
The Potential of Co-Evolution and Interactions of Gut Bacteria–Phages in Bamboo-Eating Pandas: Insights from Dietary Preference-Based Metagenomic Analysis



Abstract
1. Introduction
2. Materials and Methods
2.1. Data Collection
2.2. Metagenomic Analyses and Bioinformatics Analysis
2.3. Co-Occurrence Network Analysis
3. Results
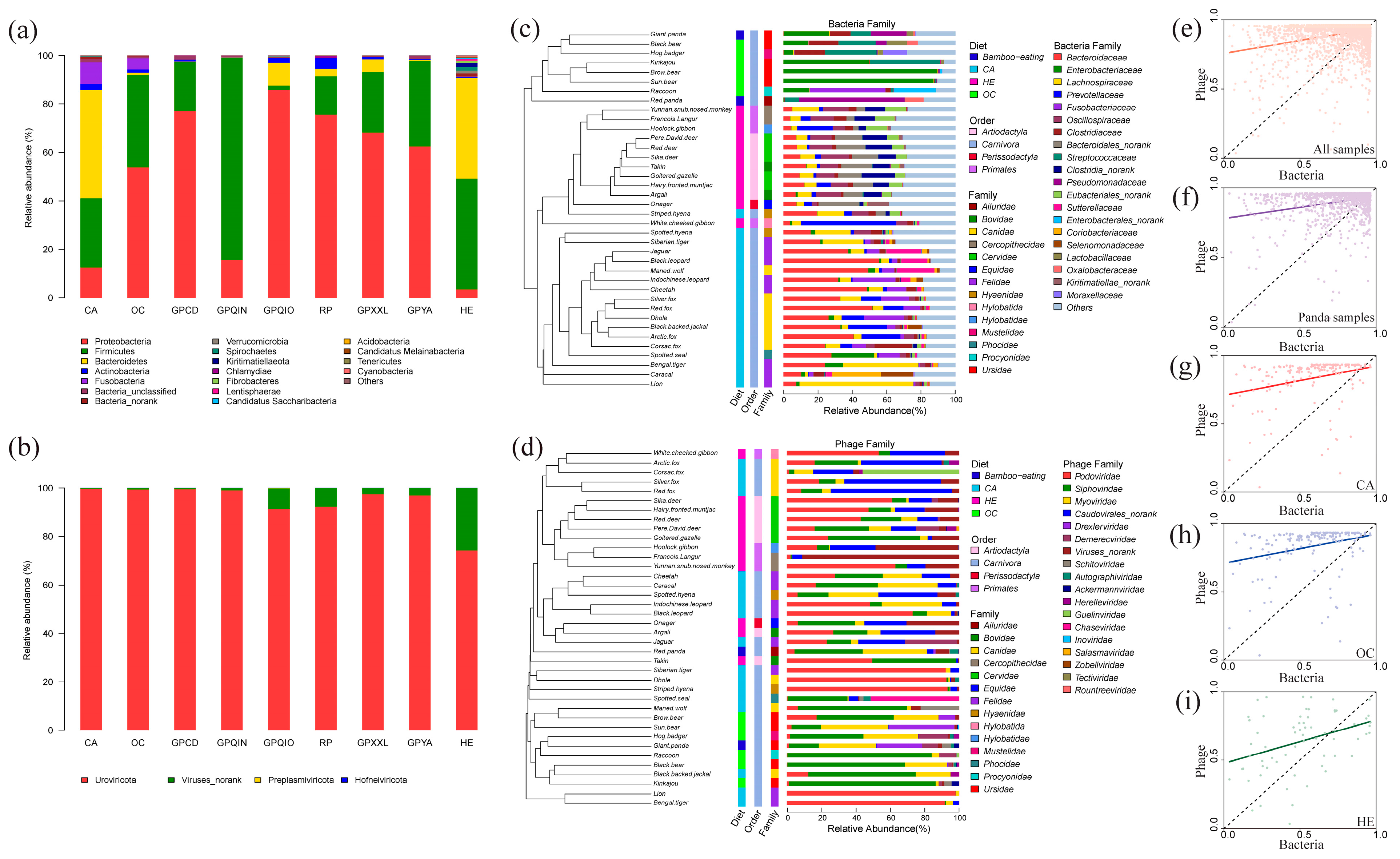
3.1. Diversity Analysis of Gut Bacterial and Phage Communities
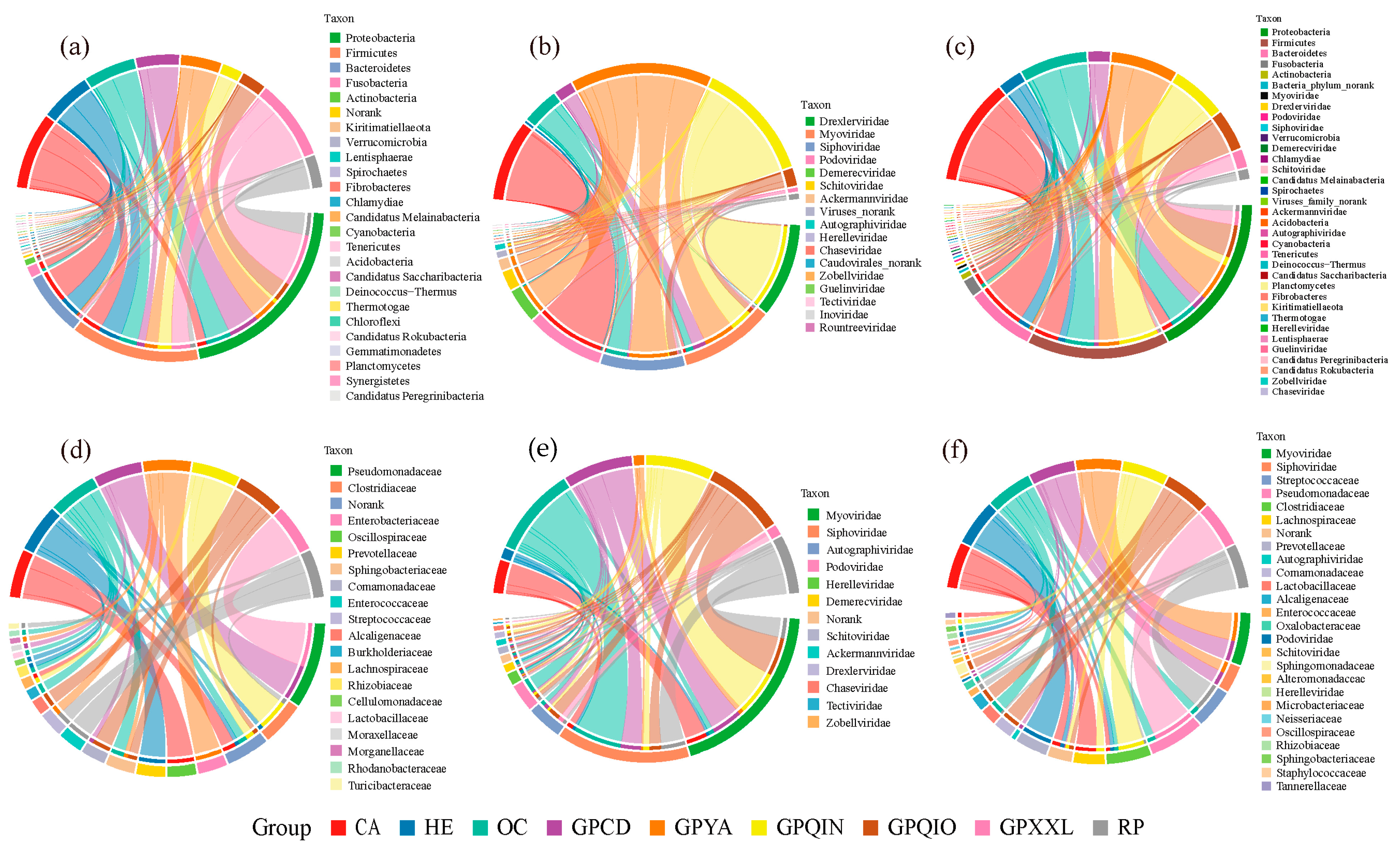
3.2. Compositions of Gut Bacterial and Phage Communities
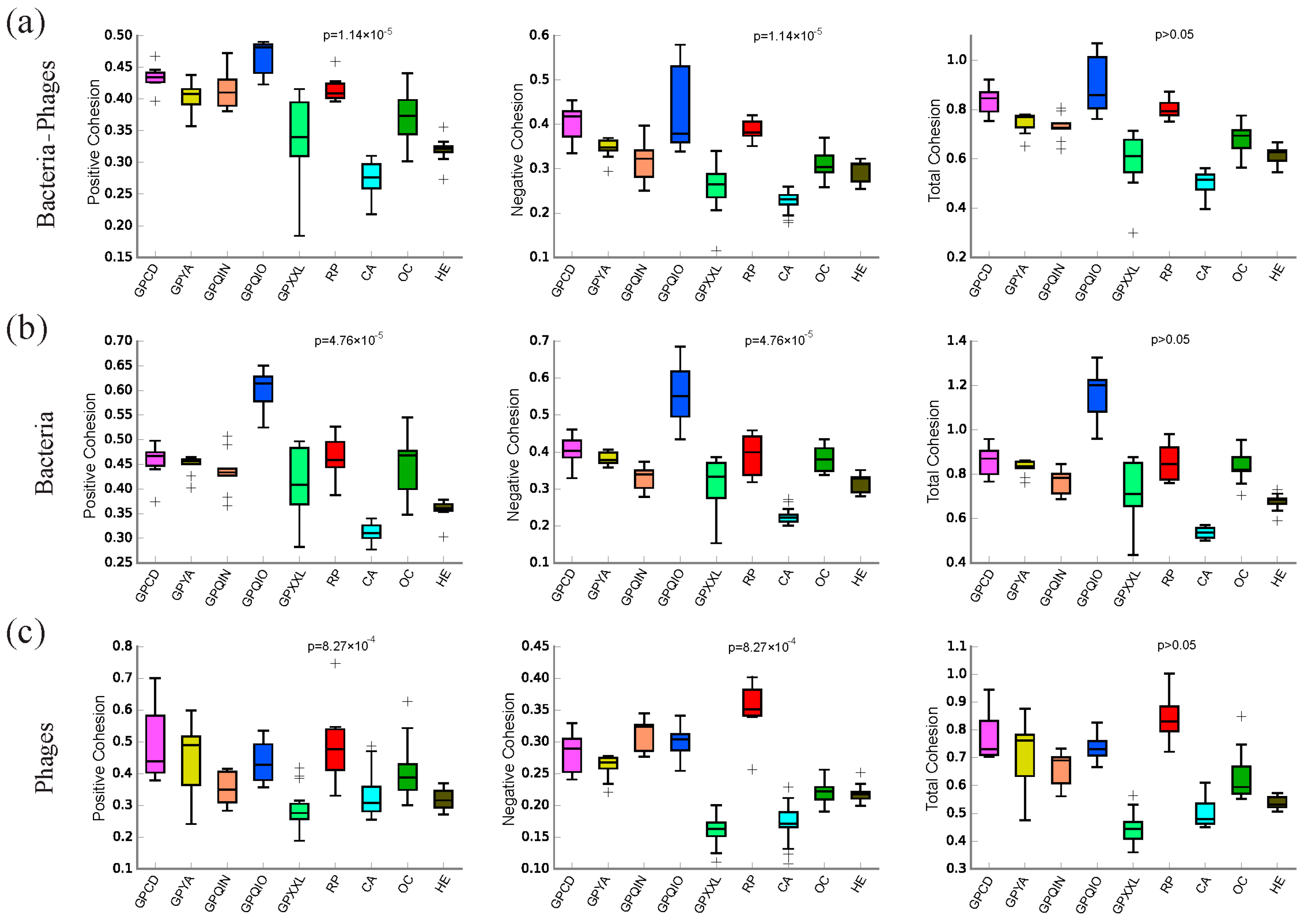
3.3. Co-Occurrence Microbial Networks Analysis of Gut Bacterial–Phage, Bacteria, and Phage Communities
3.4. Topological Features of Bacterial–Phage, Bacterial, and Phage Networks
4. Discussion
4.1. Alpha Diversity and Composition Correlation of Gut Bacterial and Phage Communities in Mammals
4.2. Diet and Phylogeny Influence the Structure of Mammalian Gut Bacterial and Phage Communities
4.3. Interactions between Gut Bacterial and Phage Communities
4.4. Potential Correlations of Gut Phages with Corresponding Bacterial Hosts
4.5. The Uniqueness of the Gut Bacterial and Phage Community in Qinling Giant Pandas
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kurilshikov, A.; Wijmenga, C.; Fu, J.; Zhernakova, A. Host Genetics and Gut Microbiome: Challenges and Perspectives. Trends Immunol. 2017, 38, 633–647. [Google Scholar] [CrossRef]
- Matijašić, M.; Meštrović, T.; Paljetak, H.; Perić, M.; Barešić, A.; Verbanac, D. Gut Microbiota Beyond Bacteria-Mycobiome, Virome, Archaeome, and Eukaryotic Parasites in IBD. Int. J. Mol. Sci. 2020, 21, 2668. [Google Scholar] [CrossRef]
- Dai, Q.; Ding, J.; Cui, X.; Zhu, Y.; Chen, H.; Zhu, L. Beyond Bacteria: Reconstructing Microorganism Connections and Deciphering the Predicted Mutualisms in Mammalian Gut Metagenomes. Ecol. Evol. 2023, 13, e9829. [Google Scholar] [CrossRef]
- Zhu, D.; Lu, L.; Zhang, Z.; Qi, D.; Zhang, M.; O’Connor, P.; Wei, F.; Zhu, Y.G. Insights into the Roles of Fungi and Protist in the Giant Panda Gut Microbiome and Antibiotic Resistome. Environ. Int. 2021, 155, 106703. [Google Scholar] [CrossRef]
- Du Toit, A. Gut phages at the centre. Nat. Rev. Microbiol. 2019, 17, 195. [Google Scholar] [CrossRef]
- Tong, Y.; Gao, H.; Qi, Q.; Liu, X.; Li, J.; Gao, J.; Li, P.; Wang, Y.; Du, L.; Wang, C. High fat diet, gut microbiome and gastrointestinal cancer. Theranostics 2021, 11, 5889–5910. [Google Scholar] [CrossRef]
- Hills, R.D., Jr.; Pontefract, B.A.; Mishcon, H.R.; Black, C.A.; Sutton, S.C.; Theberge, C.R. Gut Microbiome: Profound Implications for Diet and Disease. Nutrients 2019, 11, 1613. [Google Scholar] [CrossRef]
- Strandwitz, P. Neurotransmitter modulation by the gut microbiota. Brain Res. 2018, 1693, 128–133. [Google Scholar] [CrossRef]
- Davis, E.C.; Castagna, V.P.; Sela, D.A.; Hillard, M.A.; Lindberg, S.; Mantis, N.J.; Seppo, A.E.; Järvinen, K.M. Gut microbiome and breast-feeding: Implications for early immune development. J. Allergy Clin. Immunol. 2022, 150, 523–534. [Google Scholar] [CrossRef]
- Maky, M.A.; Ishibashi, N.; Nakayama, J.; Zendo, T. Characterization of the Biosynthetic Gene Cluster of Enterocin F4-9, a Glycosylated Bacteriocin. Microorganisms 2021, 9, 2276. [Google Scholar] [CrossRef]
- Takagi, T.; Inoue, R.; Oshima, A.; Sakazume, H.; Ogawa, K.; Tominaga, T.; Mihara, Y.; Sugaya, T.; Mizushima, K.; Uchiyama, K.; et al. Typing of the Gut Microbiota Community in Japanese Subjects. Microorganisms 2022, 10, 664. [Google Scholar] [CrossRef]
- Kassa, T. Bacteriophages Against Pathogenic Bacteria and Possibilities for Future Application in Africa. Infect. Drug Resist. 2021, 14, 17–31. [Google Scholar] [CrossRef]
- Chevallereau, A.; Pons, B.J.; van Houte, S.; Westra, E.R. Interactions between bacterial and phage communities in natural environments. Nat. Rev. Microbiol. 2022, 20, 49–62. [Google Scholar] [CrossRef]
- Federici, S.; Nobs, S.P.; Elinav, E. Phages and their potential to modulate the microbiome and immunity. Cell. Mol. Immunol. 2021, 18, 889–904. [Google Scholar] [CrossRef]
- Breitbart, M.; Rohwer, F. Here a virus, there a virus, everywhere the same virus? Trends Microbiol. 2005, 13, 278–284. [Google Scholar] [CrossRef]
- Jeong, J.; Mun, S.; Oh, Y.; Cho, C.-S.; Yun, K.; Ahn, Y.; Chung, W.-H.; Lim, M.Y.; Lee, K.E.; Hwang, T.S.; et al. A qRT-PCR Method Capable of Quantifying Specific Microorganisms Compared to NGS-Based Metagenome Profiling Data. Microorganisms 2022, 10, 324. [Google Scholar] [CrossRef]
- Camarillo-Guerrero, L.F.; Almeida, A.; Rangel-Pineros, G.; Finn, R.D.; Lawley, T.D. Massive expansion of human gut bacteriophage diversity. Cell 2021, 184, 1098–1109.e1099. [Google Scholar] [CrossRef]
- Al-Shayeb, B.; Sachdeva, R.; Chen, L.X.; Ward, F.; Munk, P.; Devoto, A.; Castelle, C.J.; Olm, M.R.; Bouma-Gregson, K.; Amano, Y.; et al. Clades of huge phages from across Earth’s ecosystems. Nature 2020, 578, 425–431. [Google Scholar] [CrossRef]
- Gogokhia, L.; Buhrke, K.; Bell, R.; Hoffman, B.; Brown, D.G.; Hanke-Gogokhia, C.; Ajami, N.J.; Wong, M.C.; Ghazaryan, A.; Valentine, J.F.; et al. Expansion of Bacteriophages Is Linked to Aggravated Intestinal Inflammation and Colitis. Cell Host Microbe 2019, 25, 285–299.e288. [Google Scholar] [CrossRef]
- Rohwer, F.; Segall, A.M. A century of phage lessons. Nature 2015, 528, 46–47. [Google Scholar] [CrossRef]
- Wommack, K.E.; Colwell, R.R. Virioplankton: Viruses in aquatic ecosystems. Microbiol. Mol. Biol. Rev. 2000, 64, 69–114. [Google Scholar] [CrossRef] [PubMed]
- Dion, M.B.; Oechslin, F.; Moineau, S. Phage diversity, genomics and phylogeny. Nat. Rev. Microbiol. 2020, 18, 125–138. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Zhang, T.; Yu, M.; Chen, Y.L.; Jin, M. The Life Cycle Transitions of Temperate Phages: Regulating Factors and Potential Ecological Implications. Viruses 2022, 14, 1904. [Google Scholar] [CrossRef] [PubMed]
- Nawel, Z.; Rima, O.; Amira, B. An overview on Vibrio temperate phages: Integration mechanisms, pathogenicity, and lysogeny regulation. Microb. Pathog. 2022, 165, 105490. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Wu, J.; Li, Y.; Zhang, Y.; Cho, W.C.; Ju, X.; van Schothorst, E.M.; Zheng, Y. Gut bacteria formation and influencing factors. FEMS Microbiol. Ecol. 2021, 97, fiab043. [Google Scholar] [CrossRef]
- Youngblut, N.D.; Reischer, G.H.; Walters, W.; Schuster, N.; Walzer, C.; Stalder, G.; Ley, R.E.; Farnleitner, A.H. Host diet and evolutionary history explain different aspects of gut microbiome diversity among vertebrate clades. Nat. Commun. 2019, 10, 2200. [Google Scholar] [CrossRef] [PubMed]
- Mallott, E.K.; Amato, K.R. Host specificity of the gut microbiome. Nat. Rev. Microbiol. 2021, 19, 639–653. [Google Scholar] [CrossRef]
- Ley, R.E.; Hamady, M.; Lozupone, C.; Turnbaugh, P.J.; Ramey, R.R.; Bircher, J.S.; Schlegel, M.L.; Tucker, T.A.; Schrenzel, M.D.; Knight, R.; et al. Evolution of mammals and their gut microbes. Science 2008, 320, 1647–1651. [Google Scholar] [CrossRef]
- Gogarten, J.F.; Rühlemann, M.; Archie, E.; Tung, J.; Akoua-Koffi, C.; Bang, C.; Deschner, T.; Muyembe-Tamfun, J.J.; Robbins, M.M.; Schubert, G.; et al. Primate phageomes are structured by superhost phylogeny and environment. Proc. Natl. Acad. Sci. USA 2021, 118, e2013535118. [Google Scholar] [CrossRef]
- Leigh, B.A.; Bordenstein, S.R.; Brooks, A.W.; Mikaelyan, A.; Bordenstein, S.R. Finer-Scale Phylosymbiosis: Insights from Insect Viromes. mSystems 2018, 3. [Google Scholar] [CrossRef]
- Minot, S.; Sinha, R.; Chen, J.; Li, H.; Keilbaugh, S.A.; Wu, G.D.; Lewis, J.D.; Bushman, F.D. The human gut virome: Inter-individual variation and dynamic response to diet. Genome Res. 2011, 21, 1616–1625. [Google Scholar] [CrossRef]
- Howe, A.; Ringus, D.L.; Williams, R.J.; Choo, Z.N.; Greenwald, S.M.; Owens, S.M.; Coleman, M.L.; Meyer, F.; Chang, E.B. Divergent responses of viral and bacterial communities in the gut microbiome to dietary disturbances in mice. ISME J. 2016, 10, 1217–1227. [Google Scholar] [CrossRef]
- Oh, J.H.; Alexander, L.M.; Pan, M.; Schueler, K.L.; Keller, M.P.; Attie, A.D.; Walter, J.; van Pijkeren, J.P. Dietary Fructose and Microbiota-Derived Short-Chain Fatty Acids Promote Bacteriophage Production in the Gut Symbiont Lactobacillus reuteri. Cell Host Microbe 2019, 25, 273–284.e276. [Google Scholar] [CrossRef] [PubMed]
- Guerin, E.; Hill, C. Shining Light on Human Gut Bacteriophages. Front. Cell. Infect. Microbiol. 2020, 10, 481. [Google Scholar] [CrossRef]
- Cani, P.D. Human gut microbiome: Hopes, threats and promises. Gut 2018, 67, 1716–1725. [Google Scholar] [CrossRef]
- Koskella, B.; Brockhurst, M.A. Bacteria-phage coevolution as a driver of ecological and evolutionary processes in microbial communities. FEMS Microbiol. Rev. 2014, 38, 916–931. [Google Scholar] [CrossRef] [PubMed]
- Marantos, A.; Mitarai, N.; Sneppen, K. From kill the winner to eliminate the winner in open phage-bacteria systems. PLoS Comput. Biol. 2022, 18, e1010400. [Google Scholar] [CrossRef]
- Korytowski, D.A.; Smith, H. Permanence and Stability of a Kill the Winner Model in Marine Ecology. Bull. Math. Biol. 2017, 79, 995–1004. [Google Scholar] [CrossRef]
- Silveira, C.B.; Rohwer, F.L. Piggyback-the-Winner in host-associated microbial communities. NPJ Biofilms Microbiomes 2016, 2, 16010. [Google Scholar] [CrossRef]
- Chen, X.; Weinbauer, M.G.; Jiao, N.; Zhang, R. Revisiting marine lytic and lysogenic virus-host interactions: Kill-the-Winner and Piggyback-the-Winner. Sci. Bull. 2021, 66, 871–874. [Google Scholar] [CrossRef]
- Brüssow, H.; Canchaya, C.; Hardt, W.D. Phages and the evolution of bacterial pathogens: From genomic rearrangements to lysogenic conversion. Microbiol. Mol. Biol. Rev. 2004, 68, 560–602. [Google Scholar] [CrossRef] [PubMed]
- Forde, S.E.; Thompson, J.N.; Holt, R.D.; Bohannan, B.J. Coevolution drives temporal changes in fitness and diversity across environments in a bacteria-bacteriophage interaction. Evol. Int. J. Org. Evol. 2008, 62, 1830–1839. [Google Scholar] [CrossRef]
- Faruque, S.M.; Mekalanos, J.J. Phage-bacterial interactions in the evolution of toxigenic Vibrio cholerae. Virulence 2012, 3, 556–565. [Google Scholar] [CrossRef] [PubMed]
- Boyd, E.F.; Brüssow, H. Common themes among bacteriophage-encoded virulence factors and diversity among the bacteriophages involved. Trends Microbiol. 2002, 10, 521–529. [Google Scholar] [CrossRef]
- Hsu, B.B.; Way, J.C.; Silver, P.A. Stable Neutralization of a Virulence Factor in Bacteria Using Temperate Phage in the Mammalian Gut. mSystems 2020, 5, e00013-20. [Google Scholar] [CrossRef]
- De Sordi, L.; Khanna, V.; Debarbieux, L. The Gut Microbiota Facilitates Drifts in the Genetic Diversity and Infectivity of Bacterial Viruses. Cell Host Microbe 2017, 22, 801–808.e803. [Google Scholar] [CrossRef]
- Scanlan, P.D. Bacteria-Bacteriophage Coevolution in the Human Gut: Implications for Microbial Diversity and Functionality. Trends Microbiol. 2017, 25, 614–623. [Google Scholar] [CrossRef]
- Tao, J.; Meng, D.; Qin, C.; Liu, X.; Liang, Y.; Xiao, Y.; Liu, Z.; Gu, Y.; Li, J.; Yin, H. Integrated network analysis reveals the importance of microbial interactions for maize growth. Appl. Microbiol. Biotechnol. 2018, 102, 3805–3818. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Kato, H.; Kawatsu, K.; Osada, Y.; Azuma, T.; Nagata, Y.; Kondoh, M. Reconstruction of a Soil Microbial Network Induced by Stress Temperature. Microbiol. Spectr. 2022, 10, e0274822. [Google Scholar] [CrossRef]
- Liu, Q.; Jiang, Y. Application of microbial network analysis to discriminate environmental heterogeneity in Fildes Peninsula, Antarctica. Mar. Pollut. Bull. 2020, 156, 111244. [Google Scholar] [CrossRef]
- Wang, Z.; Liu, F.; Li, E.; Yuan, Y.; Yang, Y.; Xu, M.; Qiu, R. Network analysis reveals microbe-mediated impacts of aeration on deep sediment layer microbial communities. Front. Microbiol. 2022, 13, 931585. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Zhang, Q.; Zhang, Q.; Chen, H.; Liu, G.; Zhu, L. The putative maintaining mechanism of gut bacterial ecosystem in giant pandas and its potential application in conservation. Evol. Appl. 2023, 16, 36–47. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.; Wang, H.; Dsouza, M.; Lou, J.; He, Y.; Dai, Z.; Brookes, P.C.; Xu, J.; Gilbert, J.A. Geographic patterns of co-occurrence network topological features for soil microbiota at continental scale in eastern China. ISME J. 2016, 10, 1891–1901. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Jiang, Y.H.; Yang, Y.; He, Z.; Luo, F.; Zhou, J. Molecular ecological network analyses. BMC Bioinform. 2012, 13, 113. [Google Scholar] [CrossRef] [PubMed]
- Centler, F.; Günnigmann, S.; Fetzer, I.; Wendeberg, A. Keystone Species and Modularity in Microbial Hydrocarbon Degradation Uncovered by Network Analysis and Association Rule Mining. Microorganisms 2020, 8, 190. [Google Scholar] [CrossRef] [PubMed]
- Matchado, M.S.; Lauber, M.; Reitmeier, S.; Kacprowski, T.; Baumbach, J.; Haller, D.; List, M. Network analysis methods for studying microbial communities: A mini review. Comput. Struct. Biotechnol. J. 2021, 19, 2687–2698. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.; Xiong, X.; Tan, L.; Deng, Y.; Du, X.; Yang, X.; Hu, Q. Soil microbial community assembly and stability are associated with potato (Solanum tuberosum L.) fitness under continuous cropping regime. Front. Plant Sci. 2022, 13, 1000045. [Google Scholar] [CrossRef]
- Zhao, Y.; Cai, J.; Zhang, P.; Qin, W.; Lou, Y.; Liu, Z.; Hu, B. Core fungal species strengthen microbial cooperation in a food-waste composting process. Environ. Sci. Ecotechnology 2022, 12, 100190. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Gao, X.; Meng, J.; Zhang, A.; Zhou, Y.; Long, M.; Li, B.; Deng, W.; Jin, L.; Zhao, S.; et al. Metagenomic Analysis of Bacteria, Fungi, Bacteriophages, and Helminths in the Gut of Giant Pandas. Front. Microbiol. 2018, 9, 1717. [Google Scholar] [CrossRef]
- Lu, J.; Wang, H.; Wang, C.; Zhao, M.; Hou, R.; Shen, Q.; Yang, S.; Ji, L.; Liu, Y.; Wang, X.; et al. Gut phageome of the giant panda (Ailuropoda melanoleuca) reveals greater diversity than relative species. mSystems 2023, 8, e0016123. [Google Scholar] [CrossRef]
- Guo, M.; Liu, G.; Chen, J.; Ma, J.; Lin, J.; Fu, Y.; Fan, G.; Lee, S.M.; Zhang, L. Dynamics of bacteriophages in gut of giant pandas reveal a potential regulation of dietary intake on bacteriophage composition. Sci. Total Environ. 2020, 734, 139424. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Gallego, J.L.; Chou, S.P.; Di Rienzi, S.C.; Goodrich, J.K.; Spector, T.D.; Bell, J.T.; Youngblut, N.D.; Hewson, I.; Reyes, A.; Ley, R.E. Virome Diversity Correlates with Intestinal Microbiome Diversity in Adult Monozygotic Twins. Cell Host Microbe 2019, 25, 261–272.e265. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Liu, W.; Hou, R.; Zhang, L.; Schmitz-Esser, S.; Sun, H.; Xie, J.; Zhang, Y.; Wang, C.; Li, L.; et al. Age-associated microbiome shows the giant panda lives on hemicelluloses, not on cellulose. ISME J. 2018, 12, 1319–1328. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Mishra, S.; Zhao, J.; Tang, J.; Zeng, B.; Kong, F.; Ning, R.; Li, M.; Zhang, H.; Zeng, Y.; et al. Metagenomic Study Suggests That the Gut Microbiota of the Giant Panda (Ailuropoda melanoleuca) May Not Be Specialized for Fiber Fermentation. Front. Microbiol. 2018, 9, 229. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Wang, X.; Ding, Y.; Hu, Y.; Nie, Y.; Wei, W.; Ma, S.; Yan, L.; Zhu, L.; Wei, F. Seasonal variation in nutrient utilization shapes gut microbiome structure and function in wild giant pandas. Proc. R. Soc. B Biol. Sci. 2017, 284, 20170955. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Yang, Z.; Yao, R.; Xu, L.; Chen, H.; Gu, X.; Wu, T.; Yang, X. Potential Mechanism of Detoxification of Cyanide Compounds by Gut Microbiomes of Bamboo-Eating Pandas. mSphere 2018, 3, e00229-18. [Google Scholar] [CrossRef]
- Zhu, L.; Wu, Q.; Deng, C.; Zhang, M.; Zhang, C.; Chen, H.; Lu, G.; Wei, F. Adaptive evolution to a high purine and fat diet of carnivorans revealed by gut microbiomes and host genomes. Environ. Microbiol. 2018, 20, 1711–1722. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Li, D.; Luo, R.; Liu, C.M.; Leung, C.M.; Ting, H.F.; Sadakane, K.; Yamashita, H.; Lam, T.W. MEGAHIT v1.0: A fast and scalable metagenome assembler driven by advanced methodologies and community practices. Methods 2016, 102, 3–11. [Google Scholar] [CrossRef]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [PubMed]
- Hyatt, D.; LoCascio, P.F.; Hauser, L.J.; Uberbacher, E.C. Gene and translation initiation site prediction in metagenomic sequences. Bioinformatics 2012, 28, 2223–2230. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [PubMed]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Bray, J.R.; Curtis, J.T. An Ordination of the Upland Forest Communities of Southern Wisconsin. Ecol. Monogr. 1957, 27, 325–349. [Google Scholar] [CrossRef]
- Dixon, P. VEGAN, a package of R functions for community ecology. J. Veg. Sci. 2003, 14, 927–930. [Google Scholar] [CrossRef]
- Beals, E.W. Bray-Curtis Ordination: An Effective Strategy for Analysis of Multivariate Ecological Data. In Advances in Ecological Research; MacFadyen, A., Ford, E.D., Eds.; Academic Press: Cambridge, MA, USA, 1984; Volume 14, pp. 1–55. [Google Scholar]
- Kenkel, N.C.; Orloci, L. Applying Metric and Nonmetric Multidimensional Scaling to Ecological Studies: Some New Results. Ecology 1986, 67, 919–928. [Google Scholar] [CrossRef]
- Li, S.; Vogtmann, E.; Graubard, B.I.; Gail, M.H.; Abnet, C.C.; Shi, J. fast.adonis: A computationally efficient non-parametric multivariate analysis of microbiome data for large-scale studies. Bioinform. Adv. 2022, 2, vbac044. [Google Scholar] [CrossRef] [PubMed]
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.; O’Hara, R.B.; Simpson, G.; Solymos, P.; et al. Vegan Community Ecology Package Version 2.5-7 November 2020. 2020. Available online: https://www.researchgate.net/publication/346579465_vegan_community_ecology_package_version_25-7_November_2020 (accessed on 20 February 2024).
- Kembel, S.W.; Cowan, P.D.; Helmus, M.R.; Cornwell, W.K.; Morlon, H.; Ackerly, D.D.; Blomberg, S.P.; Webb, C.O. Picante: R tools for integrating phylogenies and ecology. Bioinformatics 2010, 26, 1463–1464. [Google Scholar] [CrossRef] [PubMed]
- Hu, A.; Ju, F.; Hou, L.; Li, J.; Yang, X.; Wang, H.; Mulla, S.I.; Sun, Q.; Bürgmann, H.; Yu, C.P. Strong impact of anthropogenic contamination on the co-occurrence patterns of a riverine microbial community. Environ. Microbiol. 2017, 19, 4993–5009. [Google Scholar] [CrossRef]
- Xue, Y.; Chen, H.; Yang, J.R.; Liu, M.; Huang, B.; Yang, J. Distinct patterns and processes of abundant and rare eukaryotic plankton communities following a reservoir cyanobacterial bloom. ISME J. 2018, 12, 2263–2277. [Google Scholar] [CrossRef] [PubMed]
- Csárdi, G.; Nepusz, T. The Igraph Software Package for Complex Network Research. 2006. Available online: https://api.semanticscholar.org/CorpusID:16923281 (accessed on 24 February 2024).
- Du, S.; Dini-Andreote, F.; Zhang, N.; Liang, C.; Yao, Z.; Zhang, H.; Zhang, D. Divergent Co-occurrence Patterns and Assembly Processes Structure the Abundant and Rare Bacterial Communities in a Salt Marsh Ecosystem. Appl. Environ. Microbiol. 2020, 86, e00322-20. [Google Scholar] [CrossRef] [PubMed]
- Newman, M.E. Modularity and community structure in networks. Proc. Natl. Acad. Sci. USA 2006, 103, 8577–8582. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Yang, T.; Bao, Y.; He, P.; Yang, K.; Mei, X.; Wei, Z.; Xu, Y.; Shen, Q.; Banerjee, S. Network analysis and subsequent culturing reveal keystone taxa involved in microbial litter decomposition dynamics. Soil Biol. Biochem. 2021, 157, 108230. [Google Scholar] [CrossRef]
- Sun, Y.; Shi, J.; Wang, X.; Ding, C.; Wang, J. Deciphering the Mechanisms Shaping the Plastisphere Microbiota in Soil. mSystems 2022, 7, e0035222. [Google Scholar] [CrossRef] [PubMed]
- Herren, C.M.; McMahon, K.D. Cohesion: A method for quantifying the connectivity of microbial communities. ISME J. 2017, 11, 2426–2438. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, D.J.; David, A.S.; Menges, E.S.; Searcy, C.A.; Afkhami, M.E. Environmental stress destabilizes microbial networks. ISME J. 2021, 15, 1722–1734. [Google Scholar] [CrossRef] [PubMed]
- Shkoporov, A.N.; Hill, C. Bacteriophages of the Human Gut: The “Known Unknown” of the Microbiome. Cell Host Microbe 2019, 25, 195–209. [Google Scholar] [CrossRef] [PubMed]
- Chibani-Chennoufi, S.; Bruttin, A.; Dillmann, M.L.; Brüssow, H. Phage-host interaction: An ecological perspective. J. Bacteriol. 2004, 186, 3677–3686. [Google Scholar] [CrossRef]
- Cornuault, J.K.; Petit, M.A.; Mariadassou, M.; Benevides, L.; Moncaut, E.; Langella, P.; Sokol, H.; De Paepe, M. Phages infecting Faecalibacterium prausnitzii belong to novel viral genera that help to decipher intestinal viromes. Microbiome 2018, 6, 65. [Google Scholar] [CrossRef]
- Shkoporov, A.N.; Clooney, A.G.; Sutton, T.D.S.; Ryan, F.J.; Daly, K.M.; Nolan, J.A.; McDonnell, S.A.; Khokhlova, E.V.; Draper, L.A.; Forde, A.; et al. The Human Gut Virome Is Highly Diverse, Stable, and Individual Specific. Cell Host Microbe 2019, 26, 527–541.e525. [Google Scholar] [CrossRef]
- Sausset, R.; Petit, M.A.; Gaboriau-Routhiau, V.; De Paepe, M. New insights into intestinal phages. Mucosal Immunol. 2020, 13, 205–215. [Google Scholar] [CrossRef]
- De Sordi, L.; Lourenço, M.; Debarbieux, L. The Battle Within: Interactions of Bacteriophages and Bacteria in the Gastrointestinal Tract. Cell Host Microbe 2019, 25, 210–218. [Google Scholar] [CrossRef]
- Frazão, N.; Gordo, I. Ecotype formation and prophage domestication during gut bacterial evolution. BioEssays News Rev. Mol. Cell. Dev. Biol. 2023, 45, e2300063. [Google Scholar] [CrossRef]
- Shaer Tamar, E.; Kishony, R. Multistep diversification in spatiotemporal bacterial-phage coevolution. Nat. Commun. 2022, 13, 7971. [Google Scholar] [CrossRef] [PubMed]
- Shock, T.; Badang, L.; Ferguson, B.; Martinez-Guryn, K. The interplay between diet, gut microbes, and host epigenetics in health and disease. J. Nutr. Biochem. 2021, 95, 108631. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Havulinna, A.S.; Liu, Y.; Jousilahti, P.; Ritchie, S.C.; Tokolyi, A.; Sanders, J.G.; Valsta, L.; Brożyńska, M.; Zhu, Q.; et al. Combined effects of host genetics and diet on human gut microbiota and incident disease in a single population cohort. Nat. Genet. 2022, 54, 134–142. [Google Scholar] [CrossRef]
- Huang, G.; Wang, X.; Hu, Y.; Wu, Q.; Nie, Y.; Dong, J.; Ding, Y.; Yan, L.; Wei, F. Diet drives convergent evolution of gut microbiomes in bamboo-eating species. Sci. China. Life Sci. 2021, 64, 88–95. [Google Scholar] [CrossRef]
- Tarou, L.R.; Williams, J.; Powell, D.M.; Tabet, R.; Allen, M. Behavioral preferences for bamboo in a pair of captive giant pandas (Ailuropoda melanoleuca). Zoo Biol. 2005, 24, 177–183. [Google Scholar] [CrossRef]
- Li, R.; Wei, F.; Geng, T.; Zhu, H.; Lin, H.; Al, E.J.N. The sequence and de novo assembly of the giant panda genome. Nature 2010, 463, 311–317. [Google Scholar]
- Guo, W.; Mishra, S.; Wang, C.; Zhang, H.; Ning, R.; Kong, F.; Zeng, B.; Zhao, J.; Li, Y. Comparative Study of Gut Microbiota in Wild and Captive Giant Pandas (Ailuropoda melanoleuca). Genes 2019, 10, 827. [Google Scholar] [CrossRef]
- Sommer, F.; Ståhlman, M.; Ilkayeva, O.; Arnemo, J.M.; Kindberg, J.; Josefsson, J.; Newgard, C.B.; Fröbert, O.; Bäckhed, F. The Gut Microbiota Modulates Energy Metabolism in the Hibernating Brown Bear Ursus arctos. Cell Rep. 2016, 14, 1655–1661. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Wang, B.; Tan, J.; Zhu, L.; Lou, D.; Cen, X. Comparative analysis of the gut microbiota of black bears in China using high-throughput sequencing. Mol. Genet. Genom. 2017, 292, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Wang, F.; Zhang, H.; Chen, G.; Deng, Y.; Chen, J.; Yang, J.; Ge, T. Enrichment of beneficial rhizosphere microbes in Chinese wheat yellow mosaic virus-resistant cultivars. Appl. Microbiol. Biotechnol. 2021, 105, 9371–9383. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, M.; Eguíluz, V.M.; Duarte, C.M.; Voolstra, C.R. Rare symbionts may contribute to the resilience of coral–algal assemblages. ISME J. 2018, 12, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Wen, D. Archaeal and bacterial communities assembly and co-occurrence networks in subtropical mangrove sediments under Spartina alterniflora invasion. Environ. Microbiome 2021, 16, 10. [Google Scholar] [CrossRef]
- Santolini, M.; Barabási, A.L. Predicting perturbation patterns from the topology of biological networks. Proc. Natl. Acad. Sci. USA 2018, 115, E6375–E6383. [Google Scholar] [CrossRef]
- Konopka, A.; Lindemann, S.; Fredrickson, J. Dynamics in microbial communities: Unraveling mechanisms to identify principles. ISME J. 2015, 9, 1488–1495. [Google Scholar] [CrossRef]
- Shuwen, H.; Kefeng, D. Intestinal phages interact with bacteria and are involved in human diseases. Gut Microbes 2022, 14, 2113717. [Google Scholar] [CrossRef]
- Edwards, R.A.; Vega, A.A.; Norman, H.M.; Ohaeri, M.; Levi, K.; Dinsdale, E.A.; Cinek, O.; Aziz, R.K.; McNair, K.; Barr, J.J.; et al. Global phylogeography and ancient evolution of the widespread human gut virus crAssphage. Nat. Microbiol. 2019, 4, 1727–1736. [Google Scholar] [CrossRef]
- Kirsch, J.M.; Brzozowski, R.S.; Faith, D.; Round, J.L.; Secor, P.R.; Duerkop, B.A. Bacteriophage-Bacteria Interactions in the Gut: From Invertebrates to Mammals. Annu. Rev. Virol. 2021, 8, 95–113. [Google Scholar] [CrossRef]
- Scanlan, P.D.; Bischofberger, A.M.; Hall, A.R. Modification of Escherichia coli-bacteriophage interactions by surfactants and antibiotics in vitro. FEMS Microbiol. Ecol. 2017, 93, fiw211. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Dong, P.; Gao, F.; Huang, L.; Wang, S.; Wang, R.; Yan, M.; Zhang, D. Sucrose addition directionally enhances bacterial community convergence and network stability of the shrimp culture system. NPJ Biofilms Microbiomes 2022, 8, 22. [Google Scholar] [CrossRef] [PubMed]
- Guimerà, R.; Sales-Pardo, M.; Amaral, L.A. Classes of complex networks defined by role-to-role connectivity profiles. Nat. Phys. 2007, 3, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Xiong, W.; Jousset, A.; Guo, S.; Karlsson, I.; Zhao, Q.; Wu, H.; Kowalchuk, G.A.; Shen, Q.; Li, R.; Geisen, S. Soil protist communities form a dynamic hub in the soil microbiome. ISME J. 2018, 12, 634–638. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Schlaeppi, K.; van der Heijden, M.G.A. Keystone taxa as drivers of microbiome structure and functioning. Nat. Rev. Microbiol. 2018, 16, 567–576. [Google Scholar] [CrossRef]
- Guimerà, R.; Nunes Amaral, L.A. Functional cartography of complex metabolic networks. Nature 2005, 433, 895–900. [Google Scholar] [CrossRef]
- Barabási, A.-L. Scale-Free Networks: A Decade and Beyond. Science 2009, 325, 412–413. [Google Scholar] [CrossRef]
- Łusiak-Szelachowska, M.; Weber-Dąbrowska, B.; Jończyk-Matysiak, E.; Wojciechowska, R.; Górski, A. Bacteriophages in the gastrointestinal tract and their implications. Gut Pathog. 2017, 9, 44. [Google Scholar] [CrossRef]
- Zhai, J.; Wang, Y.; Tang, B.; Zheng, S.; He, S.; Zhao, W.; Chen, H.; Lin, J.; Li, F.; Bao, Y.; et al. Comparative analysis of gut DNA viromes in wild and captive Himalayan vultures. Front. Microbiol. 2023, 14, 1120838. [Google Scholar] [CrossRef]
- Chong, R.; Shi, M.; Grueber, C.E.; Holmes, E.C.; Hogg, C.J.; Belov, K.; Barrs, V.R. Fecal Viral Diversity of Captive and Wild Tasmanian Devils Characterized Using Virion-Enriched Metagenomics and Metatranscriptomics. J. Virol. 2019, 93. [Google Scholar] [CrossRef]
- Feng, Y.; Chen, Y.; Liu, S.; Hou, R.; Yan, X.; Geng, Y.; Zhong, Z.; Guo, H.; Ouyang, P.; Zhang, D.; et al. Surveillance Study of Klebsiella pneumoniae in the Giant Panda Revealed High Genetic Diversity and Antibiotic Therapy Challenge. Antibiotics 2022, 11, 473. [Google Scholar] [CrossRef]
- Jin, L.; Huang, Y.; Yang, S.; Wu, D.; Li, C.; Deng, W.; Zhao, K.; He, Y.; Li, B.; Zhang, G.; et al. Diet, habitat environment and lifestyle conversion affect the gut microbiomes of giant pandas. Sci. Total Environ. 2021, 770, 145316. [Google Scholar] [CrossRef] [PubMed]
- Shkoporov, A.N.; Turkington, C.J.; Hill, C. Mutualistic interplay between bacteriophages and bacteria in the human gut. Nat. Rev. Microbiol. 2022, 20, 737–749. [Google Scholar] [CrossRef]
- Hampton, H.G.; Watson, B.N.J.; Fineran, P.C. The arms race between bacteria and their phage foes. Nature 2020, 577, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Waller, A.S.; Yamada, T.; Kristensen, D.M.; Kultima, J.R.; Sunagawa, S.; Koonin, E.V.; Bork, P. Classification and quantification of bacteriophage taxa in human gut metagenomes. ISME J. 2014, 8, 1391–1402. [Google Scholar] [CrossRef] [PubMed]
- Reyes, A.; Wu, M.; McNulty, N.P.; Rohwer, F.L.; Gordon, J.I. Gnotobiotic mouse model of phage-bacterial host dynamics in the human gut. Proc. Natl. Acad. Sci. USA 2013, 110, 20236–20241. [Google Scholar] [CrossRef] [PubMed]
- Serwer, P.; Hayes, S.J.; Zaman, S.; Lieman, K.; Rolando, M.; Hardies, S.C. Improved isolation of undersampled bacteriophages: Finding of distant terminase genes. Virology 2004, 329, 412–424. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Xiang, Y. Structural studies of the nucleus-like assembly of jumbo bacteriophage 201φ2-1. Front. Microbiol. 2023, 14, 1170112. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.A.; Weintraub, S.T.; Hakala, K.; Serwer, P.; Hardies, S.C. Proteome of the large Pseudomonas myovirus 201 phi 2-1: Delineation of proteolytically processed virion proteins. Mol. Cell. Proteom. 2010, 9, 940–951. [Google Scholar] [CrossRef]
- Zhu, L.; Wang, J.; Bahrndorff, S. Editorial: The Wildlife Gut Microbiome and Its Implication for Conservation Biology. Front. Microbiol. 2021, 12, 697499. [Google Scholar] [CrossRef]
- Shkoporov, A.N.; Khokhlova, E.V.; Stephens, N.; Hueston, C.; Seymour, S.; Hryckowian, A.J.; Scholz, D.; Ross, R.P.; Hill, C. Long-term persistence of crAss-like phage crAss001 is associated with phase variation in Bacteroides intestinalis. BMC Biol. 2021, 19, 163. [Google Scholar] [CrossRef] [PubMed]
- Shkoporov, A.N.; Khokhlova, E.V.; Fitzgerald, C.B.; Stockdale, S.R.; Draper, L.A.; Ross, R.P.; Hill, C. ΦCrAss001 represents the most abundant bacteriophage family in the human gut and infects Bacteroides intestinalis. Nat. Commun. 2018, 9, 4781. [Google Scholar] [CrossRef] [PubMed]
- Babenko, V.V.; Millard, A.; Kulikov, E.E.; Spasskaya, N.N.; Letarova, M.A.; Konanov, D.N.; Belalov, I.S.; Letarov, A.V. The ecogenomics of dsDNA bacteriophages in feces of stabled and feral horses. Comput. Struct. Biotechnol. J. 2020, 18, 3457–3467. [Google Scholar] [CrossRef] [PubMed]
- Guerin, E.; Shkoporov, A.; Stockdale, S.R.; Clooney, A.G.; Ryan, F.J.; Sutton, T.D.S.; Draper, L.A.; Gonzalez-Tortuero, E.; Ross, R.P.; Hill, C. Biology and Taxonomy of crAss-like Bacteriophages, the Most Abundant Virus in the Human Gut. Cell Host Microbe 2018, 24, 653–664.e656. [Google Scholar] [CrossRef]
- Yutin, N.; Makarova, K.S.; Gussow, A.B.; Krupovic, M.; Segall, A.; Edwards, R.A.; Koonin, E.V. Discovery of an expansive bacteriophage family that includes the most abundant viruses from the human gut. Nat. Microbiol. 2018, 3, 38–46. [Google Scholar] [CrossRef]
- Sabar, M.A.; Honda, R.; Haramoto, E. CrAssphage as an indicator of human-fecal contamination in water environment and virus reduction in wastewater treatment. Water Res. 2022, 221, 118827. [Google Scholar] [CrossRef] [PubMed]
- Ballesté, E.; Blanch, A.R.; Mendez, J.; Sala-Comorera, L.; Maunula, L.; Monteiro, S.; Farnleitner, A.H.; Tiehm, A.; Jofre, J.; García-Aljaro, C. Bacteriophages Are Good Estimators of Human Viruses Present in Water. Front. Microbiol. 2021, 12, 619495. [Google Scholar] [CrossRef] [PubMed]
- Karkman, A.; Pärnänen, K.; Larsson, D.G.J. Fecal pollution can explain antibiotic resistance gene abundances in anthropogenically impacted environments. Nat. Commun. 2019, 10, 80. [Google Scholar] [CrossRef]
- Malla, B.; Makise, K.; Nakaya, K.; Mochizuki, T.; Yamada, T.; Haramoto, E. Evaluation of Human- and Animal-Specific Viral Markers and Application of CrAssphage, Pepper Mild Mottle Virus, and Tobacco Mosaic Virus as Potential Fecal Pollution Markers to River Water in Japan. Food Environ. Virol. 2019, 11, 446–452. [Google Scholar] [CrossRef]
- Hu, T.; Dai, Q.; Chen, H.; Zhang, Z.; Dai, Q.; Gu, X.; Yang, X.; Yang, Z.; Zhu, L. Geographic pattern of antibiotic resistance genes in the metagenomes of the giant panda. Microb. Biotechnol. 2021, 14, 186–197. [Google Scholar] [CrossRef]
- Zhao, M.; Li, Y.; Wei, W.; Zhang, Z.; Zhou, H. The distribution variation of pathogens and virulence factors in different geographical populations of giant pandas. Front. Microbiol. 2023, 14, 1264786. [Google Scholar] [CrossRef]
- Huang, G.; Qi, D.; Yang, Z.; Hou, R.; Shi, W.; Zhao, F.; Li, Z.; Yan, L.; Wei, F. Gut microbiome as a key monitoring indicator for reintroductions of captive animals. Conserv. Biol. 2023, 38, e14173. [Google Scholar] [CrossRef] [PubMed]
- Carr, A.; Diener, C.; Baliga, N.S.; Gibbons, S.M. Use and abuse of correlation analyses in microbial ecology. ISME J. 2019, 13, 2647–2655. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Ding, C.; Li, X.; Su, D.; He, J. Biotic interactions contribute more than environmental factors and geographic distance to biogeographic patterns of soil prokaryotic and fungal communities. Front. Microbiol. 2023, 14, 1134440. [Google Scholar] [CrossRef] [PubMed]
- Pinto, S.; Benincà, E.; van Nes, E.H.; Scheffer, M.; Bogaards, J.A. Species abundance correlations carry limited information about microbial network interactions. PLoS Comput. Biol. 2022, 18, e1010491. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.J.; Diener, C.; Wilson, J.; Valenzuela, J.J.; Baliga, N.S.; Gibbons, S.M. Growth phase estimation for abundant bacterial populations sampled longitudinally from human stool metagenomes. Nat. Commun. 2023, 14, 5682. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, H.; Shen, W.; Feng, K.; Li, S.; Gu, S.; Zhou, Y.; Peng, X.; Du, X.; He, Q.; et al. The Stability of Phyto-Zooplanktonic Networks Varied with Zooplanktonic Sizes in Chinese Coastal Ecosystem. mSystems 2022, 7, e0082122. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Type | Df | F | R2 | p |
|---|---|---|---|---|---|
| Bacteria | Diet | 3 | 9.4758 | 0.45537 | 0.001 |
| Family | 3 | 4.6384 | 0.7153 | 0.001 | |
| Order | 3 | 4.7941 | 0.29726 | 0.001 | |
| Phages | Diet | 3 | 2.9126 | 0.16211 | 0.001 |
| Family | 3 | 1.4877 | 0.44625 | 0.001 | |
| Order | 3 | 1.9018 | 0.14369 | 0.001 |
| Network | Group | Node | Edge | Positive Correlation | Negative Correlation | Positive Correlation Rate | Negative Correlation Rate | Average Degree | Modularity Value |
|---|---|---|---|---|---|---|---|---|---|
| Bacteria | CA | 859 | 8135 | 7880 | 255 | 96.87% | 3.13% | 18.94 | 0.627 |
| HE | 717 | 1846 | 1634 | 212 | 88.52% | 11.48% | 27.8 | 0.766 | |
| OC | 861 | 7850 | 7459 | 391 | 95.02% | 4.98% | 103.92 | 0.71 | |
| GPCD | 983 | 16,533 | 13,698 | 2835 | 82.85% | 17.15% | 83.25 | 0.708 | |
| GPYA | 656 | 2402 | 2292 | 110 | 95.42% | 4.58% | 47.02 | 0.843 | |
| GPQIN | 373 | 522 | 518 | 4 | 99.23% | 0.77% | 111.91 | 0.925 | |
| GPQIO | 646 | 6135 | 5461 | 674 | 89.01% | 10.99% | 471.74 | 0.733 | |
| GPXXL | 797 | 9882 | 9882 | 0 | 100.00% | 0.00% | 73.41 | 0.676 | |
| RP | 963 | 7041 | 5394 | 1647 | 76.61% | 23.39% | 83.39 | 0.857 | |
| Phages | CA | 273 | 1699 | 1699 | 0 | 100.00% | 0.00% | 12.45 | 0.845 |
| HE | 98 | 200 | 200 | 0 | 100.00% | 0.00% | 4.74 | 0.88 | |
| OC | 615 | 10,966 | 10,966 | 0 | 100.00% | 0.00% | 43.31 | 0.76 | |
| GPCD | 291 | 5534 | 5532 | 2 | 99.96% | 0.04% | 38.91 | 0.715 | |
| GPYA | 435 | 6167 | 6167 | 0 | 100.00% | 0.00% | 60.09 | 0.695 | |
| GPQIN | 282 | 3487 | 3487 | 0 | 100.00% | 0.00% | 28.12 | 0.524 | |
| GPQIO | 373 | 6550 | 6550 | 0 | 100.00% | 0.00% | 40.23 | 0.653 | |
| GPXXL | 386 | 9544 | 9544 | 0 | 100.00% | 0.00% | 50.46 | 0.248 | |
| RP | 245 | 4141 | 4138 | 3 | 99.93% | 0.07% | 35.16 | 0.667 | |
| Bacteria–Phages | CA | 846 | 2598 | 1210 | 1388 | 46.57% | 53.43% | 134.61 | 0.52 |
| HE | 284 | 353 | 316 | 37 | 89.52% | 10.48% | 28.4 | 0.772 | |
| OC | 1146 | 4181 | 3434 | 747 | 82.13% | 17.87% | 83.8 | 0.606 | |
| GPCD | 864 | 8406 | 8161 | 245 | 97.09% | 2.91% | 59.63 | 0.516 | |
| GPYA | 1008 | 9423 | 8331 | 1093 | 88.40% | 11.60% | 70.33 | 0.463 | |
| GPQIN | 967 | 3054 | 2085 | 969 | 68.27% | 31.73% | 67.41 | 0.667 | |
| GPQIO | 840 | 2104 | 527 | 1577 | 25.05% | 74.95% | 204.07 | 0.553 | |
| GPXXL | 177 | 173 | 173 | 0 | 100.00% | 0.00% | 40.19 | 0.867 | |
| RP | 418 | 1515 | 1335 | 180 | 88.12% | 11.88% | 23.78 | 0.585 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, M.; Zhou, Y.; Cui, X.; Zhu, L. The Potential of Co-Evolution and Interactions of Gut Bacteria–Phages in Bamboo-Eating Pandas: Insights from Dietary Preference-Based Metagenomic Analysis. Microorganisms 2024, 12, 713. https://doi.org/10.3390/microorganisms12040713
Zhang M, Zhou Y, Cui X, Zhu L. The Potential of Co-Evolution and Interactions of Gut Bacteria–Phages in Bamboo-Eating Pandas: Insights from Dietary Preference-Based Metagenomic Analysis. Microorganisms. 2024; 12(4):713. https://doi.org/10.3390/microorganisms12040713
Chicago/Turabian StyleZhang, Mingyue, Yanan Zhou, Xinyuan Cui, and Lifeng Zhu. 2024. "The Potential of Co-Evolution and Interactions of Gut Bacteria–Phages in Bamboo-Eating Pandas: Insights from Dietary Preference-Based Metagenomic Analysis" Microorganisms 12, no. 4: 713. https://doi.org/10.3390/microorganisms12040713
APA StyleZhang, M., Zhou, Y., Cui, X., & Zhu, L. (2024). The Potential of Co-Evolution and Interactions of Gut Bacteria–Phages in Bamboo-Eating Pandas: Insights from Dietary Preference-Based Metagenomic Analysis. Microorganisms, 12(4), 713. https://doi.org/10.3390/microorganisms12040713