G.L. Trichomonacidal activity of human polymorphonuclear neutrophils: Killing by disruption and fragmentation. J. Infect. Dis. 1980, 142, 575–585. [Google Scholar] [CrossRef]
- Styrt, B.; Sugarman, B.; Mummaw, N.; White, J.C. Chemorepulsion of trichomonads by products of neutrophil oxidative metabolism. J. Infect. Dis. 1991, 163, 176–179. [Google Scholar] [CrossRef]
- Nohgawa, M.; Sasada, M.; Maeda, A.; Asagoe, K.; Harakawa, N.; Takano, K.; Yamamoto, K.; Okuma, M. Leukotriene B4-activated human endothelial cells promote transendothelial neutrophil migration. J. Leukoc. Biol. 1997, 62, 203–209. [Google Scholar] [CrossRef][Green Version]
- Shaio, M.F.; Lin, P.R.; Lee, C.S.; Hou, S.C.; Tang, P.; Yang, K.D. A novel neutrophil-activating factor released by Trichomonas vaginalis. Infect. Immun. 1992, 60, 4475–4482. [Google Scholar] [CrossRef]
- Shaio, M.F.; Lin, P.R. Influence of humoral immunity on leukotriene B4 production by neutrophils in response to Trichomonas vaginalis stimulation. Parasite Immunol. 1995, 17, 127–133. [Google Scholar] [CrossRef]
- Irimia, D. Neutrophil swarms are more than the accumulation of cells. Microbiol. Insights 2020, 13, 1178636120978272. [Google Scholar] [CrossRef]
- Demirezen, S.; Safi, Z.; Beksaç, S. The interaction of trichomonas vaginalis with epithelial cells, polymorphonuclear leucocytes and erythrocytes on vaginal smears: Light microscopic observation. Cytopathology 2000, 11, 326–332. [Google Scholar] [CrossRef]
- Mercer, F.; Ng, S.H.; Brown, T.M.; Boatman, G.; Johnson, P.J. Neutrophils kill the parasite Trichomonas vaginalis using trogocytosis. PLoS Biol. 2018, 16, e2003885. [Google Scholar] [CrossRef]
- Ryu, J.S.; Kang, J.H.; Jung, S.Y.; Shin, M.H.; Kim, J.M.; Park, H.; Min, D.Y. Production of interleukin-8 by human neutrophils stimulated with Trichomonas vaginalis. Infect. Immun. 2004, 72, 1326–1332. [Google Scholar] [CrossRef]
- Shaio, M.F.; Lin, P.R.; Liu, J.Y.; Tang, K.D. Monocyte-derived interleukin-8 involved in the recruitment of neutrophils induced by Trichomonas vaginalis infection. J. Infect. Dis. 1994, 170, 1638–1640. [Google Scholar] [CrossRef]
- Shaio, M.F.; Lin, P.R. Leucotriene B4 levels in the vaginal discharges from cases of trichomoniasis. Ann. Trop. Med. Parasitol. 1995, 89, 85–88. [Google Scholar] [CrossRef]
- Liew, P.X.; Kubes, P. The neutrophil’s role during health and disease. Physiol. Rev. 2019, 99, 1223–1248. [Google Scholar] [CrossRef]
- Ralston, K.S. Chew on this: Amoebic trogocytosis and host cell killing by Entamoeba histolytica. Trends Parasitol. 2015, 31, 442–452. [Google Scholar] [CrossRef]
- Ralston, K.S.; Solga, M.D.; Mackey-Lawrence, N.M.; Somlata; Bhattacharya, A.; Petri, W.A., Jr. Trogocytosis by Entamoeba histolytica contributes to cell killing and tissue invasion. Narure 2014, 508, 526–530. [Google Scholar] [CrossRef]
- Lin, W.C.; Chang, W.T.; Chang, T.Y.; Shin, J.W. The pathogenesis of human cervical epithelium cells induced by interacting with Trichomonas vaginalis. PLoS ONE 2015, 10, e0124087. [Google Scholar] [CrossRef]
- Ortiz, S.F.D.N.; Verdan, R.; Fortes, F.D.S.A.; Benchimol, M. Trichomonas vaginalis: Monolayer and cluster formation-ultrastructural aspects using high-resolution scanning electron microscopy. Pathogens 2023, 12, 1381. [Google Scholar] [CrossRef]
- Coceres, V.M.; Alonso, A.M.; Nievas, Y.R.; Midlej, V.; Frontera, L.; Benchimol, M.; Johnson, P.J.; de Miguel, N. The C-terminal tail of tetraspanin proteins regulates their intracellular distribution in the parasite Trichomonas vaginalis. Cell. Microbiol. 2015, 17, 1217–1229. [Google Scholar] [CrossRef]
- Pachano, T.; Nievas, Y.R.; Lizarraga, A.; Johnson, P.J.; Strobl-Mazzulla, P.H.; de Miguel, N. Epigenetics regulates transcription and pathogenesis in the parasite Trichomonas vaginalis. Cell. Microbiol. 2017, 19, e12716. [Google Scholar] [CrossRef]
- McCracken, J.M.; Allen, L.A. Regulation of human neutrophil apoptosis and lifespan in health and disease. J. Cell Death 2014, 7, 15–23. [Google Scholar] [CrossRef]
- Kang, J.H.; Song, H.O.; Ryu, J.S.; Shin, M.H.; Kim, J.M.; Cho, Y.S.; Alderete, J.F.; Ahn, M.H.; Min, D.Y. Trichomonas vaginalis promotes apoptosis of human neutrophils by activating caspase-3 and reducing Mcl-1 expression. Parasite Immunol. 2006, 28, 439–446. [Google Scholar] [CrossRef]
- Song, H.O.; Shin, M.H.; Ahn, M.H.; Min, D.Y.; Kim, Y.S.; Ryu, J.S. Trichomonas vaginalis: Reactive oxygen species mediates caspase-3 dependent apoptosis of human neutrophils. Exp. Parasitol. 2008, 118, 59–65. [Google Scholar] [CrossRef]
- Gardai, S.; Whitlock, B.B.; Helgason, C.; Ambruso, D.; Fadok, V.; Bratton, D.; Henson, P.M. Activation of SHIP by NADPH oxidase-stimulated Lyn leads to enhanced apoptosis in neutrophils. J. Biol. Chem. 2002, 277, 5236–5246. [Google Scholar] [CrossRef]
- Burza, S.; Croft, S.L.; Boelaert, M. Leishmaniasis. Lancet 2018, 392, 951–970. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Leishmaniasis. Available online: https://www.who.int/health-topics/leishmaniasis#tab=tab_1 (accessed on 10 July 2023).
- Alvar, J.; Vélez, I.D.; Bern, C.; Herrero, M.; Desjeux, P.; Cano, J.; Jannin, J.; den Boer, M.; WHO Leishmaniasis Control Team. Leishmaniasis worldwide and global estimates of its incidence. PLoS ONE 2012, 7, e35671. [Google Scholar] [CrossRef]
- Pace, D. Leishmaniasis. J. Infect. 2014, 69, S10–S18. [Google Scholar] [CrossRef]
- Sakthianandeswaren, A.; Foote, S.J.; Handman, E. The role of host genetics in leishmaniasis. Trends Parasitol. 2009, 25, 383–391. [Google Scholar] [CrossRef]
- McGwire, B.S.; Satoskar, A.R. Leishmaniasis: Clinical syndromes and treatment. QJM Mon. J. Assoc. Physicians 2014, 107, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Bates, P.A.; Depaquit, J.; Galati, E.A.; Kamhawi, S.; Maroli, M.; McDowell, M.A.; Picado, A.; Ready, P.D.; Salomón, O.D.; Shaw, J.J.; et al. Recent advances in phlebotomine sand fly research related to leishmaniasis control. Parasites Vectors 2015, 8, 131. [Google Scholar] [CrossRef]
- Chauhan, P.; Shukla, D.; Chattopadhyay, D.; Saha, B. Redundant and regulatory roles for Toll-like receptors in Leishmania infection. Clin. Exp. Immunol. 2017, 190, 167–186. [Google Scholar] [CrossRef]
- Ronet, C.; Passelli, K.; Charmoy, M.; Scarpellino, L.; Myburgh, E.; Hauyon La Torre, Y.; Turco, S.; Mottram, J.C.; Fasel, N.; Luther, S.A.; et al. TLR2 signaling in skin nonhematopoietic cells induces early neutrophil recruitment in response to Leishmania major infection. J. Investig. Dermatol. 2019, 139, 1318–1328. [Google Scholar] [CrossRef] [PubMed]
- Guimarães-Costa, A.B.; Shannon, J.P.; Waclawiak, I.; Oliveira, J.; Meneses, C.; de Castro, W.; Wen, X.; Brzostowski, J.; Serafim, T.D.; Andersen, J.F.; et al. A sand fly salivary protein acts as a neutrophil chemoattractant. Nat. Commun. 2021, 12, 3213. [Google Scholar] [CrossRef] [PubMed]
- Giraud, E.; Lestinova, T.; Derrick, T.; Martin, O.; Dillon, R.J.; Volf, P.; Műller, I.; Bates, P.A.; Rogers, M.E. Leishmania proteophosphoglycans regurgitated from infected sand flies accelerate dermal wound repair and exacerbate leishmaniasis via insulin-like growth factor 1-dependent signalling. PLoS Pathog. 2018, 14, e1006794. [Google Scholar] [CrossRef] [PubMed]
- Sousa, L.M.; Carneiro, M.B.; Resende, M.E.; Martins, L.S.; Dos Santos, L.M.; Vaz, L.G.; Mello, P.S.; Mosser, D.M.; Oliveira, M.A.; Vieira, L.Q. Neutrophils have a protective role during early stages of Leishmania amazonensis infection in BALB/c mice. Parasite Immunol. 2014, 36, 13–31. [Google Scholar] [CrossRef] [PubMed]
- Hurrell, B.P.; Schuster, S.; Grün, E.; Coutaz, M.; Williams, R.A.; Held, W.; Malissen, B.; Malissen, M.; Yousefi, S.; Simon, H.U.; et al. Rapid sequestration of Leishmania mexicana by neutrophils contributes to the development of chronic lesion. PLoS Pathog. 2015, 11, e1004929. [Google Scholar] [CrossRef] [PubMed]
- Faria, M.S.; Reis, F.C.; Lima, A.P. Toll-like receptors in leishmania infections: Guardians or promoters? J. Parasitol. Res. 2012, 2012, 930257. [Google Scholar] [CrossRef] [PubMed]
- Talamás-Rohana, P.; Wright, S.D.; Lennartz, M.R.; Russell, D.G. Lipophosphoglycan from Leishmania mexicana promastigotes binds to members of the CR3, p150,95 and LFA-1 family of leukocyte integrins. J. Immunol. 1990, 144, 4817–4824. [Google Scholar] [CrossRef] [PubMed]
- Tavares, N.M.; Araújo-Santos, T.; Afonso, L.; Nogueira, P.M.; Lopes, U.G.; Soares, R.P.; Bozza, P.T.; Bandeira-Melo, C.; Borges, V.M.; Brodskyn, C. Understanding the mechanisms controlling Leishmania amazonensis infection in vitro: The role of LTB4 derived from human neutrophils. J. Infect. Dis. 2014, 210, 656–666. [Google Scholar] [CrossRef] [PubMed]
- Noronha, L.P.; Martins, M.D.A.; Castro-Junior, A.B.; Thorstenberg, M.L.; Costa-Soares, L.; Rangel, T.P.; Carvalho-Gondim, F.; Rossi-Bergmann, B.; Savio, L.E.B.; Canetti, C.A.; et al. Cysteinyl-leukotrienes promote cutaneous Leishmaniasis control. Front. Cell. Infect. Microbiol. 2023, 13, 1192800. [Google Scholar] [CrossRef] [PubMed]
- Lämmermann, T.; Afonso, P.V.; Angermann, B.R.; Wang, J.M.; Kastenmüller, W.; Parent, C.A.; Germain, R.N. Neutrophil swarms require LTB4 and integrins at sites of cell death in vivo. Narure 2013, 498, 371–375. [Google Scholar] [CrossRef]
- Oualha, R.; Barhoumi, M.; Marzouki, S.; Harigua-Souiai, E.; Ben Ahmed, M.; Guizani, I. Infection of human neutrophils with Leishmania infantum or Leishmania major strains triggers activation and differential cytokines release. Front. Cell. Infect. Microbiol. 2019, 9, 153. [Google Scholar] [CrossRef]
- D’Alessandro, S.; Parapini, S.; Corbett, Y.; Frigerio, R.; Delbue, S.; Modenese, A.; Gramiccia, M.; Ferrante, P.; Taramelli, D.; Basilico, N. Leishmania promastigotes enhance neutrophil recruitment through the production of CXCL8 by endothelial cells. Pathogens 2021, 10, 1380. [Google Scholar] [CrossRef] [PubMed]
- Carlsen, E.D.; Jie, Z.; Liang, Y.; Henard, C.A.; Hay, C.; Sun, J.; de Matos Guedes, H.; Soong, L. Interactions between neutrophils and Leishmania braziliensis amastigotes facilitate cell activation and parasite clearance. J. Innate Immun. 2015, 7, 354–363. [Google Scholar] [CrossRef] [PubMed]
- McFarlane, E.; Perez, C.; Charmoy, M.; Allenbach, C.; Carter, K.C.; Alexander, J.; Tacchini-Cottier, F. Neutrophils contribute to development of a protective immune response during onset of infection with Leishmania donovani. Infect. Immun. 2008, 76, 532–541. [Google Scholar] [CrossRef] [PubMed]
- Carlsen, E.D.; Hay, C.; Henard, C.A.; Popov, V.; Garg, N.J.; Soong, L. Leishmania amazonensis amastigotes trigger neutrophil activation but resist neutrophil microbicidal mechanisms. Infect. Immun. 2013, 81, 3966–3974. [Google Scholar] [CrossRef] [PubMed]
- Hurrell, B.P.; Beaumann, M.; Heyde, S.; Regli, I.B.; Müller, A.J.; Tacchini-Cottier, F. Frontline science: Leishmania mexicana amastigotes can replicate within neutrophils. J. Leukoc. Biol. 2017, 102, 1187–1198. [Google Scholar] [CrossRef]
- van Zandbergen, G.; Klinger, M.; Mueller, A.; Dannenberg, S.; Gebert, A.; Solbach, W.; Laskay, T. Cutting edge: Neutrophil granulocyte serves as a vector for Leishmania entry into macrophages. J. Immunol. 2004, 173, 6521–6525. [Google Scholar] [CrossRef] [PubMed]
- Peters, N.C.; Egen, J.G.; Secundino, N.; Debrabant, A.; Kimblin, N.; Kamhawi, S.; Lawyer, P.; Fay, M.P.; Germain, R.N.; Sacks, D. In vivo imaging reveals an essential role for neutrophils in leishmaniasis transmitted by sand flies. Science 2008, 321, 970–974. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Coll, H.; Cardona-Castro, N.; Ramos, A.P.; Llanos-Cuentas, A. Innate immune response: Ally or enemy in cutaneous leishmaniasis? Pathog. Dis. 2021, 79, ftab028. [Google Scholar] [CrossRef] [PubMed]
- Passelli, K.; Billion, O.; Tacchini-Cottier, F. The impact of neutrophil recruitment to the skin on the pathology induced by Leishmania infection. Front. Immunol. 2021, 12, 649348. [Google Scholar] [CrossRef]
- Niedergang, F.; Grinstein, S. How to build a phagosome: New concepts for an old process. Curr. Opin. Cell Biol. 2018, 50, 57–63. [Google Scholar] [CrossRef]
- Uribe-Querol, E.; Rosales, C. Control of phagocytosis by microbial pathogens. Front. Immunol. 2017, 8, 1368. [Google Scholar] [CrossRef] [PubMed]
- Vinet, A.F.; Fukuda, M.; Turco, S.J.; Descoteaux, A. The Leishmania donovani lipophosphoglycan excludes the vesicular proton-ATPase from phagosomes by impairing the recruitment of synaptotagmin V. PLoS Pathog. 2009, 5, e1000628. [Google Scholar] [CrossRef] [PubMed]
- Matheoud, D.; Moradin, N.; Bellemare-Pelletier, A.; Shio, M.T.; Hong, W.J.; Olivier, M.; Gagnon, E.; Desjardins, M.; Descoteaux, A. Leishmania evades host immunity by inhibiting antigen cross-presentation through direct cleavage of the SNARE VAMP8. Cell Host Microbe 2013, 14, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Desjardins, M.; Descoteaux, A. Inhibition of phagolysosomal biogenesis by the Leishmania lipophosphoglycan. J. Exp. Med. 1997, 185, 2061–2068. [Google Scholar] [CrossRef] [PubMed]
- Courret, N.; Fréhel, C.; Gouhier, N.; Pouchelet, M.; Prina, E.; Roux, P.; Antoine, J.C. Biogenesis of Leishmania-harbouring parasitophorous vacuoles following phagocytosis of the metacyclic promastigote or amastigote stages of the parasites. J. Cell Sci. 2002, 115, 2303–2316. [Google Scholar] [CrossRef] [PubMed]
- Real, F.; Mortara, R.A. The diverse and dynamic nature of Leishmania parasitophorous vacuoles studied by multidimensional imaging. PLoS Neglected Trop. Dis. 2012, 6, e1518. [Google Scholar] [CrossRef] [PubMed]
- Ndjamen, B.; Kang, B.H.; Hatsuzawa, K.; Kima, P.E. Leishmania parasitophorous vacuoles interact continuously with the host cell’s endoplasmic reticulum; parasitophorous vacuoles are hybrid compartments. Cell. Microbiol. 2010, 12, 1480–1494. [Google Scholar] [CrossRef] [PubMed]
- Mollinedo, F.; Janssen, H.; de la Iglesia-Vicente, J.; Villa-Pulgarin, J.A.; Calafat, J. Selective fusion of azurophilic granules with Leishmania-containing phagosomes in human neutrophils. J. Biol. Chem. 2010, 285, 34528–34536. [Google Scholar] [CrossRef]
- Jankowski, A.; Scott, C.C.; Grinstein, S. Determinants of the phagosomal pH in neutrophils. J. Biol. Chem. 2002, 277, 6059–6066. [Google Scholar] [CrossRef]
- Piacenza, L.; Trujillo, M.; Radi, R. Reactive species and pathogen antioxidant networks during phagocytosis. J. Exp. Med. 2019, 216, 501–516. [Google Scholar] [CrossRef]
- Roma, E.H.; Macedo, J.P.; Goes, G.R.; Gonçalves, J.L.; Castro, W.; Cisalpino, D.; Vieira, L.Q. Impact of reactive oxygen species (ROS) on the control of parasite loads and inflammation in Leishmania amazonensis infection. Parasites Vectors 2016, 9, 193. [Google Scholar] [CrossRef] [PubMed]
- Carneiro, M.B.H.; Roma, E.H.; Ranson, A.J.; Doria, N.A.; Debrabant, A.; Sacks, D.L.; Vieira, L.Q.; Peters, N.C. NOX2-derived reactive oxygen species control inflammation during Leishmania amazonensis infection by mediating infection-induced neutrophil apoptosis. J. Immunol. 2018, 200, 196–208. [Google Scholar] [CrossRef] [PubMed]
- Guimarães-Costa, A.B.; Nascimento, M.T.; Froment, G.S.; Soares, R.P.; Morgado, F.N.; Conceição-Silva, F.; Saraiva, E.M. Leishmania amazonensis promastigotes induce and are killed by neutrophil extracellular traps. Proc. Natl. Acad. Sci. USA 2009, 106, 6748–6753. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, Y.; Xin, L.; Beverley, S.M.; Carlsen, E.D.; Popov, V.; Chang, K.P.; Wang, M.; Soong, L. Differential microbicidal effects of human histone proteins H2A and H2B on Leishmania promastigotes and amastigotes. Infect. Immun. 2011, 79, 1124–1133. [Google Scholar] [CrossRef] [PubMed]
- Rochael, N.C.; Guimarães-Costa, A.B.; Nascimento, M.T.; DeSouza-Vieira, T.S.; Oliveira, M.P.; Garcia e Souza, L.F.; Oliveira, M.F.; Saraiva, E.M. Classical ROS-dependent and early/rapid ROS-independent release of Neutrophil Extracellular Traps triggered by Leishmania parasites. Sci. Rep. 2015, 5, 18302. [Google Scholar] [CrossRef] [PubMed]
- Guimarães-Costa, A.B.; DeSouza-Vieira, T.S.; Paletta-Silva, R.; Freitas-Mesquita, A.L.; Meyer-Fernandes, J.R.; Saraiva, E.M. 3’-nucleotidase/nuclease activity allows Leishmania parasites to escape killing by neutrophil extracellular traps. Infect. Immun. 2014, 82, 1732–1740. [Google Scholar] [CrossRef] [PubMed]
- Manfredi, A.A.; Ramirez, G.A.; Rovere-Querini, P.; Maugeri, N. The neutrophil’s choice: Phagocytose vs make neutrophil extracellular traps. Front. Immunol. 2018, 9, 288. [Google Scholar] [CrossRef] [PubMed]
- Hochberg, N.S.; Montgomery, S.P. Chagas disease. Ann. Intern. Med. 2023, 176, ITC17–ITC32. [Google Scholar] [CrossRef]
- Martín-Escolano, J.; Medina-Carmona, E.; Martín-Escolano, R. Chagas disease: Current view of an ancient and global chemotherapy challenge. ACS Infect. Dis. 2020, 6, 2830–2843. [Google Scholar] [CrossRef]
- Lidani, K.C.F.; Andrade, F.A.; Bavia, L.; Damasceno, F.S.; Beltrame, M.H.; Messias-Reason, I.J.; Sandri, T.L. Chagas disease: From discovery to a worldwide health problem. Front. Public Health 2019, 7, 166. [Google Scholar] [CrossRef]
- World Health Organization. Chagas Disease (American Trypanosomiasis). Available online: https://www.who.int/news-room/facts-in-pictures/detail/chagas-disease (accessed on 29 March 2024).
- World Health Organization. Promising Progress on Neglected Tropical Diseases in Africa. Available online: https://www.afro.who.int/news/promising-progress-neglected-tropical-diseases-africa (accessed on 29 March 2024).
- Miles, M.A. The discovery of Chagas disease: Progress and prejudice. Infect. Dis. Clin. N. Am. 2004, 18, 247–260. [Google Scholar] [CrossRef] [PubMed]
- Macaluso, G.; Grippi, F.; Di Bella, S.; Blanda, V.; Gucciardi, F.; Torina, A.; Guercio, A.; Cannella, V. A review on the immunological response against Trypanosoma cruzi. Pathogens 2023, 12, 282. [Google Scholar] [CrossRef] [PubMed]
- Monteon, V. Trypanosoma cruzi: The early contact between insect-derived metacyclic trypomastigotes and the mammalian cells. Ann. Parasitol. 2019, 65, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Zuma, A.A.; Dos Santos Barrias, E.; de Souza, W. Basic biology of Trypanosoma cruzi. Curr. Pharm. Des. 2021, 27, 1671–1732. [Google Scholar] [CrossRef] [PubMed]
- Magalhães, L.M.D.; Gollob, K.J.; Zingales, B.; Dutra, W.O. Pathogen diversity, immunity, and the fate of infections: Lessons learned from Trypanosoma cruzi human-host interactions. Lancet. Microbe 2022, 3, e711–e722. [Google Scholar] [CrossRef] [PubMed]
- Zingales, B. Trypanosoma cruzi genetic diversity: Something new for something known about Chagas disease manifestations, serodiagnosis and drug sensitivity. Acta Trop. 2018, 184, 38–52. [Google Scholar] [CrossRef] [PubMed]
- Echavarría, N.G.; Echeverría, L.E.; Stewart, M.; Gallego, C.; Saldarriaga, C. Chagas disease: Chronic Chagas cardiomyopathy. Curr. Probl. Cardiol. 2021, 46, 100507. [Google Scholar] [CrossRef] [PubMed]
- Martín-Escolano, J.; Marín, C.; Rosales, M.J.; Tsaousis, A.D.; Medina-Carmona, E.; Martín-Escolano, R. An updated view of the Trypanosoma cruzi life cycle: Intervention points for an effective treatment. ACS Infect. Dis. 2022, 8, 1107–1115. [Google Scholar] [CrossRef]
- Sales Junior, P.A.; Molina, I.; Fonseca Murta, S.M.; Sánchez-Montalvá, A.; Salvador, F.; Corrêa-Oliveira, R.; Carneiro, C.M. Experimental and clinical treatment of Chagas disease: A review. Am. J. Trop. Med. Hyg. 2017, 97, 1289–1303. [Google Scholar] [CrossRef]
- De Fuentes-Vicente, J.A.; Santos-Hernández, N.G.; Ruiz-Castillejos, C.; Espinoza-Medinilla, E.E.; Flores-Villegas, A.L.; de Alba-Alvarado, M.; Cabrera-Bravo, M.; Moreno-Rodríguez, A.; Vidal-López, D.G. What do you need to know before studying Chagas disease? A beginner’s guide. Trop. Med. Infect. Dis. 2023, 8, 360. [Google Scholar] [CrossRef]
- Ferri, G.; Edreira, M.M. All roads lead to cytosol: Trypanosoma cruzi multi-strategic approach to invasion. Front. Cell. Infect. Microbiol. 2021, 11, 634793. [Google Scholar] [CrossRef]
- Acevedo, G.R.; Girard, M.C.; Gómez, K.A. The unsolved jigsaw puzzle of the immune response in Chagas disease. Front. Immunol. 2018, 9, 1929. [Google Scholar] [CrossRef] [PubMed]
- Guimarães-Pinto, K.; Ferreira, J.R.M.; da Costa, A.L.A.; Morrot, A.; Freire-de-Lima, L.; Decote-Ricardo, D.; Freire-de-Lima, C.G.; Filardy, A.A. Cellular stress and senescence induction during Trypanosoma cruzi infection. Trop. Med. Infect. Dis. 2022, 7, 129. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.S.; Machado, F.S.; Martins, G.A. The role of nitric oxide in the pathogenesis of Chagas disease. Front. Biosci. 2003, 8, s314–s325. [Google Scholar] [CrossRef] [PubMed]
- Molina, H.A.; Kierszenbaum, F. A study of human myocardial tissue in Chagas’ disease: Distribution and frequency of inflammatory cell types. Int. J. Parasitol. 1987, 17, 1297–1305. [Google Scholar] [CrossRef] [PubMed]
- Molina, H.A.; Kierszenbaum, F. Interaction of human eosinophils or neutrophils with Trypanosoma cruzi in vitro causes bystander cardiac cell damage. Immunology 1989, 66, 289–295. [Google Scholar] [PubMed]
- Kipnis, T.L.; James, S.L.; Sher, A.; David, J.R. Cell-mediated cytotoxicity to Trypanosoma cruzi. II. Antibody-dependent killing of bloodstream forms by mouse eosinophils and neutrophils. Am. J. Trop. Med. Hyg. 1981, 30, 47–53. [Google Scholar] [CrossRef]
- Villalta, F.; Kierszenbaum, F. Role of polymorphonuclear cells in Chagas’ disease. I. Uptake and mechanisms of destruction of intracellular (amastigote) forms of Trypanosoma cruzi by human neutrophils. J. Immunol. 1983, 131, 1504–1510. [Google Scholar] [CrossRef]
- Tosello Boari, J.; Amezcua Vesely, M.C.; Bermejo, D.A.; Ramello, M.C.; Montes, C.L.; Cejas, H.; Gruppi, A.; Acosta Rodríguez, E.V. IL-17RA signaling reduces inflammation and mortality during Trypanosoma cruzi infection by recruiting suppressive IL-10-producing neutrophils. PLoS Pathog. 2012, 8, e1002658. [Google Scholar] [CrossRef]
- Chen, L.; Watanabe, T.; Watanabe, H.; Sendo, F. Neutrophil depletion exacerbates experimental Chagas’ disease in BALB/c, but protects C57BL/6 mice through modulating the Th1/Th2 dichotomy in different directions. Eur. J. Immunol. 2001, 31, 265–275. [Google Scholar] [CrossRef]
- Luna-Gomes, T.; Filardy, A.A.; Rocha, J.D.; Decote-Ricardo, D.; LaRocque-de-Freitas, I.F.; Morrot, A.; Bozza, P.T.; Castro-Faria-Neto, H.C.; DosReis, G.A.; Nunes, M.P.; et al. Neutrophils increase or reduce parasite burden in Trypanosoma cruzi-infected macrophages, depending on host strain: Role of neutrophil elastase. PLoS ONE 2014, 9, e90582. [Google Scholar] [CrossRef]
- Miyazaki, Y.; Hamano, S.; Wang, S.; Shimanoe, Y.; Iwakura, Y.; Yoshida, H. IL-17 is necessary for host protection against acute-phase Trypanosoma cruzi infection. J. Immunol. 2010, 185, 1150–1157. [Google Scholar] [CrossRef]
- Medeiros, N.I.; Fares, R.C.; Franco, E.P.; Sousa, G.R.; Mattos, R.T.; Chaves, A.T.; Nunes, M.D.; Dutra, W.O.; Correa-Oliveira, R.; Rocha, M.O.; et al. Differential expression of matrix metalloproteinases 2, 9 and cytokines by neutrophils and monocytes in the clinical forms of Chagas disease. PLoS Neglected Trop. Dis. 2017, 11, e0005284. [Google Scholar] [CrossRef]
- Dhiman, M.; Garg, N.J. NADPH oxidase inhibition ameliorates Trypanosoma cruzi-induced myocarditis during Chagas disease. J. Pathol. 2011, 225, 583–596. [Google Scholar] [CrossRef]
- Muniz-Junqueira, M.I.; Mota, L.M.; Aires, R.B.; Junqueira Júnior, L.F. Differing phagocytic function of monocytes and neutrophils in Chagas’ cardiopathy according to the presence or absence of congestive heart failure. Rev. Soc. Bras. Med. Trop. 2004, 37, 447–453. [Google Scholar] [CrossRef]
- Gomes, J.; Campi-Azevedo, A.C.; Teixeira-Carvalho, A.; Silveira-Lemos, D.; Vitelli-Avelar, D.; Sathler-Avelar, R.; Peruhype-Magalhães, V.; Silvestre, K.F.; Batista, M.A.; Schachnik, N.C.; et al. Impaired phagocytic capacity driven by downregulation of major phagocytosis-related cell surface molecules elicits an overall modulatory cytokine profile in neutrophils and monocytes from the indeterminate clinical form of Chagas disease. Immunobiology 2012, 217, 1005–1016. [Google Scholar] [CrossRef]
- Paiva, C.N.; Feijó, D.F.; Dutra, F.F.; Carneiro, V.C.; Freitas, G.B.; Alves, L.S.; Mesquita, J.; Fortes, G.B.; Figueiredo, R.T.; Souza, H.S.; et al. Oxidative stress fuels Trypanosoma cruzi infection in mice. J. Clin. Investig. 2012, 122, 2531–2542. [Google Scholar] [CrossRef]
- Santiago, H.C.; Gonzalez Lombana, C.Z.; Macedo, J.P.; Utsch, L.; Tafuri, W.L.; Campagnole-Santos, M.J.; Alves, R.O.; Alves-Filho, J.C.; Romanha, A.J.; Cunha, F.Q.; et al. NADPH phagocyte oxidase knockout mice control Trypanosoma cruzi proliferation, but develop circulatory collapse and succumb to infection. PLoS Neglected Trop. Dis. 2012, 6, e1492. [Google Scholar] [CrossRef]
- Sousa-Rocha, D.; Thomaz-Tobias, M.; Diniz, L.F.; Souza, P.S.; Pinge-Filho, P.; Toledo, K.A. Trypanosoma cruzi and its soluble antigens induce NET release by stimulating Toll-like receptors. PLoS ONE 2015, 10, e0139569. [Google Scholar] [CrossRef]
- de Buhr, N.; Bonilla, M.C.; Jimenez-Soto, M.; von Köckritz-Blickwede, M.; Dolz, G. Extracellular trap formation in response to Trypanosoma cruzi infection in granulocytes isolated from dogs and common opossums, natural reservoir hosts. Front. Microbiol. 2018, 9, 966. [Google Scholar] [CrossRef]
- Czaikoski, P.G.; Mota, J.M.; Nascimento, D.C.; Sônego, F.; Castanheira, F.V.; Melo, P.H.; Scortegagna, G.T.; Silva, R.L.; Barroso-Sousa, R.; Souto, F.O.; et al. Neutrophil extracellular traps induce organ damage during experimental and clinical sepsis. PLoS ONE 2016, 11, e0148142. [Google Scholar] [CrossRef]
- Guillén, N. Pathogenicity and virulence of Entamoeba histolytica, the agent of amoebiasis. Virulence 2023, 14, 2158656. [Google Scholar] [CrossRef]
- Carrero, J.C.; Reyes-López, M.; Serrano-Luna, J.; Shibayama, M.; Unzueta, J.; León-Sicairos, N.; de la Garza, M. Intestinal amoebiasis: 160 years of its first detection and still remains as a health problem in developing countries. Int. J. Med. Microbiol. 2020, 310, 151358. [Google Scholar] [CrossRef]
- GBD 2016 Diarrhoeal Disease Collaborators. Estimates of the global, regional, and national morbidity, mortality, and aetiologies of diarrhoea in 195 countries: A systematic analysis for the Global Burden of Disease Study 2016. Lancet. Infect. Dis. 2018, 18, 1211–1228. [Google Scholar] [CrossRef]
- Fu, X.; Zhong, Y.; Chen, L.; Ge, M.; Yu, M.; Sun, Y.; Shen, L. Global burden and trends of the Entamoeba infection-associated diseases from 1990 to 2019: An observational trend study. Acta Trop. 2023, 240, 106866. [Google Scholar] [CrossRef]
- Oliveira, F.M.; Neumann, E.; Gomes, M.A.; Caliari, M.V. Entamoeba dispar: Could it be pathogenic. Trop. Parasitol. 2015, 5, 9–14. [Google Scholar] [CrossRef]
- Haque, R.; Huston, C.D.; Hughes, M.; Houpt, E.; Petri, W.A., Jr. Amebiasis. N. Engl. J. Med. 2003, 348, 1565–1573. [Google Scholar] [CrossRef]
- Moonah, S.N.; Jiang, N.M.; Petri, W.A., Jr. Host immune response to intestinal amebiasis. PLoS Pathog. 2013, 9, e1003489. [Google Scholar] [CrossRef]
- Prakash, V.; Jackson-Akers, J.Y.; Oliver, T.I. Amebic Liver Abscess. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2020. Available online: https://www.ncbi.nlm.nih.gov/books/NBK430832/ (accessed on 7 November 2019).
- Acuna-Soto, R.; Maguire, J.H.; Wirth, D.F. Gender distribution in asymptomatic and invasive amebiasis. Am. J. Gastroenterol. 2000, 95, 1277–1283. [Google Scholar] [CrossRef]
- Bansal, Y.; Maurya, V.; Tak, V.; Bohra, G.K.; Kumar, D.; Goel, A.D.; Yadav, T.; Nag, V.L. Clinical and laboratory profile of patients with amoebic liver abscess. Trop. Parasitol. 2022, 12, 113–118. [Google Scholar] [CrossRef]
- Snow, M.; Chen, M.; Guo, J.; Atkinson, J.; Stanley, S.L., Jr. Differences in complement-mediated killing of Entamoeba histolytica between men and women--an explanation for the increased susceptibility of men to invasive amebiasis? Am. J. Trop. Med. Hyg. 2008, 78, 922–923. [Google Scholar] [CrossRef]
- Lotter, H.; Helk, E.; Bernin, H.; Jacobs, T.; Prehn, C.; Adamski, J.; González-Roldán, N.; Holst, O.; Tannich, E. Testosterone increases susceptibility to amebic liver abscess in mice and mediates inhibition of IFNγ secretion in natural killer T cells. PLoS ONE 2013, 8, e55694. [Google Scholar] [CrossRef]
- Er-Lukowiak, M.; Hänzelmann, S.; Rothe, M.; Moamenpour, D.T.; Hausmann, F.; Khatri, R.; Hansen, C.; Boldt, J.; Bärreiter, V.A.; Honecker, B.; et al. Testosterone affects type I/type II interferon response of neutrophils during hepatic amebiasis. Front. Immunol. 2023, 14, 1279245. [Google Scholar] [CrossRef]
- Uribe-Querol, E.; Rosales, C. Immune response to the enteric parasite Entamoeba histolytica. Physiology 2020, 35, 244–260. [Google Scholar] [CrossRef]
- Marie, C.; Petri, W.A., Jr. Regulation of virulence of Entamoeba histolytica. Annu. Rev. Microbiol. 2014, 68, 493–520. [Google Scholar] [CrossRef]
- Cornick, S.; Chadee, K. Entamoeba histolytica: Host parasite interactions at the colonic epithelium. Tissue Barriers 2017, 5, e1283386. [Google Scholar] [CrossRef]
- Ximénez, C.; Morán, P.; Rojas, L.; Valadez, A.; Gómez, A.; Ramiro, M.; Cerritos, R.; González, E.; Hernández, E.; Oswaldo, P. Novelties on amoebiasis: A neglected tropical disease. J. Glob. Infect. Dis. 2011, 3, 166–174. [Google Scholar] [CrossRef]
- Krishnan, D.; Ghosh, S.K. Cellular events of multinucleated giant cells formation during the encystation of Entamoeba invadens. Front. Cell. Infect. Microbiol. 2018, 8, 262. [Google Scholar] [CrossRef]
- Leon-Coria, A.; Kumar, M.; Chadee, K. The delicate balance between Entamoeba histolytica, mucus and microbiota. Gut Microbes 2020, 11, 118–125. [Google Scholar] [CrossRef]
- Uddin, M.J.; Leslie, J.L.; Petri, W.A., Jr. Host protective mechanisms to intestinal amebiasis. Trends Parasitol. 2021, 37, 165–175. [Google Scholar] [CrossRef]
- Singh, A.; Banerjee, T.; Kumar, R.; Shukla, S.K. Prevalence of cases of amebic liver abscess in a tertiary care centre in India: A study on risk factors, associated microflora and strain variation of Entamoeba histolytica. PLoS ONE 2019, 14, e0214880. [Google Scholar] [CrossRef]
- Ngobeni, R.; Samie, A.; Moonah, S.; Watanabe, K.; Petri, W.A., Jr.; Gilchrist, C. Entamoeba species in South Africa: Correlations with the host microbiome, parasite burdens, and first description of Entamoeba bangladeshi outside of Asia. J. Infect. Dis. 2017, 216, 1592–1600. [Google Scholar] [CrossRef]
- Haghighi, A.; Kobayashi, S.; Takeuchi, T.; Masuda, G.; Nozaki, T. Remarkable genetic polymorphism among Entamoeba histolytica isolates from a limited geographic area. J. Clin. Microbiol. 2002, 40, 4081–4090. [Google Scholar] [CrossRef]
- Ramos, F.; García, G.; Valadez, A.; Morán, P.; González, E.; Gómez, A.; Melendro, E.I.; Valenzuela, O.; Ximénez, C.E. dispar strain: Analysis of polymorphism as a tool for study of geographic distribution. Mol. Biochem. Parasitol. 2005, 141, 175–177. [Google Scholar] [CrossRef]
- Thibeaux, R.; Weber, C.; Hon, C.C.; Dillies, M.A.; Avé, P.; Coppée, J.Y.; Labruyère, E.; Guillén, N. Identification of the virulence landscape essential for Entamoeba histolytica invasion of the human colon. PLoS Pathog. 2013, 9, e1003824. [Google Scholar] [CrossRef]
- Tillack, M.; Nowak, N.; Lotter, H.; Bracha, R.; Mirelman, D.; Tannich, E.; Bruchhaus, I. Increased expression of the major cysteine proteinases by stable episomal transfection underlines the important role of EhCP5 for the pathogenicity of Entamoeba histolytica. Mol. Biochem. Parasitol. 2006, 149, 58–64. [Google Scholar] [CrossRef]
- Petri, W.A., Jr.; Haque, R.; Mann, B.J. The bittersweet interface of parasite and host: Lectin-carbohydrate interactions during human invasion by the parasite Entamoeba histolytica. Annu. Rev. Microbiol. 2002, 56, 39–64. [Google Scholar] [CrossRef]
- Betanzos, A.; Bañuelos, C.; Orozco, E. Host invasion by pathogenic amoebae: Epithelial disruption by parasite proteins. Genes 2019, 10, 618. [Google Scholar] [CrossRef]
- Ghosh, S.; Padalia, J.; Moonah, S. Tissue destruction caused by Entamoeba histolytica parasite: Cell death, inflammation, invasion, and the gut microbiome. Curr. Clin. Microbiol. Rep. 2019, 6, 51–57. [Google Scholar] [CrossRef]
- Mortimer, L.; Moreau, F.; Cornick, S.; Chadee, K. The NLRP3 inflammasome is a pathogen sensor for invasive Entamoeba histolytica via activation of α5β1 integrin at the macrophage-amebae intercellular junction. PLoS Pathog. 2015, 11, e1004887. [Google Scholar] [CrossRef]
- Dey, I.; Chadee, K. Prostaglandin E2 produced by Entamoeba histolytica binds to EP4 receptors and stimulates interleukin-8 production in human colonic cells. Infect. Immun. 2008, 76, 5158–5163. [Google Scholar] [CrossRef]
- Campos-Rodríguez, R.; Gutiérrez-Meza, M.; Jarillo-Luna, R.A.; Drago-Serrano, M.E.; Abarca-Rojano, E.; Ventura-Juárez, J.; Cárdenas-Jaramillo, L.M.; Pacheco-Yepez, J. A review of the proposed role of neutrophils in rodent amebic liver abscess models. Parasite 2016, 23, 6. [Google Scholar] [CrossRef]
- Espinosa-Cantellano, M.; Martínez-Palomo, A. Pathogenesis of intestinal amebiasis: From molecules to disease. Clin. Microbiol. Rev. 2000, 13, 318–331. [Google Scholar] [CrossRef]
- Guerrant, R.L.; Brush, J.; Ravdin, J.I.; Sullivan, J.A.; Mandell, G.L. Interaction between Entamoeba histolytica and human polymorphonuclear neutrophils. J. Infect. Dis. 1981, 143, 83–93. [Google Scholar] [CrossRef]
- Denis, M.; Chadee, K. Human neutrophils activated by interferon-gamma and tumour necrosis factor-alpha kill Entamoeba histolytica trophozoites in vitro. J. Leukoc. Biol. 1989, 46, 270–274. [Google Scholar] [CrossRef]
- Estrada-Figueroa, L.A.; Ramírez-Jiménez, Y.; Osorio-Trujillo, C.; Shibayama, M.; Navarro-García, F.; García-Tovar, C.; Talamás-Rohana, P. Absence of CD38 delays arrival of neutrophils to the liver and innate immune response development during hepatic amoebiasis by Entamoeba histolytica. Parasite Immunol. 2011, 33, 661–668. [Google Scholar] [CrossRef]
- Jarillo-Luna, R.A.; Campos-Rodríguez, R.; Tsutsumi, V. Entamoeba histolytica: Immunohistochemical study of hepatic amoebiasis in mouse. Neutrophils and nitric oxide as possible factors of resistance. Exp. Parasitol. 2002, 101, 40–56. [Google Scholar] [CrossRef]
- Naylor, C.; Burgess, S.; Madan, R.; Buonomo, E.; Razzaq, K.; Ralston, K.; Petri, W.A., Jr. Leptin receptor mutation results in defective neutrophil recruitment to the colon during Entamoeba histolytica infection. mBio 2014, 5, e02046-14. [Google Scholar] [CrossRef]
- Velazquez, C.; Shibayama-Salas, M.; Aguirre-Garcia, J.; Tsutsumi, V.; Calderon, J. Role of neutrophils in innate resistance to Entamoeba histolytica liver infection in mice. Parasite Immunol. 1998, 20, 255–262. [Google Scholar] [CrossRef]
- Watanabe, K.; Gilchrist, C.A.; Uddin, M.J.; Burgess, S.L.; Abhyankar, M.M.; Moonah, S.N.; Noor, Z.; Donowitz, J.R.; Schneider, B.N.; Arju, T.; et al. Microbiome-mediated neutrophil recruitment via CXCR2 and protection from amebic colitis. PLoS Pathog. 2017, 13, e1006513. [Google Scholar] [CrossRef]
- Dickson-Gonzalez, S.M.; de Uribe, M.L.; Rodriguez-Morales, A.J. Polymorphonuclear neutrophil infiltration intensity as consequence of Entamoeba histolytica density in amebic colitis. Surg. Infect. 2009, 10, 91–97. [Google Scholar] [CrossRef]
- Olivos-García, A.; Carrero, J.C.; Ramos, E.; Nequiz, M.; Tello, E.; Montfort, I.; Pérez-Tamayo, R. Late experimental amebic liver abscess in hamster is inhibited by cyclosporine and N-acetylcysteine. Exp. Mol. Pathol. 2007, 82, 310–315. [Google Scholar] [CrossRef]
- Pérez-Tamayo, R.; Montfort, I.; García, A.O.; Ramos, E.; Ostria, C.B. Pathogenesis of acute experimental liver amebiasis. Arch. Med. Res. 2006, 37, 203–209. [Google Scholar] [CrossRef]
- Kienle, K.; Lämmermann, T. Neutrophil swarming: An essential process of the neutrophil tissue response. Immunol. Rev. 2016, 273, 76–93. [Google Scholar] [CrossRef]
- Palomino-Segura, M.; Hidalgo, A. Immunity: Neutrophil quorum at the wound. Curr. Biol. 2020, 30, R828–R830. [Google Scholar] [CrossRef]
- Fonseca, Z.; Uribe-Querol, E.; Díaz-Godínez, C.; Carrero, J.C.; Rosales, C. Pathogenic Entamoeba histolytica, but not Entamoeba dispar, induce neutrophil extracellular trap (NET) formation. J. Leukoc. Biol. 2019, 105, 1167–1181. [Google Scholar] [CrossRef]
- Babatunde, K.A.; Wang, X.; Hopke, A.; Lannes, N.; Mantel, P.Y.; Irimia, D. Chemotaxis and swarming in differentiated HL-60 neutrophil-like cells. Sci. Rep. 2021, 11, 778. [Google Scholar] [CrossRef]
- Hopke, A.; Irimia, D. Ex vivo human neutrophil swarming against live microbial targets. Methods Mol. Biol. 2020, 2087, 107–116. [Google Scholar] [CrossRef]
- Biller, L.; Matthiesen, J.; Kühne, V.; Lotter, H.; Handal, G.; Nozaki, T.; Saito-Nakano, Y.; Schümann, M.; Roeder, T.; Tannich, E.; et al. The cell surface proteome of Entamoeba histolytica. Mol. Cell. Proteom. 2014, 13, 132–144. [Google Scholar] [CrossRef]
- Choi, M.H.; Sajed, D.; Poole, L.; Hirata, K.; Herdman, S.; Torian, B.E.; Reed, S.L. An unusual surface peroxiredoxin protects invasive Entamoeba histolytica from oxidant attack. Mol. Biochem. Parasitol. 2005, 143, 80–89. [Google Scholar] [CrossRef]
- Ghosh, A.S.; Dutta, S.; Raha, S. Hydrogen peroxide-induced apoptosis-like cell death in Entamoeba histolytica. Parasitol. Int. 2010, 59, 166–172. [Google Scholar] [CrossRef]
- Davis, P.H.; Zhang, X.; Guo, J.; Townsend, R.R.; Stanley, S.L., Jr. Comparative proteomic analysis of two Entamoeba histolytica strains with different virulence phenotypes identifies peroxiredoxin as an important component of amoebic virulence. Mol. Microbiol. 2006, 61, 1523–1532. [Google Scholar] [CrossRef]
- Cruz-Baquero, A.; Jarillo-Luna, R.A.; Cárdenas-Jaramillo, L.M.; Drago-Serrano, M.E.; Serrano-Luna, J.J.; Pacheco-Yépez, J. Ascorbic acid ameriolates liver damage by myeloperoxidase oxidative products in a hamster model of amoebic liver abscess. Front. Cell. Infect. Microbiol. 2022, 12, 855822. [Google Scholar] [CrossRef]
- Higuera-Martínez, G.; Arciniega-Martínez, I.M.; Jarillo-Luna, R.A.; Cárdenas-Jaramillo, L.M.; Levaro-Loquio, D.; Velásquez-Torres, M.; Abarca-Rojano, E.; Reséndiz-Albor, A.A.; Pacheco-Yépez, J. Apocynin, an NADPH oxidase enzyme inhibitor, prevents amebic liver abscess in hamster. Biomedicines 2023, 11, 2322. [Google Scholar] [CrossRef]
- Díaz-Godínez, C.; Fonseca, Z.; Néquiz, M.; Laclette, J.P.; Rosales, C.; Carrero, J.C. Entamoeba histolytica trophozoites induce a rapid non-classical NETosis mechanism independent of NOX2-derived reactive oxygen species and PAD4 activity. Front. Cell. Infect. Microbiol. 2018, 8, 184. [Google Scholar] [CrossRef]
- Fonseca, Z.; Díaz-Godínez, C.; Mora, N.; Alemán, O.R.; Uribe-Querol, E.; Carrero, J.C.; Rosales, C. Entamoeba histolytica induce signaling via Raf/MEK/ERK for neutrophil extracellular trap (NET) formation. Front. Cell. Infect. Microbiol. 2018, 8, 226. [Google Scholar] [CrossRef]
- Ávila, E.E.; Salaiza, N.; Pulido, J.; Rodríguez, M.C.; Díaz-Godínez, C.; Laclette, J.P.; Becker, I.; Carrero, J.C. Entamoeba histolytica trophozoites and lipopeptidophosphoglycan trigger human neutrophil extracellular traps. PLoS ONE 2016, 11, e0158979. [Google Scholar] [CrossRef]
- Díaz-Godínez, C.; Jorge-Rosas, J.F.; Néquiz, M.; Martínez-Calvillo, S.; Laclette, J.P.; Rosales, C.; Carrero, J.C. New insights on NETosis induced by Entamoeba histolytica: Dependence on ROS from amoebas and extracellular MPO activity. Antioxidants 2021, 10, 974. [Google Scholar] [CrossRef]
- Díaz-Godínez, C.; Ríos-Valencia, D.G.; García-Aguirre, S.; Martínez-Calvillo, S.; Carrero, J.C. Immunomodulatory effect of extracellular vesicles from Entamoeba histolytica trophozoites: Regulation of NETs and respiratory burst during confrontation with human neutrophils. Front. Cell. Infect. Microbiol. 2022, 12, 1018314. [Google Scholar] [CrossRef]
- Sharma, M.; Lozano-Amado, D.; Chowdhury, D.; Singh, U. Extracellular vesicles and their impact on the biology of protozoan parasites. Trop. Med. Infect. Dis. 2023, 8, 448. [Google Scholar] [CrossRef]
- Cruz-Baquero, A.; Cárdenas Jaramillo, L.M.; Gutiérrez-Meza, M.; Jarillo-Luna, R.A.; Campos-Rodríguez, R.; Rivera-Aguilar, V.; Miliar-García, A.; Pacheco-Yepez, J. Different behavior of myeloperoxidase in two rodent amoebic liver abscess models. PLoS ONE 2017, 12, e0182480. [Google Scholar] [CrossRef] [PubMed]
- Contis Montes de Oca, A.; Cruz Baquero, A.; Campos Rodríguez, R.; Cárdenas Jaramillo, L.M.; Aguayo Flores, J.E.; Rojas Hernández, S.; Olivos García, A.; Pacheco Yepez, J. Neutrophil extracellular traps and MPO in models of susceptibility and resistance against Entamoeba histolytica. Parasite Immunol. 2020, 42, e12714. [Google Scholar] [CrossRef] [PubMed]
- Pacheco-Yépez, J.; Rivera-Aguilar, V.; Barbosa-Cabrera, E.; Rojas Hernández, S.; Jarillo-Luna, R.A.; Campos-Rodríguez, R. Myeloperoxidase binds to and kills Entamoeba histolytica trophozoites. Parasite Immunol. 2011, 33, 255–264. [Google Scholar] [CrossRef]
- Sim, S.; Yong, T.S.; Park, S.J.; Im, K.I.; Kong, Y.; Ryu, J.S.; Min, D.Y.; Shin, M.H. NADPH oxidase-derived reactive oxygen species-mediated activation of ERK1/2 is required for apoptosis of human neutrophils induced by Entamoeba histolytica. J. Immunol. 2005, 174, 4279–4288. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Uribe-Querol, E.; Rosales, C. Neutrophils versus Protozoan Parasites: Plasmodium, Trichomonas, Leishmania, Trypanosoma, and Entameoba. Microorganisms 2024, 12, 827. https://doi.org/10.3390/microorganisms12040827
Uribe-Querol E, Rosales C. Neutrophils versus Protozoan Parasites: Plasmodium, Trichomonas, Leishmania, Trypanosoma, and Entameoba. Microorganisms. 2024; 12(4):827. https://doi.org/10.3390/microorganisms12040827
Chicago/Turabian StyleUribe-Querol, Eileen, and Carlos Rosales. 2024. "Neutrophils versus Protozoan Parasites: Plasmodium, Trichomonas, Leishmania, Trypanosoma, and Entameoba" Microorganisms 12, no. 4: 827. https://doi.org/10.3390/microorganisms12040827
APA StyleUribe-Querol, E., & Rosales, C. (2024). Neutrophils versus Protozoan Parasites: Plasmodium, Trichomonas, Leishmania, Trypanosoma, and Entameoba. Microorganisms, 12(4), 827. https://doi.org/10.3390/microorganisms12040827