Abstract
Shiga toxin-producing Escherichia coli are zoonotic pathogens that cause food-borne human disease. Among these, the O157:H7 serotype has evolved from an enteropathogenic O55:H7 ancestor through the displacement of the somatic gene cluster and recurrent toxigenic conversion by Shiga toxin-converting bacteriophages. However, atypical strains that lack the Shiga toxin, the characteristic virulence hallmark, are circulating in this lineage. For this study, we analyzed the pathogenome and virulence inventories of the stx+ strain, TT12A, isolated from a patient with hemorrhagic colitis, and its respective co-isolated stx− strain, TT12B. Sequencing the genomes to closure proved critical to the cataloguing of subtle strain differentiating sequence and structural polymorphisms at a high-level of phylogenetic accuracy and resolution. Phylogenomic profiling revealed SNP and MLST profiles similar to the near clonal outbreak isolates. Their prophage inventories, however, were notably different. The attenuated atypical non-shigatoxigenic status of TT12B is explained by the absence of both the ΦStx1a- and ΦStx2a-prophages carried by TT12A, and we also recorded further alterations in the non-Stx prophage complement. Phenotypic characterization indicated that culture growth was directly impacted by the strains’ distinct lytic phage complement. Altogether, our phylogenomic and phenotypic analyses show that these intimately related isogenic strains are on divergent Stx(+/stx−) evolutionary paths.
1. Introduction
Shiga toxin-producing Escherichia coli (STEC) are causative agents of severe foodborne human disease [1]. Among these, the O157:H7 lineage has emerged as one of the globally predominant pathogenic serotypes [2,3,4,5,6,7,8]. Infections with STEC O157:H7 can cause hemorrhagic colitis (HC) [9,10,11], which may lead to life-threatening complications such as the hemolytic uremic syndrome (HUS), ultimately resulting in renal failure [12,13,14,15,16].
Notable virulence determinants in STEC are the Shiga toxins [17], the lineage-specific pO157 virulence plasmid [18], and the locus of enterocyte effacement (LEE) [19,20,21], responsible for the characteristic attaching and effacing lesions [22,23]. The LEE pathogenicity island contains type III secretion system (T3SS) along with various LEE and non-LEE effectors [22,24,25]. The defining virulence hallmark is the production of the phage-borne Shiga toxin, a potent protein synthesis inhibitor [26,27], which is sufficient to cause disease [28]. This toxin is cytopathic for eukaryotic cells and specifically toxigenic to renal cells [29,30,31,32,33,34,35]. Single or multiple ΦStx-prophages can be carried, with the toxin expressed and produced after phage mobilization during the phage lytic cycle [31,36,37,38,39,40]. Various suballeles of the major stx alleles 1 and 2 have been described [41,42,43,44]. Among these, Stx2a is highly cytopathic, with up to 400x increased toxicity in mice as compared to Stx1a [11,28,45,46,47,48,49].
Serotype O157:H7 evolved from an stx-negative enteropathogenic E. coli (EPEC) O55:H7 progenitor through the toxigenic conversion of strains by Stx-phages [50,51,52,53,54,55,56,57,58,59,60,61]. However, atypical non-shigatoxigenic strains have been described in diverse STEC serotypes [52,54,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76,77]. Complex genomic alterations can result in the disruption, confinement, or complete loss of the stx locus or entire ΦStx-prophages [68]. Such alterations may occur during routine culturing or intentionally in the laboratory through the addition of phage-mobilizing agents to the growth media [69,70,74,75,77,78].
This study analyzes the clinical O157:H7 isolate, TT12, which originated from a patient presenting with hemorrhagic colitis. When grown on selective media, the original study found that the isolates exhibited two distinct colony morphologies, designated as TT12A and TT12B [62]. Subsequent stx PCR-interrogation indicated that TT12A and TT12B were Stx(+) and Stx(−), respectively. Further molecular analyses suggested that these strains were isogenic, but the results were not definitive, and not informed by genome sequences. For this study, we investigated the isolates’ presumed isogenic status from a whole genome perspective making use of high-resolution comparative genomics techniques. The generation of high-quality closed genomes provided the basis for in-depth phylogenomic comparisons and allowed us to catalogue subtle strain-differentiating sequence and structural polymorphisms, which explain the atypical, non-shigatoxigenic status of strain TT12B.
2. Materials and Methods
2.1. Bacterial Strains Analyzed in This Study
Strain-associated metadata for TT12A and TT12B, along with other O157:H7 strains investigated in this study, can be found in Supplemental Table S1. The genome of TT12A was sequenced to closure in this study, while the co-isolated TT12B genome was previously sequenced by our group [68].
2.2. Genome Sequencing, Assembly, and Annotation
TT12A strain was cultured overnight at 37 °C with shaking at 220 rpm in lysogeny broth (LB) (Thermo Fisher Scientific, Asheville, NC, USA). The culture was then diluted to an OD600 of 0.03 in fresh LB medium and grown at 37 °C with shaking at 220 rpm to mid-log phase (OD600~0.5). Total genomic DNA (gDNA) was extracted using the QIAamp DNA Mini Kit (Qiagen, Inc., Valencia, CA, USA) according to the manufacturer’s instructions. Genomic DNA preparation was subjected to both long-read (Pacific Biosciences, Menlo Park, CA, USA) and short-read (Illumina, San Diego, CA, USA) sequencing. For long-read sequencing on the PacBio RS II platform, gDNA was sheared into 20 kb fragments using g-TUBE (Covaris, Inc., Woburn, MA, USA). The library was prepared based on the 20 kb PacBio sample preparation protocol and sequenced using P6/C4 chemistry on four single-molecule real-time (SMRT) cells with a 240 min collection time. The continuous long-read data were de novo assembled using the PacBio hierarchical genome assembly process (HGAP v.3.0) with the default parameters in SMRT Analysis (v.2.3.0), including consensus polishing with Quiver [79]. Long-reads were complemented with Illumina short-reads generated on the MiSeq platform. Paired-end libraries were prepared with the NxSeq AmpFREE Low DNA Library Kit (Lucigen, Middleton, WI, USA) with a 250 bp read length and sequenced using the MiSeq Reagent kit (v2) (500-cycle). Sequencing reads in the fastq format were imported into Galaxy [80], and the default software parameters were used for all analysis unless specified otherwise. FastQC (v.0.74 + Galaxy0) (http://www.bioinformatics.babraham.ac.uk/projects/fastqc, accessed on 10 December 2023) and Trim Galore (https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/, accessed on 10 December 2023) were used to determine read quality. Illumina reads were utilized for PacBio sequence error correction using Pilon (v.1.23) [81], and read-based SNP discovery as described below. The resulting contigs were evaluated with QUAST (v.5.2.0 + Galaxy1) [82]. The chromosomal oriC (http://tubic.tju.edu.cn/Ori-Finder/, accessed on 10 December 2023) [83] and plasmid repA genes were designated as the zero point of the closed molecules, prior to annotation, using the NCBI Prokaryotic Genome Annotation Pipeline (PGAP) [84].
2.3. Pathogenome Make-Up and Virulence Complement
Chromosomes and plasmids of the strains, TT12A and TT12B, were comprehensively analyzed and visualized in Blast Ring Image Generator BRIG (v.0.95) [85]. Serotypes in the assembled genomes were confirmed in silico using the EcOH database [86] in ABRicate (Galaxy v.1.0.1); (https://github.com/tseemann/ABRicate, accessed on 10 December 2023) with the options –minid 80 –mincov 80 in Galaxy [80]. The average nucleotide identities (ANI) for the chromosomes and pO157 plasmids using the E. coli strain TT12A as designated reference were calculated with FastANI (Galaxy v.1.3), based on MinHash mapping [87]. Clade typing was performed according to [88]. Clades and subgroups were assigned by in silico interrogation of the allelic status of 89 core genome-single nucleotide polymorphisms (cgSNPs) in the assembled genomes using a custom workflow on Galaxy [80], which was informed by eight definitive polymorphic positions [89,90]. Lineages were assigned according to published protocols [91]. Chromosomal repeats were identified with FindRepeats (v.1.8.2 + Galaxy1) [92,93]. Virulence and antibiotic resistance genes were identified using VFDB [94] and ResFinder (https://cge.cbs.dtu.dk/services/ResFinder/, accessed on 10 December 2023) [95], respectively. Prophage regions including intact, partial, or remnant prophages were identified using PHASTER [96,97]. The mechanism of phage insertion can create direct repeats (DR), hence insertion sites were investigated for DR and attachments sites (att) using NUCmer (v.4.0.0rc1 + Galaxy2) [98] and BLASTn [99]. Prophages of interest were manually curated with BLASTn/p against the non-redundant NCBI databases [99] and visualized in Easyfig (v.2.2.2) [100]. Insertion sequence (IS) elements were identified and curated using ISEScan (v.1.7.2.3 + Galaxy0) [101]. Genomic islands (GI) were detected with IslandViewer4 [102,103,104]. Plasmid incompatibility groups were identified and analyzed with MOB-Typer (v.3.0.3 + Galaxy0) [105].
2.4. Core Genome SNP Phylogeny
To place strains TT12A and TT12B into their phylogenomic context, a custom-built cgSNP discovery pipeline [68,106,107], implemented on Galaxy [80], was applied. The chromosomal core genome is defined as a set of genic and intragenic regions that are not repeated and do not contain mobile elements, such as phages, genomic islands, IS elements, or plasmids, which evolve at different rates and are not indicative of evolutionary relationships. These regions were determined in the designated reference chromosome of E. coli strain EC4115 (GenBank accession: CP001164) [106] as described above. All mobile genetic elements were excluded from SNP discovery. Illumina reads were used for read-based SNP discovery. The modular pipeline contained the following workflow steps: (i) SNP discovery and typing Illumina reads were used for read-based SNP discovery and aligned to the designated reference with BWA–MEM (Galaxy v.0.7.17.2) [108]. The resulting alignments were processed with FreeBayes (Galaxy v.0.4.1.0) [109] with the following threshold settings: mapping quality 30, base quality 30, coverage 10, and allelic frequency 0.75. For contig-based discovery, assemblies were aligned to the O157:H7 strain EC4115 reference molecules using NUCmer [98], followed by SNP prediction with delta-filter (v.4.0.0rc1 + Galaxy2) and show-snps distributed with the MUMmer package (v.4.0.0rc1 + Galaxy2) [98,110]. The resulting SNP panel for each of the query genomes was used for further processing; (ii) SNP validation and filtering Catalogued SNPs from each genome were merged into a single SNP panel and SNPs located within the identified excluded regions were removed, as well as low-quality alignments or misalignments, non-uniformly distributed regions, and variant insertions and deletions (InDels), as previously described [106,107,111]. SNPs were further curated by extracting the surrounding 40 nucleotides (nt) for each predicted SNP in the reference genome, followed by BLASTn of these fragments against the query genomes. SNPs with missing information (“no hits”) or multiple hits were filtered out as well as ambiguous nucleotides; (iii) SNP annotation and chromosomal distribution The functional effects of SNPs were inferred from the reference genome annotation. Identified SNPs were classified into genic or intergenic by mapping the SNPs to the reference genome. The SNP-matrix tables were manipulated with Query Tabular Tool (Galaxy v.3.3.0] [112]; (iv) SNP phylogeny The curated panel of high-quality SNPs served as a basis for phylogenetic reconstruction by maximum parsimony with PAUP (v.4.0) [113] with 100,000 bootstrap replicates. The majority-rule consensus SNP tree was visualized in Geneious (v.2022.2) [114] and decorated in iTol (v.6.5.8) [115]. Calculation of the consistency index (CI) in Mesquite (v.3.6) [116] for each SNP allowed us to identify parsimony-informative SNPs and flag homoplastic SNPs as previously described [68,106,107,111,117,118]. Locally collinear blocks between TT12A and TT12B chromosomes and plasmids were identified with progressive Mauve (v.2.4.1) [119] with the default settings in Geneious (v.2022.2) [114]. Subtle sequence disambiguities comprising SNPs and InDels between the respective molecules were identified using “Find Variations/SNPs” in Geneious. A prediction of protein stability changes for single-site mutations was analyzed with MuPro (v.1.0) [120].
2.5. Core Genome MLST
The closed genomes of the representative O157:H7 strains (Supplemental Table S1) were imported into SeqSphere+ (v.8.3) (Ridom GmbH, Münster, Germany) for gene-by-gene alignment, allele calling, and comparison [121]. A core genome MLST (cgMLST) schema was developed using the closed chromosome of E. coli K-12 substrain, MG1655, (GenBank accession U00096) [122] as a seed and queried against 11 closed genomes representing the 9 distinct O157:H7 phylogenetic clades [68,88,106,107]. Core and accessory MLST targets were identified according to the inclusion/exclusion criteria of the SeqSphere+ Target Definer. The allele information from the defined core genome gene of the queried strains was used to establish phylogenetic hypotheses using the minimum-spanning method [123,124] with default settings in Ridom SeqSphere+ (v.8.3).
2.6. Bacterial Growth, Phage Induction, and Cell Viability
All experiments were executed with two biological replicates. TT12A and TT12B strains were cultured overnight (o/n) at 37 °C with shaking (220 rpm) in LB medium. Bacterial o/n cultures were diluted to an OD600 of 0.03 in fresh LB medium and grown at 37 °C with shaking (220 rpm) to early-log phase (OD600~0.3) and then divided into two subcultures, LB and LB + Mitomycin C (MMC). Triggering the RecA-dependent SOS response with MMC constitutes a major pathway of Stx phage induction and mobilization [125]. Subculture LB + MMC was supplemented with MMC (Sigma-Aldrich, Saint Louis, MO, USA) at a final concentration of 0.5 μg/mL to mobilize carried prophages, while subculture LB was used to evaluate spontaneous prophage mobilization. Growth curves were recorded in a 96-well plate (Corning 3370, Corning Inc., Corning, NY, USA) at OD600 on a BioTek Synergy H1 plate reader (BioTek Instruments, Inc., Winooski, VT, USA) for 16 hrs at 10 min intervals to assess prophage-induced bacterial lysis.
2.7. Prophage Profiling and Gene Expressions
PCR primer sequences, conditions, and amplicon lengths are provided in Supplemental Table S2. PCR was performed on gDNA preparations using the boiling extraction method [126] for strains TT12A and TT122B, processing three cultures each for the characteristic TT12A- and TT12B-specific morphology [62]. To determine the orientation of an inversion in the shared Enterobacteria phage SfI-PP2, primers were designed with the NCBI primer design tool [127]. PCR-amplicons were separated on a 1.5% agarose gel at 120V and examined in the GelDoc EZ Gel Imaging System and ImageLab (v.6.1) (BioRad, Hercules, CA, USA).
2.8. Mobilization of TT12A-Specific Prophages
To assess TT12A-specific phage mobilization upon MMC-induction, LB and LB + MMC subcultures were grown for 6 hrs at 37 °C with shaking (220 rpm) and then centrifuged at 5000× g for 10 min. Supernatants were filtered through low-protein-binding 0.22 μm pore size membrane filters (Millex-GP; Merck Millipore Ltd., Burlington, MA, USA) and treated with DNase I (Invitrogen, Waltham, MA, USA) for 15 min to remove bacterial gDNA. Phage DNA was extracted from the lysate using the QIAamp DNA Mini Kit (Qiagen Inc., Valencia, CA, USA), and eluted with 50 μL nuclease-free water. Phage mobilization was determined by qPCR targeting the phage-borne toxin genes stx1 and stx2 as well as ΦPP10-carried nleL gene on the StepOne Real-Time PCR System software (v 2.3) (Applied Biosystems, Foster City, CA, USA). Statistical significance was determined using Prism (v.9.5.0) (GraphPad Software, San Diego, CA, USA) with two-way ANOVA with Sidak’s multiple comparisons test to compare non-induced to MMC-induced conditions.
2.9. Gene Expressions
Transcripts were quantified relative to the endogenous tufA gene by RT-qPCR [128]. Cultures were grown in LB and LB + MMC for 6 hrs at 37 °C with shaking (220 rpm) then centrifuged (5000× g, 10 min). Cell pellets were used for total RNA purification using the PureLink RNA Mini kit (Invitrogen, Waltham, MA, USA). RNA quantity and quality were measured with the NanoDrop ND-1000 Spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA). Total RNA was treated with amplification grade DNase I (Invitrogen, Waltham, MA, USA), and reverse transcribed using the RevertAid H Minus First Strand cDNA Synthesis Kit (Thermo Fisher Scientific, Waltham, MA, USA). Targets for qPCR were the toxin genes stx1 and stx2 and the SOS-regulator recA. PCR reactions were performed on the StepOne Real-Time PCR System (Applied Biosystems, Foster City, CA, USA) using GoTaq qPCR Master Mix (Promega, Madison, WI, USA). Primer sequences, RT-qPCR conditions, and amplicon lengths are provided in Supplemental Table S2. Statistical significance was determined using Prism (v.9.5.0) (GraphPad Software, San Diego, CA, USA) with two-way ANOVA with Sidak’s multiple comparisons test to compare results of non-induced to MMC-induced conditions for each strain.
3. Results and Discussion
3.1. Pathogenome Architectures and Mobilome Inventories of TT12A and TT12B
The proposed isogenic status for the O157:H7 isolates TT12A (stx+) and TT12B (stx−), was originally inferred from the PCR-based interrogation of the stx locus, along with a similar PFGE fragmentation pattern [62]. To reassess the isolates’ relationship, their closed genomes were subjected to high-resolution whole genome sequence typing (WGST) [107,129]. Due to the homogeneity of prophage content and other repeats, E. coli O157:H7 genomes are known to assemble into fragmented drafts when only short-read sequencing technologies are applied [130,131]. In response, we used PacBioRS long-read sequencing followed by error correction with Illumina short-reads. The resulting high-quality genomes allowed us to catalogue strain-differentiating sequence and structural polymorphisms at a high degree of phylogenetic resolution. Strain-associated metadata and genome statistics for the chromosomes and carried lineage-specific pO157 virulence plasmids are provided in Supplemental Table S1. Major drivers of the pathogenome diversification in the O157:H7 lineage are mobile genome elements and more prominently, the individual prophage contents [106,107,132,133,134,135,136]. As evident in the chromosome comparison in Figure 1, the TT12A and TT12B backbones are largely conserved and syntenic with an average nucleotide identity (ANI) of 99.99%. In comparison to non-pathogenic E. coli K12-type strains, O157:H7 acquired ΦStx and non-Stx prophages resulting in widespread genetic mosaicism [132,137,138,139]. Bacteriophages target conserved chromosomal loci and undergo evolutionary acquisition, loss, and dissemination, which collectively shape a strain’s individual gene inventory and pathogenicity traits [140,141,142]. A substantial proportion of the TT12A and TT12B chromosomes is made up of prophages, accounting for 16% and 13.1%, respectively, of the total chromosomal sequence information. 21 phages are shared; however, strain TT12A (5,501,009 bp) is distinguished from strain TT12B (5,335,866 bp) by the presence of three additional phages, ΦStx1a, ΦStx2a, and non-Stx Enterobacteria phage ΦBP-4795 prophage. The latter is partly homologous to the 57,930 bp ΦStx1-prophage carried by the E. coli O84:H4 strain with an average nucleotide identity of 45.3% [143]. The direct evolutionary relationship of these Stx/non-Stx phages is unknown; however, alterations of the stx locus, resulting in confined loss or deletion of the entire phage, has been described in diverse STEC lineages [68]. The absence of these three prophages, ΦStx1a, ΦStx2a, and non-Stx Enterobacter phage ΦBP-4795, in the strain TT12B results in a 165,143 bp larger genome of strain TT12A (Figure 1, Supplemental Table S3). The three strain-differentiating prophages along with an inversion in the shared ΦPP2 are visualized in their chromosomal context in Figure 2. In analogy to the chromosomes, the carried pO157 plasmids have an ANI of 99.98%, and differ in size by only 3 bp, again indicative of a close phylogenetic relationship of these strains (Figure 3).
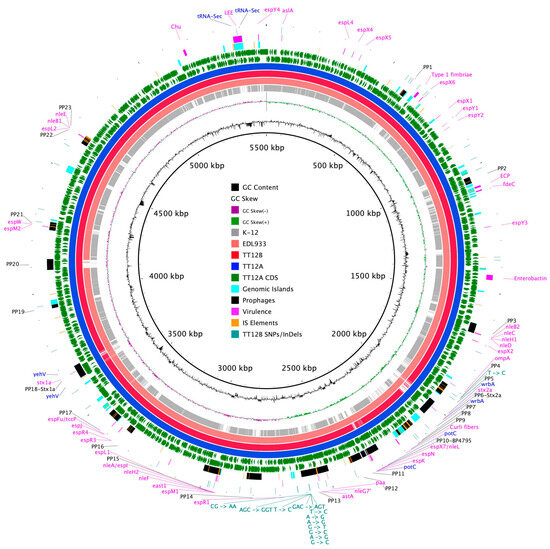
Figure 1.
Chromosome comparison of TT12A and TT12B BRIG comparison of the Stx(+) TT12A and Stx(−) TT12B genome architecture and gene content referenced to the larger Stx(+) genome. The comparison is further extended to include the EDL933 strain, clade 3.12, and the K-12 E. coli strain. CDSs are presented as arrows on the +/− strands, and functional annotations for the shared and three TT12A-specific prophages, virulence, and resistance genes, along with polymorphisms differentiating the two strains, are highlighted as shown in the legend. Chromosomal synteny in K-12 is disrupted by multiple prophages and other MGEs.
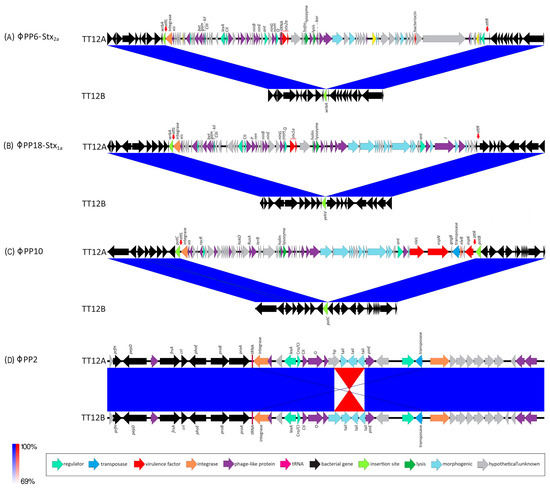
Figure 2.
Prophages and Stx-status of TT12A and TT12B BLASTn-based comparison of the prophage inventory and polymorphisms visualized in Easyfig. Strains are differentiated by the presence of three additional ΦStx- and Φnon-Stx prophages in the larger (stx+) TT12A strain at the following loci: (A) ΦStx2a-phage at wrbA: This locus is unoccupied in TT12B, while in strain A, a 63,250 kb ΦStx2a-prophage is inserted at wrbA, a preferred target locus for ΦStx2a-phage insertion in O157:H7. (B) ΦStx1a-phage at yehV: This locus is unoccupied in TT12B, while a 53,637 kb ΦStx1-prophage is inserted at yehV, a preferred target locus for ΦStx1a-phage insertion in O157:H7. (C) ΦPP10-Enterobacteria phage BP-4795 at potC: This locus is unoccupied in TT12B, while a 49,073 kb prophage is inserted at potC, a known target for phage insertion in O157:H7. (D) ΦPP2-Enterobacteria SfI phage: Chromosome assemblies feature an inversion within the shared ΦPP2-Enterobacteria SfI prophage.
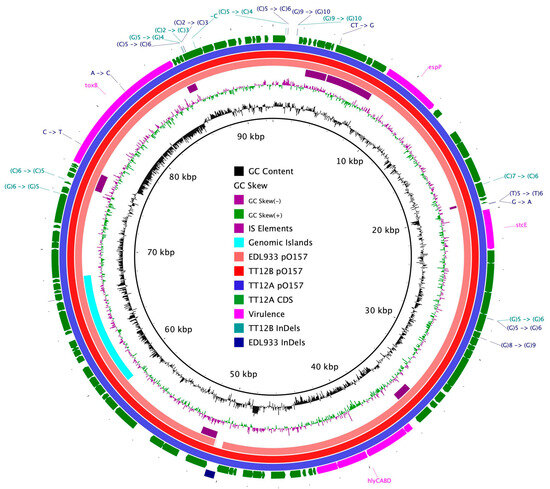
Figure 3.
Comparison of clade 3 pO157 plasmids BRIG comparison of the pO157 plasmid architecture and gene content. CDSs are presented as arrows on the +/− strands, and functional annotations for notable virulence determinants, such as EHEC hemolysin (hlyCABD), the serine protease (espP) along with plasmid differentiating polymorphisms, including SNPs and InDels, are highlighted as shown in the legend.
3.2. Phylogenomic Position of TT12A and TT12B within the O157:H7 Lineage
In silico genotyping classified TT12A Stx(+) and its co-isolated TT12B Stx(−) strains as Sequence Type ST11, Lineage I, Clade 3.12 isolates (Supplemental Table S1) [88,91,121,144]. To place the strains into the broader context of O157:H7/NM evolution [51,52,53,54,55,56], we constructed a phylogenetic hypothesis based on reference-based cgSNP discovery including representative O157:H7 strains for the nine distinct clades [68,88,106,107] (Supplemental Table S4). As evident in Figure 4, the overall tree topology reflects the general understanding of O157:H7/NM evolution from an enteropathogenic E. coli (EPEC) O55:H7 progenitor [51,52,53,54,55,56,145]. Clade 3.12 strains TT12A, TT12B, and EDL933 form a cluster, and strains TT12A and TT12B were found to be indistinguishable on the chromosomal cgSNP level [62]. This intimate relationship is further mirrored in the cgMLST analysis, with no allelic changes observed (Supplemental Figure S1, Supplemental Table S4), and along with the SNP information being consistent with an isogenic status. The strains’ intimate relationship is also reflected in the IS element profiles. In total, ISEScan detected 87 IS elements and categorized them into 14 families and 27 clusters (Supplemental Table S3). The strains feature identical chromosomal or plasmid-borne profiles, not considering the two phage-borne IS elements that are part of the TT12A-specific Enterobacteria phage ΦBP-4795-PP10 (Figure 1, Supplemental Table S3), supporting an intimate relationship between these two strains. To build the clade-inclusive cgSNP phylogeny (Figure 4), we excluded SNPs within mobile elements and repeats, as the alignment of homologous regions of different evolutionary origins can introduce false positive signals and ultimately, phylogenetic inaccuracies [106,107]. We thus used an alternative approach for these closely related isolates and processed all collinear blocks in their genomes to record the SNPs and InDels. This resulted in the detection of 16 chromosomal intragenic SNPs, all situated within two prophages, while no SNPs were recorded on the pO157 (Supplemental Table S4). In this context, we note that plasticity in the ΦStx-phage complement of O157:H7 and other STEC serotypes is well established [138]. Particular variations within the ΦStx2a-phages have been associated with altered toxin production capabilities [146,147,148,149]. Exploring the effects of such phage-to-phage variants on phage–host interactions or pathogenesis could deepen our understanding of the evolution of O157: H7 virulence. A single nonsynonymous (ns) SNP was detected in the shared ΦPP4-phage, while 15 SNPs were located within a single gene on the carried TT12A-ΦPP13 and TT12B-ΦPP11 prophages that code for the host specificity protein J [150]. Though we cannot delineate the physiological effects of these variants, we note that 80% of SNPs are non-synonymous and are predicted to decrease protein stability in TT12B [120] with potential impacts on phage biology.
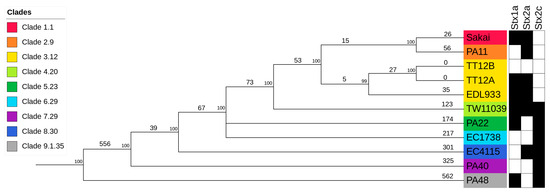
Figure 4.
Phylogenomic position of TT12A and TT12B within the O157:H7 serotype Genome comparisons of TT12A and TT12B and the representative strains from clades 1 to 9 O157:H7 genomes yielded a total of 2,654 SNPs when referenced to strain EC4115. The tree shown is the majority-consensus tree of three equally parsimonious trees with a CI of 0.998. Trees were recovered using a heuristic search in PAUP with 100,000 bootstrap replicates. The tree is rooted to the clade 9 strain, PA48, visualized and decorated in iTol. Nodes are color-coded according to clade, and numbers of separating SNPs are shown. The prevalence of Stx-subtypes is indicated by black (stx+) and white (stx−) boxes.
In addition, we detected 65 strain-differentiating InDels, 56 of which are chromosomal (Supplemental Table S4, Figure 1 and Figure 3). The majority, 38 chromosomal and 8 plasmid-borne InDels, are located within homopolymer repeats, which are prone to dynamic expansion or shrinkage during short-term evolutionary terms [151]. We also note here that 37 of the chromosomal and 1 of the plasmid InDels are associated with mobile genetic elements, including prophages, IS elements, and genomic islands (Supplemental Table S4).
3.3. Comparison of Virulence and Resistance Determinants
We surveyed the TT12A and TT12B genomes for chromosomal and plasmid-borne virulence and resistance loci. Neither strain carries antibiotic resistance genes, other than the chromosomal broad-spectrum multidrug resistance efflux pump, MdfA [152], commonly found in E. coli [153,154,155]. Not taking into account the TT12A-specific phage inventory, comprising the ΦStx1/2a-phages and several T3SS effectors nleL, espN, and espK on the Enterobacteria phage ΦBP-4795-PP10 (Figure 1), the strains’ virulence profiles are indistinguishable, showing a characteristic O157:H7 virulence repertoire [156,157] (Figure 5, Supplemental Table S3). The virulence factors include the locus of enterocyte effacement (LEE) at the tRNA-Sec (Figure 1) [23,158,159,160,161] which causes attaching and effacing (A/E) lesions [22,23]. This pathogenicity island encodes T3SS components, such as regulators, chaperones, and LEE/non-LEE effectors [22,24,25]. Among the LEE-encoded proteins is intimin (eae-γ1) [162,163], an outer membrane adhesin that facilitates intimate bacterial attachment to the host’s intestinal cells [24,158,164,165,166,167] (Figure 5). Variation in the architecture and coding capabilities of the lineage-specific virulence pO157 plasmid has provided insight into O157:H7 evolution [68,168,169,170,171]. The TT12A (92,710 bp) and TT12B (92,707 bp) plasmids are highly conserved with a size difference of only 3 bp, supporting an intimate relationship between these two strains [18,68,168,172,173] (Figure 3 and Figure 5). Loss of pO157 has been linked to diminished virulence in O157:H7 strains [174]. Characteristic plasmid-borne virulence determinants are a type II secretion system metalloprotease (stcE), hemolysin (hlyCABD), enterohemolysin (ehxA), type V-secreted serine protease (espP), and adhesin (toxB) [18,172,173,175,176,177] (Supplemental Table S3).

Figure 5.
Virulence determinants in TT12A and TT12B A heatmap visualizing the percentage identities for each virulence determinant identified in TT12A and TT12B. Apart from Stx-phage contributed toxins and nleL, espN, and espK genes on ΦPP10 Enterobacteria phage BP-4795 only present in TT12A, the strains’ virulence profiles are similar.
3.4. Prophage Content and Genomic Basis of the Attenuated Stx(−) Status
Toxin production and organ toxicity is dependent on the carried stx suballeles [48,178,179,180]. Diverse stx-phage combinations are found in the O157:H7 lineage [68,107,133,147]. A genetically altered nonstandard stx-locus, such as through IS element disruption [68], might not amplify with generic PCR primers and thus can create false-negatives [42,62,143,181]. The availability of closed genomes is thus critical to describe the genomic basis of the absence of stx genes in TT12B. Strain TT12A carries two Stx-prophages, ΦStx2a-PP6 (63,250 bp) (Figure 2A) and ΦStx1-PP18 (53,637 bp) (Figure 2B) inserted into wrbA and yehV [182,183] (Figure 1), respectively, which are the preferred insertion targets of these particular Stx-phage subtypes [68,106,138]. In mice, Stx2a confers up to 400x times higher toxicity than Stx1 [11,28,45,46,47,48,49]. Both Stx B-subunits bind to the Globotriaosylceramide receptor, Gb3, on the eukaryotic cell. The Stx2a–Gb3 binding, however, is weaker in comparison to Stx1a–Gb3 [147,184]. Consequently, the Stx1/2a toxin status of TT12A may indicate lower toxicity in comparison to stx2a-only strains [147,180,184,185,186]. Furthermore, we quantified the expression levels of the co-carried toxin variants. In cultures of TT12A treated with MMC, both phages were responsive, and we recorded a 4.4-fold and a 3.4-fold increase in stx2a and stx1a transcripts, respectively (Supplemental Figure S2). The ΦStx2a phage is of the Eru-2 type, characterized by the absence of the lambdoid O and P genes, while the ΦStx1a identifies as the lambdoid–Eru type [148,149] (Figure 2). The ΦStx1a phage features a γ-replicase, when applying a replicase P-informed subtyping schema [146]. However, the ΦStx2a phage is not typable, showing only 53% identity to β-replicase subtype. TT12A is further distinguished by the presence of Enterobacteria phage ΦBP-4795-PP10 (49,073 bp) at potC (Figure 2C), a known phage insertion site in O157:H7 and other STEC serotypes [187,188,189]. In silico comparison further identified an 1833 bp inversion within the shared incomplete ΦPP2-prophage (Figure 2D, Supplemental Table S3). Further PCR interrogation, however, revealed that this polymorphism is not strain-specific; and both orientations were detected in cultures of TT12A and B (Supplemental Figure S3). This inversion affects a single gene, annotated as a phage tail protein in the TT12B assembly (locus tag: E5F07-04800) and a hypothetical protein in TT12A (E5F08-04795) (Figure 2D).
3.5. Differences in Growth Phenotypes and Impact of the Prophage Inventories
We recorded growth in LB and under phage-mobilizing conditions in LB + MMC to determine the degree of prophage-induced lysis in TT12A and TT12B. Increased expression of recA post MMC treatment confirmed the successful SOS response activation in both cultures (Supplemental Figure S4A) [125,190,191,192]. Culture growth of TT12A is considerably affected through phage lysis, unlike strain TT12B (Supplemental Figure S4B). The different growth phenotypes in the MMC-treated cultures thus seem to be mediated by the three TT12A-specific ΦStx1a/2a/non-Stx phages, all of which are known or predicted to be lytic in the case of Enterobacteria phage ΦBP-4795-PP10 (Figure 2, Supplemental Table S3). Even though another seven lytic phages are predicted as part of the shared phage complement (Supplemental Table S3), these phages are likely not mobilized through the SOS response pathway activation, as evident in the similar growth of TT12B in LB and LB + MMC media (Supplemental Figure S4B).
4. Conclusions
Whole genome sequence typing has proven to be invaluable for the identification and strain attribution of near clonal E. coli pathogen populations [40,107,193,194]. Availability of high-resolution closed genomes allowed us to record subtle strain-level sequence and structural polymorphisms with high phylogenetic accuracy, demonstrating an isogenic relationship of these Stx(+/−) TT12A and TT12B isolates. The number of strain-differentiating SNPs is similar to the range reported for clonal O157:H7 outbreak strains [40,106,107,111,195]. However, SNP data on its own are clearly insufficient to infer clonality in microbes without further assessing changes in genome structure and content. Dynamic phage acquisition and loss resulted in the strain-specific ΦStx and non-ΦStx prophage content, which may have occurred in a single or separate evolutionary events likely triggered by the mobilization and ultimately, loss of these phages. STECs have been intentionally cured from their Stx-phages by antibiotic or MMC addition to the growth medium [63,68,196]. In this context, it is noteworthy that excised copies of the three TT12A-differentiating prophages ΦStx/non-Stx phages, absent in TT12B, were significantly increased when grown in the phage-inducing LB + MMC media (Supplemental Figure S5). Additionally, none of the insertion sites occupied in TT12A showed scarring that would indicate a former phage presence in TT12B. Alternative evolutionary scenarios can explain the Stx-phage absence in atypical O157:H7 strains, such as TT12B [51,68,197]. We can only speculate about the events that gave rise to the TT12A and TT12B variants. Given the fact that these strains originate from a patient suffering from hemorrhagic colitis [62] along with the strains’ established intimate phylogenetic relationship, the secondary loss of both ΦStx1/2 prophages along with Enterobacteria phage ΦBP-4795-PP10 in TT12B during the course of infection, in a singular or in multiple events, seems likely. The ratio of Stx(+) to non-shigatoxigenic isolates is not known; however, dynamic loss of Stx-phages and potential re-acquisition may cause transitional stx(+/−) shifts in pathogenic potential [68,69,74]. Bacteriophages control diverse bacterial biological functions. Stx has a dual role in human disease and spontaneous low-level Stx production is considered a form of bacterial altruism, promoting the toxin-dependent killing of eukaryotic predators and macrophages [29,198,199,200,201,202,203,204,205]. The ΦStx phage carriage has been associated with a number of virulence and fitness traits beyond Stx-production. The characterization of laboratory-engineered Stx-lysogens, often recovered in E. coli K12 backgrounds, indicated an impact on acid resistance, type III secretion, motility, and metabolism [206,207,208,209,210,211,212,213,214,215]. The analyzed Stx(+/−) isogen cultures and genomes provide an excellent model to further investigate the impact of ΦStx-phage carriage in a native O157:H7 genome background that is not accounted for in the K12-engineered Stx-lysogens [206,208,210,215]. Altogether, the genomic and phenotypic comparisons show that this isogen pair is intimately related, yet perhaps on a divergent evolutionary path. The role of the catalogued sequence and architectural polymorphisms, however, cannot be simply inferred from static genome comparison. Further investigations using transcriptomic and phenotypic profiling may provide greater insight into the role these variants may play in the physiology and pathogenicity of strains TT12A and TT12B.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/microorganisms12040699/s1, Figure S1: Core genome MLST, Figure S2: Expression of stx genes, Figure S3: PCR interrogation of inversion boundaries and directionality in ΦPP2, Figure S4: Growth phenotypes, Figure S5: Mobilization pattern of TT12A-specific phages; Table S1: Strain-associated metadata and sequence accessions, Table S2: Primers used in this study, Table S3: Genome features and contents, Table S4: Core genome SNP and MLST loci.
Author Contributions
Conceptualization, M.E.; Data Curation, A.A.K., J.M.B., J.L.B. and M.E.; Formal Analysis, A.A.K., S.S.K.K., P.F., J.M.B., J.L.B. and M.E.; Funding Acquisition, M.E.; Investigation, A.A.K., S.S.K.K., P.F., J.M.B., J.L.B. and M.E.; Methodology, A.A.K.; Project Administration, M.E.; Resources, J.M.B., J.L.B. and M.E.; Software, M.E.; Supervision, M.E.; Validation, A.A.K. and M.E.; Visualization, A.A.K. and M.E.; Writing—Original Draft, M.E.; Writing—Review and Editing, A.A.K., S.S.K.K., P.F., J.M.B., J.L.B. and M.E. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Institute of General Medical Sciences of the National Institutes of Health, under Award Number SC1GM135110, and the South Texas Center for Emerging Infectious Diseases (STCEID). The contributions of JLB and JMB were funded by the Agricultural Research Service of the U.S. Department of Agriculture (ARS-USDA).
Data Availability Statement
The sequence data sets generated and analyzed in this study have been deposited in the Sequence Read Archive (SRA) and GenBank at NCBI under BioProject PRJNA530317. Accessions for the reads, assembled annotated chromosomes, and pO157 plasmids of strains TT12A (CP038496, CP038497) and TT12B (CP038494, CP038495) [68] are provided in Supplemental Table S1. This table also includes strain-associated metadata and representative O157:H7 strains retrieved from NCBI in support of comprehensive analyses.
Acknowledgments
We would like to thank Felix Borrego for the assistance with figure preparation. This work received computational support from the high-performance computing cluster operated by Tech Solutions at UTSA. The use of product and company names is necessary to accurately report the methods and results; however, the United States Department of Agriculture (USDA) neither guarantees nor warrants the standard of the products, and the use of names by the USDA does not imply approval of the product to the exclusion of others that may also be suitable. The USDA is an equal opportunity provider and employer.
Conflicts of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Karmali, M.A. Emerging Public Health Challenges of Shiga Toxin-Producing Escherichia coli Related to Changes in the Pathogen, the Population, and the Environment. Clin. Infect. Dis. 2017, 64, 371–376. [Google Scholar] [CrossRef] [PubMed]
- Pennington, H. Escherichia coli O157. Lancet 2010, 376, 1428–1435. [Google Scholar] [CrossRef] [PubMed]
- Sanjar, F.; Hazen, T.H.; Shah, S.M.; Koenig, S.S.; Agrawal, S.; Daugherty, S.; Sadzewicz, L.; Tallon, L.J.; Mammel, M.K.; Feng, P.; et al. Genome Sequence of Escherichia coli O157:H7 Strain 2886-75, Associated with the First Reported Case of Human Infection in the United States. Genome Announc. 2014, 2, e01120-13. [Google Scholar] [CrossRef] [PubMed]
- Rangel, J.M.; Sparling, P.H.; Crowe, C.; Griffin, P.M.; Swerdlow, D.L. Epidemiology of Escherichia coli O157:H7 outbreaks, United States, 1982–2002. Emerg. Infect. Dis. 2005, 11, 603–609. [Google Scholar] [CrossRef] [PubMed]
- Heiman, K.E.; Mody, R.K.; Johnson, S.D.; Griffin, P.M.; Gould, L.H. Escherichia coli O157 Outbreaks in the United States, 2003–2012. Emerg. Infect. Dis. 2015, 21, 1293–1301. [Google Scholar] [CrossRef] [PubMed]
- Omer, M.K.; Álvarez-Ordoñez, A.; Prieto, M.; Skjerve, E.; Asehun, T.; Alvseike, O.A. A Systematic Review of Bacterial Foodborne Outbreaks Related to Red Meat and Meat Products. Foodborne Pathog. Dis. 2018, 15, 598–611. [Google Scholar] [CrossRef] [PubMed]
- Tack, D.M.; Kisselburgh, H.M.; Richardson, L.C.; Geissler, A.; Griffin, P.M.; Payne, D.C.; Gleason, B.L. Shiga Toxin-Producing Escherichia coli Outbreaks in the United States, 2010–2017. Microorganisms 2021, 9, 1529. [Google Scholar] [CrossRef]
- Marquezini, M.G.; da Costa, L.H.; Bromberg, R. Occurrence of the Seven Most Common Serotypes of Shiga Toxin-Producing Escherichia coli in Beef Cuts Produced in Meat Processing Plants in the State of São Paulo, Brazil. J. Food Prot. 2022, 85, 261–265. [Google Scholar] [CrossRef] [PubMed]
- Riley, L.W.; Remis, R.S.; Helgerson, S.D.; McGee, H.B.; Wells, J.G.; Davis, B.R.; Hebert, R.J.; Olcott, E.S.; Johnson, L.M.; Hargrett, N.T.; et al. Hemorrhagic colitis associated with a rare Escherichia coli serotype. N. Engl. J. Med. 1983, 308, 681–685. [Google Scholar] [CrossRef] [PubMed]
- Joseph, A.; Cointe, A.; Mariani Kurkdjian, P.; Rafat, C.; Hertig, A. Shiga Toxin-Associated Hemolytic Uremic Syndrome: A Narrative Review. Toxins 2020, 12, 67. [Google Scholar] [CrossRef] [PubMed]
- Boerlin, P.; McEwen, S.A.; Boerlin-Petzold, F.; Wilson, J.B.; Johnson, R.P.; Gyles, C.L. Associations between virulence factors of Shiga toxin-producing Escherichia coli and disease in humans. J. Clin. Microbiol. 1999, 37, 497–503. [Google Scholar] [CrossRef] [PubMed]
- Karmali, M.A.; Petric, M.; Lim, C.; Fleming, P.C.; Steele, B.T. Escherichia coli cytotoxin, haemolytic-uraemic syndrome, and haemorrhagic colitis. Lancet 1983, 2, 1299–1300. [Google Scholar] [CrossRef] [PubMed]
- Karmali, M.A. Infection by Shiga toxin-producing Escherichia coli: An overview. Mol. Biotechnol. 2004, 26, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Davis, T.K.; Van De Kar, N.; Tarr, P.I. Shiga Toxin/Verocytotoxin-Producing Escherichia coli Infections: Practical Clinical Perspectives. Microbiol. Spectr. 2014, 2, Ehec-0025-2014. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.X.; Suri, R.S.; Barrowman, N.; Rehman, F.; Matsell, D.; Rosas-Arellano, M.P.; Salvadori, M.; Haynes, R.B.; Clark, W.F. Long-term renal prognosis of diarrhea-associated hemolytic uremic syndrome: A systematic review, meta-analysis, and meta-regression. Jama 2003, 290, 1360–1370. [Google Scholar] [CrossRef] [PubMed]
- Mele, C.; Remuzzi, G.; Noris, M. Hemolytic uremic syndrome. Semin. Immunopathol. 2014, 36, 399–420. [Google Scholar] [CrossRef] [PubMed]
- Melton-Celsa, A.R. Shiga Toxin (Stx) Classification, Structure, and Function. Microbiol. Spectr. 2014, 2, Ehec-0024-2013. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.Y.; Yoon, J.; Hovde, C.J. A brief overview of Escherichia coli O157:H7 and its plasmid O157. J. Microbiol. Biotechnol. 2010, 20, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Kaper, J.B. Cloning and characterization of the eae gene of enterohaemorrhagic Escherichia coli O157:H7. Mol. Microbiol. 1992, 6, 411–417. [Google Scholar] [CrossRef] [PubMed]
- Donnenberg, M.S.; Tzipori, S.; McKee, M.L.; O’Brien, A.D.; Alroy, J.; Kaper, J.B. The role of the eae gene of enterohemorrhagic Escherichia coli in intimate attachment in vitro and in a porcine model. J. Clin. Investig. 1993, 92, 1418–1424. [Google Scholar] [CrossRef] [PubMed]
- Kaper, J.B. The locus of enterocyte effacement pathogenicity island of Shiga toxin-producing Escherichia coli O157:H7 and other attaching and effacing E. coli. Jpn. J. Med. Sci. Biol. 1998, 51 (Suppl. 1), S101–S107. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.A. LEEways: Tales of EPEC, ATEC and EHEC. Cell. Microbiol. 2010, 12, 1544–1552. [Google Scholar] [CrossRef] [PubMed]
- Stevens, M.P.; Frankel, G.M. The Locus of Enterocyte Effacement and Associated Virulence Factors of Enterohemorrhagic Escherichia coli. Microbiol. Spectr. 2014, 2, Ehec-0007-2013. [Google Scholar] [CrossRef] [PubMed]
- Mellies, J.L.; Elliott, S.J.; Sperandio, V.; Donnenberg, M.S.; Kaper, J.B. The Per regulon of enteropathogenic Escherichia coli: Identification of a regulatory cascade and a novel transcriptional activator, the locus of enterocyte effacement (LEE)-encoded regulator (Ler). Mol. Microbiol. 1999, 33, 296–306. [Google Scholar] [CrossRef] [PubMed]
- Kirsch, P.; Jores, J.; Wieler, L.H. Plasticity of bacterial genomes: Pathogenicity islands and the locus of enterocyte effacement (LEE). Berl. Munch. Tierarztl. Wochenschr. 2004, 117, 116–129. [Google Scholar] [PubMed]
- Trofa, A.F.; Ueno-Olsen, H.; Oiwa, R.; Yoshikawa, M. Dr. Kiyoshi Shiga: Discoverer of the dysentery bacillus. Clin. Infect. Dis. 1999, 29, 1303–1306. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, A.D.; Lively, T.A.; Chang, T.W.; Gorbach, S.L. Purification of Shigella dysenteriae 1 (Shiga)-like toxin from Escherichia coli O157:H7 strain associated with haemorrhagic colitis. Lancet 1983, 2, 573. [Google Scholar] [CrossRef] [PubMed]
- Del Cogliano, M.E.; Pinto, A.; Goldstein, J.; Zotta, E.; Ochoa, F.; Fernandez-Brando, R.J.; Muniesa, M.; Ghiringhelli, P.D.; Palermo, M.S.; Bentancor, L.V. Relevance of Bacteriophage 933W in the Development of Hemolytic Uremic Syndrome (HUS). Front. Microbiol. 2018, 9, 3104. [Google Scholar] [CrossRef] [PubMed]
- Mauro, S.A.; Koudelka, G.B. Shiga toxin: Expression, distribution, and its role in the environment. Toxins 2011, 3, 608–625. [Google Scholar] [CrossRef]
- Melton-Celsa, A.; Mohawk, K.; Teel, L.; O’Brien, A. Pathogenesis of Shiga-toxin producing Escherichia coli. Curr. Top. Microbiol. Immunol. 2012, 357, 67–103. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, Y.; Sperandio, V. Enterohemorrhagic E. coli (EHEC) pathogenesis. Front. Cell. Infect. Microbiol. 2012, 2, 90. [Google Scholar] [CrossRef] [PubMed]
- Johannes, L.; Romer, W. Shiga toxins—From cell biology to biomedical applications. Nat. Rev. Microbiol. 2010, 8, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Eaton, K.A.; Friedman, D.I.; Francis, G.J.; Tyler, J.S.; Young, V.B.; Haeger, J.; Abu-Ali, G.; Whittam, T.S. Pathogenesis of renal disease due to enterohemorrhagic Escherichia coli in germ-free mice. Infect. Immun. 2008, 76, 3054–3063. [Google Scholar] [CrossRef] [PubMed]
- Shin, I.S.; Ishii, S.; Shin, J.S.; Sung, K.I.; Park, B.S.; Jang, H.Y.; Kim, B.W. Globotriaosylceramide (Gb3) content in HeLa cells is correlated to Shiga toxin-induced cytotoxicity and Gb3 synthase expression. Bmb Rep. 2009, 42, 310–314. [Google Scholar] [CrossRef] [PubMed]
- Kiyokawa, N.; Taguchi, T.; Mori, T.; Uchida, H.; Sato, N.; Takeda, T.; Fujimoto, J. Induction of apoptosis in normal human renal tubular epithelial cells by Escherichia coli Shiga toxins 1 and 2. J. Infect. Dis. 1998, 178, 178–184. [Google Scholar] [CrossRef] [PubMed]
- Baumler, A.J.; Sperandio, V. Interactions between the microbiota and pathogenic bacteria in the gut. Nature 2016, 535, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Tyler, J.S.; Beeri, K.; Reynolds, J.L.; Alteri, C.J.; Skinner, K.G.; Friedman, J.H.; Eaton, K.A.; Friedman, D.I. Prophage induction is enhanced and required for renal disease and lethality in an EHEC mouse model. PLoS Pathog. 2013, 9, e1003236. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanian, S.; Osburne, M.S.; BrinJones, H.; Tai, A.K.; Leong, J.M. Prophage induction, but not production of phage particles, is required for lethal disease in a microbiome-replete murine model of enterohemorrhagic E. coli infection. PLoS Pathog. 2019, 15, e1007494. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Rubio, L.; Haarmann, N.; Schwidder, M.; Muniesa, M.; Schmidt, H. Bacteriophages of Shiga Toxin-Producing Escherichia coli and Their Contribution to Pathogenicity. Pathogens 2021, 10, 404. [Google Scholar] [CrossRef] [PubMed]
- Eppinger, M.; Almería, S.; Allué-Guardia, A.; Bagi, L.K.; Kalalah, A.A.; Gurtler, J.B.; Fratamico, P.M. Genome Sequence Analysis and Characterization of Shiga Toxin 2 Production by Escherichia coli O157:H7 Strains Associated With a Laboratory Infection. Front. Cell. Infect. Microbiol. 2022, 12, 888568. [Google Scholar] [CrossRef] [PubMed]
- Carrillo, C.D.; Koziol, A.G.; Mathews, A.; Goji, N.; Lambert, D.; Huszczynski, G.; Gauthier, M.; Amoako, K.; Blais, B.W. Comparative Evaluation of Genomic and Laboratory Approaches for Determination of Shiga Toxin Subtypes in Escherichia coli. J. Food Prot. 2016, 79, 2078–2085. [Google Scholar] [CrossRef] [PubMed]
- Scheutz, F.; Teel, L.D.; Beutin, L.; Piérard, D.; Buvens, G.; Karch, H.; Mellmann, A.; Caprioli, A.; Tozzoli, R.; Morabito, S.; et al. Multicenter evaluation of a sequence-based protocol for subtyping Shiga toxins and standardizing Stx nomenclature. J. Clin. Microbiol. 2012, 50, 2951–2963. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.; Fu, S.; Zhang, J.; Fan, R.; Xu, Y.; Sun, H.; He, X.; Xu, J.; Xiong, Y. Identification and pathogenomic analysis of an Escherichia coli strain producing a novel Shiga toxin 2 subtype. Sci. Rep. 2018, 8, 6756. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Bai, X.; Zhang, J.; Sun, H.; Fu, S.; Fan, R.; He, X.; Scheutz, F.; Matussek, A.; Xiong, Y. Escherichia coli strains producing a novel Shiga toxin 2 subtype circulate in China. Int. J. Med. Microbiol. 2020, 310, 151377. [Google Scholar] [CrossRef] [PubMed]
- Tesh, V.L.; Burris, J.A.; Owens, J.W.; Gordon, V.M.; Wadolkowski, E.A.; O’Brien, A.D.; Samuel, J.E. Comparison of the relative toxicities of Shiga-like toxins type I and type II for mice. Infect. Immun. 1993, 61, 3392–3402. [Google Scholar] [CrossRef] [PubMed]
- Orth, D.; Grif, K.; Khan, A.B.; Naim, A.; Dierich, M.P.; Wurzner, R. The Shiga toxin genotype rather than the amount of Shiga toxin or the cytotoxicity of Shiga toxin in vitro correlates with the appearance of the hemolytic uremic syndrome. Diagn. Microbiol. Infect. Dis. 2007, 59, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Russo, L.M.; Melton-Celsa, A.R.; Smith, M.A.; Smith, M.J.; O’Brien, A.D. Oral intoxication of mice with Shiga toxin type 2a (Stx2a) and protection by anti-Stx2a monoclonal antibody 11E10. Infect. Immun. 2014, 82, 1213–1221. [Google Scholar] [CrossRef] [PubMed]
- Fuller, C.A.; Pellino, C.A.; Flagler, M.J.; Strasser, J.E.; Weiss, A.A. Shiga toxin subtypes display dramatic differences in potency. Infect. Immun. 2011, 79, 1329–1337. [Google Scholar] [CrossRef] [PubMed]
- Hauser, J.R.; Atitkar, R.R.; Petro, C.D.; Lindsey, R.L.; Strockbine, N.; O’Brien, A.D.; Melton-Celsa, A.R. The Virulence of Escherichia coli O157:H7 Isolates in Mice Depends on Shiga Toxin Type 2a (Stx2a)-Induction and High Levels of Stx2a in Stool. Front. Cell. Infect. Microbiol. 2020, 10, 62. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Li, X.; Liu, B.; Beutin, L.; Xu, J.; Ren, Y.; Feng, L.; Lan, R.; Reeves, P.R.; Wang, L. Derivation of Escherichia coli O157:H7 from its O55:H7 precursor. PLoS ONE 2010, 5, e8700. [Google Scholar] [CrossRef] [PubMed]
- Kyle, J.L.; Cummings, C.A.; Parker, C.T.; Quinones, B.; Vatta, P.; Newton, E.; Huynh, S.; Swimley, M.; Degoricija, L.; Barker, M.; et al. Escherichia coli serotype O55:H7 diversity supports parallel acquisition of bacteriophage at Shiga toxin phage insertion sites during evolution of the O157:H7 lineage. J. Bacteriol. 2012, 194, 1885–1896. [Google Scholar] [CrossRef] [PubMed]
- Rump, L.V.; Strain, E.A.; Cao, G.; Allard, M.W.; Fischer, M.; Brown, E.W.; Gonzalez-Escalona, N. Draft genome sequences of six Escherichia coli isolates from the stepwise model of emergence of Escherichia coli O157:H7. J. Bacteriol. 2011, 193, 2058–2059. [Google Scholar] [CrossRef] [PubMed]
- Feng, P.; Lampel, K.A.; Karch, H.; Whittam, T.S. Genotypic and phenotypic changes in the emergence of Escherichia coli O157:H7. J. Infect. Dis. 1998, 177, 1750–1753. [Google Scholar] [CrossRef] [PubMed]
- Jenke, C.; Leopold, S.R.; Weniger, T.; Rothganger, J.; Harmsen, D.; Karch, H.; Mellmann, A. Identification of intermediate in evolutionary model of enterohemorrhagic Escherichia coli O157. Emerg. Infect. Dis. 2012, 18, 582–588. [Google Scholar] [CrossRef] [PubMed]
- Leopold, S.R.; Shaikh, N.; Tarr, P.I. Further evidence of constrained radiation in the evolution of pathogenic Escherichia coli O157:H7. Infect. Genet. Evol. J. Mol. Epidemiol. Evol. Genet. Infect. Dis. 2010, 10, 1282–1285. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wick, L.M.; Qi, W.; Lacher, D.W.; Whittam, T.S. Evolution of genomic content in the stepwise emergence of Escherichia coli O157:H7. J. Bacteriol. 2005, 187, 1783–1791. [Google Scholar] [CrossRef] [PubMed]
- Tozzoli, R.; Grande, L.; Michelacci, V.; Fioravanti, R.; Gally, D.; Xu, X.; La Ragione, R.; Anjum, M.; Wu, G.; Caprioli, A.; et al. Identification and characterization of a peculiar vtx2-converting phage frequently present in verocytotoxin-producing Escherichia coli O157 isolated from human infections. Infect. Immun. 2014, 82, 3023–3032. [Google Scholar] [CrossRef] [PubMed]
- Dallman, T.J.; Ashton, P.M.; Byrne, L.; Perry, N.T.; Petrovska, L.; Ellis, R.J.; Allison, L.; Hanson, M.; Holmes, A.; Gunn, G.J.; et al. Applying phylogenomics to understand the emergence of Shiga-toxin-producing Escherichia coli O157:H7 strains causing severe human disease in the UK. Microb. Genom. 2015, 1, e000029. [Google Scholar] [CrossRef]
- Cooper, K.K.; Mandrell, R.E.; Louie, J.W.; Korlach, J.; Clark, T.A.; Parker, C.T.; Huynh, S.; Chain, P.S.; Ahmed, S.; Carter, M.Q. Comparative genomics of enterohemorrhagic Escherichia coli O145:H28 demonstrates a common evolutionary lineage with Escherichia coli O157:H7. BMC Genom. 2014, 15, 17. [Google Scholar] [CrossRef] [PubMed]
- Ogura, Y.; Ooka, T.; Iguchi, A.; Toh, H.; Asadulghani, M.; Oshima, K.; Kodama, T.; Abe, H.; Nakayama, K.; Kurokawa, K.; et al. Comparative genomics reveal the mechanism of the parallel evolution of O157 and non-O157 enterohemorrhagic Escherichia coli. Proc. Natl. Acad. Sci. USA 2009, 106, 17939–17944. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, S.C.; Gonzalez-Escalona, N.; Kotewicz, M.L.; Fischer, M.; Kase, J.A. Genome sequencing and comparative genomics of enterohemorrhagic Escherichia coli O145:H25 and O145:H28 reveal distinct evolutionary paths and marked variations in traits associated with virulence & colonization. Bmc Microbiol. 2017, 17, 183. [Google Scholar]
- Feng, P.; Dey, M.; Abe, A.; Takeda, T. Isogenic strain of Escherichia coli O157:H7 that has lost both Shiga toxin 1 and 2 genes. Clin. Diagn. Lab. Immunol. 2001, 8, 711–717. [Google Scholar] [CrossRef] [PubMed]
- Bielaszewska, M.; Middendorf, B.; Kock, R.; Friedrich, A.W.; Fruth, A.; Karch, H.; Schmidt, M.A.; Mellmann, A. Shiga toxin-negative attaching and effacing Escherichia coli: Distinct clinical associations with bacterial phylogeny and virulence traits and inferred in-host pathogen evolution. Clin. Infect. Dis. 2008, 47, 208–217. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, A.W.; Zhang, W.; Bielaszewska, M.; Mellmann, A.; Kock, R.; Fruth, A.; Tschape, H.; Karch, H. Prevalence, virulence profiles, and clinical significance of Shiga toxin-negative variants of enterohemorrhagic Escherichia coli O157 infection in humans. Clin. Infect. Dis. 2007, 45, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Stephan, R.; Zhang, W.; Bielaszewska, M.; Mellmann, A.; Karch, H. Phenotypic and genotypic traits of Shiga toxin-negative E. coli O157:H7/H(-) bovine and porcine strains. Foodborne Pathog. Dis. 2009, 6, 235–243. [Google Scholar] [CrossRef]
- Themphachana, M.; Nakaguchi, Y.; Nishibuchi, M.; Seto, K.; Rattanachuay, P.; Singkhamanan, K.; Sukhumungoon, P. First report in Thailand of a stx-negative Escherichia coli 0157 strain from a patient with diarrhea. S. Asian J. Trop. Med. Public Health 2014, 45, 881–889. [Google Scholar]
- Uhlich, G.A.; Paoli, G.C.; Zhang, X.; Andreozzi, E. Whole-Genome Sequence of Escherichia coli Serotype O157:H7 Strain ATCC 43888. Microbiol. Resour. Announc. 2019, 8, e00906-19. [Google Scholar] [CrossRef] [PubMed]
- Nyong, E.C.; Zaia, S.R.; Allué-Guardia, A.; Rodriguez, A.L.; Irion-Byrd, Z.; Koenig, S.S.K.; Feng, P.; Bono, J.L.; Eppinger, M. Pathogenomes of Atypical Non-shigatoxigenic Escherichia coli NSF/SF O157:H7/NM: Comprehensive Phylogenomic Analysis Using Closed Genomes. Front. Microbiol. 2020, 11, 619. [Google Scholar] [CrossRef] [PubMed]
- Ferdous, M.; Zhou, K.; Mellmann, A.; Morabito, S.; Croughs, P.D.; de Boer, R.F.; Kooistra-Smid, A.M.; Rossen, J.W.; Friedrich, A.W. Is Shiga Toxin-Negative Escherichia coli O157:H7 Enteropathogenic or Enterohemorrhagic Escherichia coli? Comprehensive Molecular Analysis Using Whole-Genome Sequencing. J. Clin. Microbiol. 2015, 53, 3530–3538. [Google Scholar] [CrossRef] [PubMed]
- Bielaszewska, M.; Prager, R.; Köck, R.; Mellmann, A.; Zhang, W.; Tschäpe, H.; Tarr, P.I.; Karch, H. Shiga toxin gene loss and transfer in vitro and in vivo during enterohemorrhagic Escherichia coli O26 infection in humans. Appl. Environ. Microbiol. 2007, 73, 3144–3150. [Google Scholar] [CrossRef] [PubMed]
- Wetzel, A.N.; Lejeune, J.T. Isolation of Escherichia coli O157:H7 strains that do not produce Shiga toxin from bovine, avian and environmental sources. Lett. Appl. Microbiol. 2007, 45, 504–507. [Google Scholar] [CrossRef] [PubMed]
- Bettelheim, K. Re: Isolation of Escherichia coli O157:H7 strains that do not produce Shiga toxin from bovine, avian and environmental sources. Lett. Appl. Microbiol. 2008, 46, 281. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, H.; Scheef, J.; Huppertz, H.I.; Frosch, M.; Karch, H. Escherichia coli O157:H7 and O157:H(-) strains that do not produce Shiga toxin: Phenotypic and genetic characterization of isolates associated with diarrhea and hemolytic-uremic syndrome. J. Clin. Microbiol. 1999, 37, 3491–3496. [Google Scholar] [CrossRef] [PubMed]
- Mellmann, A.; Lu, S.; Karch, H.; Xu, J.G.; Harmsen, D.; Schmidt, M.A.; Bielaszewska, M. Recycling of Shiga toxin 2 genes in sorbitol-fermenting enterohemorrhagic Escherichia coli O157:NM. Appl. Environ. Microbiol. 2008, 74, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Senthakumaran, T.; Brandal, L.T.; Lindstedt, B.-A.; Jørgensen, S.B.; Charnock, C.; Tunsjø, H.S. Implications of stx loss for clinical diagnostics of Shiga toxin-producing Escherichia coli. Eur. J. Clin. Microbiol. Infect. Dis. 2018, 37, 2361–2370. [Google Scholar] [CrossRef] [PubMed]
- Koitabashi, T.; Vuddhakul, V.; Radu, S.; Morigaki, T.; Asai, N.; Nakaguchi, Y.; Nishibuchi, M. Genetic characterization of Escherichia coli O157: H7/- strains carrying the stx2 gene but not producing Shiga toxin 2. Microbiol. Immunol. 2006, 50, 135–148. [Google Scholar] [CrossRef] [PubMed]
- Mellmann, A.; Bielaszewska, M.; Zimmerhackl, L.B.; Prager, R.; Harmsen, D.; Tschäpe, H.; Karch, H. Enterohemorrhagic Escherichia coli in human infection: In vivo evolution of a bacterial pathogen. Clin. Infect. Dis. 2005, 41, 785–792. [Google Scholar] [CrossRef]
- Pinto, G.; Sampaio, M.; Dias, O.; Almeida, C.; Azeredo, J.; Oliveira, H. Insights into the genome architecture and evolution of Shiga toxin encoding bacteriophages of Escherichia coli. BMC Genom. 2021, 22, 366. [Google Scholar] [CrossRef] [PubMed]
- Chin, C.-S.; Alexander, D.H.; Marks, P.; Klammer, A.A.; Drake, J.; Heiner, C.; Clum, A.; Copeland, A.; Huddleston, J.; Eichler, E.E.; et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods 2013, 10, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Community, T.G. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2022 update. Nucleic Acids Res. 2022, 50, W345–W351. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef]
- Mikheenko, A.; Prjibelski, A.; Saveliev, V.; Antipov, D.; Gurevich, A. Versatile genome assembly evaluation with QUAST-LG. Bioinformatics 2018, 34, i142–i150. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Zhang, C.T. Ori-Finder: A web-based system for finding oriCs in unannotated bacterial genomes. BMC Bioinform. 2008, 9, 79. [Google Scholar] [CrossRef] [PubMed]
- Tatusova, T.; DiCuccio, M.; Badretdin, A.; Chetvernin, V.; Nawrocki, E.P.; Zaslavsky, L.; Lomsadze, A.; Pruitt, K.D.; Borodovsky, M.; Ostell, J. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 2016, 44, 6614–6624. [Google Scholar] [CrossRef] [PubMed]
- Alikhan, N.F.; Petty, N.K.; Ben Zakour, N.L.; Beatson, S.A. BLAST Ring Image Generator (BRIG): Simple prokaryote genome comparisons. BMC Genom. 2011, 12, 402. [Google Scholar] [CrossRef] [PubMed]
- Ingle, D.J.; Valcanis, M.; Kuzevski, A.; Tauschek, M.; Inouye, M.; Stinear, T.; Levine, M.M.; Robins-Browne, R.M.; Holt, K.E. In silico serotyping of E. coli from short read data identifies limited novel O-loci but extensive diversity of O:H serotype combinations within and between pathogenic lineages. Microb. Genom. 2016, 2, e000064. [Google Scholar] [CrossRef] [PubMed]
- Jain, C.; Rodriguez-R, L.M.; Phillippy, A.M.; Konstantinidis, K.T.; Aluru, S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 2018, 9, 5114. [Google Scholar] [CrossRef]
- Manning, S.D.; Motiwala, A.S.; Springman, A.C.; Qi, W.; Lacher, D.W.; Ouellette, L.M.; Mladonicky, J.M.; Somsel, P.; Rudrik, J.T.; Dietrich, S.E.; et al. Variation in virulence among clades of Escherichia coli O157:H7 associated with disease outbreaks. Proc. Natl. Acad. Sci. USA 2008, 105, 4868–4873. [Google Scholar] [CrossRef] [PubMed]
- Riordan, J.T.; Viswanath, S.B.; Manning, S.D.; Whittam, T.S. Genetic differentiation of Escherichia coli O157:H7 clades associated with human disease by real-time PCR. J. Clin. Microbiol. 2008, 46, 2070–2073. [Google Scholar] [CrossRef]
- Yokoyama, E.; Hirai, S.; Hashimoto, R.; Uchimura, M. Clade analysis of enterohemorrhagic Escherichia coli serotype O157:H7/H- strains and hierarchy of their phylogenetic relationships. Infect. Genet. Evol. 2012, 12, 1724–1728. [Google Scholar] [CrossRef]
- Yang, Z.; Kovar, J.; Kim, J.; Nietfeldt, J.; Smith, D.R.; Moxley, R.A.; Olson, M.E.; Fey, P.D.; Benson, A.K. Identification of common subpopulations of non-sorbitol-fermenting, beta-glucuronidase-negative Escherichia coli O157:H7 from bovine production environments and human clinical samples. Appl. Environ. Microbiol. 2004, 70, 6846–6854. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kurtz, S.; Phillippy, A.; Delcher, A.L.; Smoot, M.; Shumway, M.; Antonescu, C.; Salzberg, S.L. Versatile and open software for comparing large genomes. Genome Biol. 2004, 5, R12. [Google Scholar] [CrossRef] [PubMed]
- Petkau, A.; Mabon, P.; Sieffert, C.; Knox, N.C.; Cabral, J.; Iskander, M.; Iskander, M.; Weedmark, K.; Zaheer, R.; Katz, L.S.; et al. SNVPhyl: A single nucleotide variant phylogenomics pipeline for microbial genomic epidemiology. Microb. Genom. 2017, 3, e000116. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Zheng, D.; Zhou, S.; Chen, L.; Yang, J. VFDB 2022: A general classification scheme for bacterial virulence factors. Nucleic Acids Res. 2022, 50, D912–D917. [Google Scholar] [CrossRef] [PubMed]
- Florensa, A.F.; Kaas, R.S.; Clausen, P.; Aytan-Aktug, D.; Aarestrup, F.M. ResFinder-an open online resource for identification of antimicrobial resistance genes in next-generation sequencing data and prediction of phenotypes from genotypes. Microb. Genom. 2022, 8, 000748. [Google Scholar] [CrossRef] [PubMed]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Liang, Y.; Lynch, K.H.; Dennis, J.J.; Wishart, D.S. PHAST: A fast phage search tool. Nucleic Acids Res. 2011, 39, W347–W352. [Google Scholar] [CrossRef] [PubMed]
- Marçais, G.; Delcher, A.L.; Phillippy, A.M.; Coston, R.; Salzberg, S.L.; Zimin, A. MUMmer4: A fast and versatile genome alignment system. PLoS Comput. Biol. 2018, 14, e1005944. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef]
- Sullivan, M.J.; Petty, N.K.; Beatson, S.A. Easyfig: A genome comparison visualizer. Bioinformatics 2011, 27, 1009–1010. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Tang, H. ISEScan: Automated identification of insertion sequence elements in prokaryotic genomes. Bioinformatics 2017, 33, 3340–3347. [Google Scholar] [CrossRef] [PubMed]
- Bertelli, C.; Laird, M.R.; Williams, K.P.; Simon Fraser University Research Computing, G.; Lau, B.Y.; Hoad, G.; Winsor, G.L.; Brinkman, F.S.L. IslandViewer 4: Expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 2017, 45, W30–W35. [Google Scholar] [CrossRef]
- Bertelli, C.; Brinkman, F.S.L. Improved genomic island predictions with IslandPath-DIMOB. Bioinformatics 2018, 34, 2161–2167. [Google Scholar] [CrossRef] [PubMed]
- Bertelli, C.; Tilley, K.E.; Brinkman, F.S.L. Microbial genomic island discovery, visualization and analysis. Brief. Bioinform. 2018, 20, 1685–1698. [Google Scholar] [CrossRef] [PubMed]
- Robertson, J.; Nash, J.H.E. MOB-suite: Software tools for clustering, reconstruction and typing of plasmids from draft assemblies. Microb. Genom. 2018, 4, e000206. [Google Scholar] [CrossRef] [PubMed]
- Eppinger, M.; Mammel, M.K.; Leclerc, J.E.; Ravel, J.; Cebula, T.A. Genomic anatomy of Escherichia coli O157:H7 outbreaks. Proc. Natl. Acad. Sci. USA 2011, 108, 20142–20147. [Google Scholar] [CrossRef] [PubMed]
- Rusconi, B.; Sanjar, F.; Koenig, S.S.; Mammel, M.K.; Tarr, P.I.; Eppinger, M. Whole Genome Sequencing for Genomics-Guided Investigations of Escherichia coli O157:H7 Outbreaks. Front. Microbiol. 2016, 7, 985. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Garrison, E.; Marth, G. Haplotype-based variant detection from short-read sequencing. arXiv 2012, arXiv:1207.3907. [Google Scholar]
- Delcher, A.L.; Salzberg, S.L.; Phillippy, A.M. Using MUMmer to identify similar regions in large sequence sets. Curr. Protoc. Bioinform. 2003, 10, 10–13. [Google Scholar] [CrossRef] [PubMed]
- Eppinger, M.; Pearson, T.; Koenig, S.S.; Pearson, O.; Hicks, N.; Agrawal, S.; Sanjar, F.; Galens, K.; Daugherty, S.; Crabtree, J.; et al. Genomic epidemiology of the Haitian cholera outbreak: A single introduction followed by rapid, extensive, and continued spread characterized the onset of the epidemic. mBio 2014, 5, e01721. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.E.; Kumar, P.; Easterly, C.; Esler, M.; Mehta, S.; Eschenlauer, A.C.; Hegeman, A.D.; Jagtap, P.D.; Griffin, T.J. Improve your Galaxy text life: The Query Tabular Tool. F1000Res 2018, 7, 1604. [Google Scholar] [CrossRef] [PubMed]
- Wilgenbusch, J.C.; Swofford, D. Inferring evolutionary trees with PAUP*. Curr. Protoc. Bioinform. 2003, 6, 6–14. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef] [PubMed]
- Maddison, W.P.; Maddison, D.R. Mesquite: A Modular System for Evolutionary Analysis. 2016. Available online: http://www.mesquiteproject.org/ (accessed on 10 December 2023).
- Eppinger, M.; Worsham, P.L.; Nikolich, M.P.; Riley, D.R.; Sebastian, Y.; Mou, S.; Achtman, M.; Lindler, L.E.; Ravel, J. Genome sequence of the deep-rooted Yersinia pestis strain Angola reveals new insights into the evolution and pangenome of the plague bacterium. J. Bacteriol. 2010, 192, 1685–1699. [Google Scholar] [CrossRef] [PubMed]
- Hau, S.J.; Allue-Guardia, A.; Rusconi, B.; Haan, J.S.; Davies, P.R.; Frana, T.S.; Eppinger, M.; Nicholson, T.L. Single Nucleotide Polymorphism Analysis Indicates Genetic Distinction and Reduced Diversity of Swine-Associated Methicillin Resistant Staphylococcus aureus (MRSA) ST5 Isolates Compared to Clinical MRSA ST5 Isolates. Front. Microbiol. 2018, 9, 2078. [Google Scholar] [CrossRef] [PubMed]
- Darling, A.E.; Mau, B.; Perna, N.T. progressiveMauve: Multiple genome alignment with gene gain, loss and rearrangement. PLoS ONE 2010, 5, e11147. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Randall, A.; Baldi, P. Prediction of protein stability changes for single-site mutations using support vector machines. Proteins 2006, 62, 1125–1132. [Google Scholar] [CrossRef] [PubMed]
- Jünemann, S.; Sedlazeck, F.J.; Prior, K.; Albersmeier, A.; John, U.; Kalinowski, J.; Mellmann, A.; Goesmann, A.; von Haeseler, A.; Stoye, J.; et al. Updating benchtop sequencing performance comparison. Nat. Biotechnol. 2013, 31, 294–296. [Google Scholar] [CrossRef] [PubMed]
- Riley, M.; Abe, T.; Arnaud, M.B.; Berlyn, M.K.; Blattner, F.R.; Chaudhuri, R.R.; Glasner, J.D.; Horiuchi, T.; Keseler, I.M.; Kosuge, T.; et al. Escherichia coli K-12: A cooperatively developed annotation snapshot—2005. Nucleic Acids Res. 2006, 34, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kruskal, J.B. On the Shortest Spanning Subtree of a Graph and the Traveling Salesman Problem. Proc. Am. Math. Soc. 1956, 7, 48–50. [Google Scholar] [CrossRef]
- Francisco, A.P.; Bugalho, M.; Ramirez, M.; Carriço, J.A. Global optimal eBURST analysis of multilocus typing data using a graphic matroid approach. BMC Bioinform. 2009, 10, 152. [Google Scholar] [CrossRef] [PubMed]
- Kimmitt, P.T.; Harwood, C.R.; Barer, M.R. Toxin gene expression by shiga toxin-producing Escherichia coli: The role of antibiotics and the bacterial SOS response. Emerg. Infect. Dis. 2000, 6, 458–465. [Google Scholar] [CrossRef] [PubMed]
- Sambrook, J.; Fritsch, E.F.; Maniatis, T. Molecular Cloning: A Laboratory Manual; Cold Spring Harbor Laboratory: Laurel Hollow, NY, USA, 1989. [Google Scholar]
- Ye, J.; Coulouris, G.; Zaretskaya, I.; Cutcutache, I.; Rozen, S.; Madden, T.L. Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinform. 2012, 13, 134. [Google Scholar] [CrossRef] [PubMed]
- Gobert, A.P.; Vareille, M.; Glasser, A.L.; Hindré, T.; de Sablet, T.; Martin, C. Shiga toxin produced by enterohemorrhagic Escherichia coli inhibits PI3K/NF-kappaB signaling pathway in globotriaosylceramide-3-negative human intestinal epithelial cells. J. Immunol. 2007, 178, 8168–8174. [Google Scholar] [CrossRef] [PubMed]
- Rusconi, B.; Eppinger, M. Whole genome sequence typing strategies for enterohaemorrhagic Escherichia coli of the O157: H7 serotype. In The Handbook of Microbial Bioresources; CABI: Wallingford, UK, 2016; pp. 616–633. [Google Scholar]
- Goldstein, S.; Beka, L.; Graf, J.; Klassen, J.L. Evaluation of strategies for the assembly of diverse bacterial genomes using MinION long-read sequencing. BMC Genom. 2019, 20, 23. [Google Scholar] [CrossRef] [PubMed]
- Jaudou, S.; Tran, M.L.; Vorimore, F.; Fach, P.; Delannoy, S. Evaluation of high molecular weight DNA extraction methods for long-read sequencing of Shiga toxin-producing Escherichia coli. PLoS ONE 2022, 17, e0270751. [Google Scholar] [CrossRef] [PubMed]
- Asadulghani, M.; Ogura, Y.; Ooka, T.; Itoh, T.; Sawaguchi, A.; Iguchi, A.; Nakayama, K.; Hayashi, T. The defective prophage pool of Escherichia coli O157: Prophage-prophage interactions potentiate horizontal transfer of virulence determinants. PLoS Pathog. 2009, 5, e1000408. [Google Scholar] [CrossRef] [PubMed]
- Shaaban, S.; Cowley, L.A.; McAteer, S.P.; Jenkins, C.; Dallman, T.J.; Bono, J.L.; Gally, D.L. Evolution of a zoonotic pathogen: Investigating prophage diversity in enterohaemorrhagic Escherichia coli O157 by long-read sequencing. Microb. Genom. 2016, 2, e000096. [Google Scholar] [CrossRef] [PubMed]
- Stanton, E.; Park, D.; Dopfer, D.; Ivanek, R.; Kaspar, C.W. Phylogenetic characterization of Escherichia coli O157: H7 based on IS629 distribution and Shiga toxin genotype. Microbiology 2014, 160, 502–513. [Google Scholar] [CrossRef] [PubMed]
- Toro, M.; Rump, L.V.; Cao, G.; Meng, J.; Brown, E.W.; Gonzalez-Escalona, N. Simultaneous presence of insertion sequence-excision enhancer (IEE) and insertion sequence IS629 correlates with increased diversity and virulence in Shiga-toxin producing Escherichia coli (STEC). J. Clin. Microbiol. 2015, 53, 3466–3473. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, E.; Hashimoto, R.; Etoh, Y.; Ichihara, S.; Horikawa, K.; Uchimura, M. Biased distribution of IS629 among strains in different lineages of enterohemorrhagic Escherichia coli serovar O157. Infect. Genet. Evol. J. Mol. Epidemiol. Evol. Genet. Infect. Dis. 2011, 11, 78–82. [Google Scholar] [CrossRef] [PubMed]
- Ohnishi, M.; Kurokawa, K.; Hayashi, T. Diversification of Escherichia coli genomes: Are bacteriophages the major contributors? Trends Microbiol. 2001, 9, 481–485. [Google Scholar] [CrossRef] [PubMed]
- Kruger, A.; Lucchesi, P.M. Shiga toxins and stx phages: Highly diverse entities. Microbiology 2015, 161, 451–462. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Aljaro, C.; Muniesa, M.; Jofre, J.; Blanch, A.R. Genotypic and phenotypic diversity among induced, stx2-carrying bacteriophages from environmental Escherichia coli strains. Appl. Environ. Microbiol. 2009, 75, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Canchaya, C.; Fournous, G.; Brussow, H. The impact of prophages on bacterial chromosomes. Mol. Microbiol. 2004, 53, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Fortier, L.C.; Sekulovic, O. Importance of prophages to evolution and virulence of bacterial pathogens. Virulence 2013, 4, 354–365. [Google Scholar] [CrossRef] [PubMed]
- Tozzoli, R.; Grande, L.; Michelacci, V.; Ranieri, P.; Maugliani, A.; Caprioli, A.; Morabito, S. Shiga toxin-converting phages and the emergence of new pathogenic Escherichia coli: A world in motion. Front. Cell. Infect. Microbiol. 2014, 4, 80. [Google Scholar] [CrossRef] [PubMed]
- Creuzburg, K.; Köhler, B.; Hempel, H.; Schreier, P.; Jacobs, E.; Schmidt, H. Genetic structure and chromosomal integration site of the cryptic prophage CP-1639 encoding Shiga toxin 1. Microbiology 2005, 151, 941–950. [Google Scholar] [CrossRef] [PubMed]
- Clermont, O.; Gordon, D.; Denamur, E. Guide to the various phylogenetic classification schemes for Escherichia coli and the correspondence among schemes. Microbiology 2015, 161, 980–988. [Google Scholar] [CrossRef] [PubMed]
- Leopold, S.R.; Magrini, V.; Holt, N.J.; Shaikh, N.; Mardis, E.R.; Cagno, J.; Ogura, Y.; Iguchi, A.; Hayashi, T.; Mellmann, A.; et al. A precise reconstruction of the emergence and constrained radiations of Escherichia coli O157 portrayed by backbone concatenomic analysis. Proc. Natl. Acad. Sci. USA 2009, 106, 8713–8718. [Google Scholar] [CrossRef] [PubMed]
- Ogura, Y.; Mondal, S.I.; Islam, M.R.; Mako, T.; Arisawa, K.; Katsura, K.; Ooka, T.; Gotoh, Y.; Murase, K.; Ohnishi, M.; et al. The Shiga toxin 2 production level in enterohemorrhagic Escherichia coli O157:H7 is correlated with the subtypes of toxin-encoding phage. Sci. Rep. 2015, 5, 16663. [Google Scholar] [CrossRef] [PubMed]
- Miyata, T.; Taniguchi, I.; Nakamura, K.; Gotoh, Y.; Yoshimura, D.; Itoh, T.; Hirai, S.; Yokoyama, E.; Ohnishi, M.; Iyoda, S.; et al. Alteration of a Shiga toxin-encoding phage associated with a change in toxin production level and disease severity in Escherichia coli. Microb. Genom. 2023, 9, 000935. [Google Scholar] [CrossRef] [PubMed]
- Fagerlund, A.; Aspholm, M.; Węgrzyn, G.; Lindbäck, T. High diversity in the regulatory region of Shiga toxin encoding bacteriophages. BMC Genom. 2022, 23, 230. [Google Scholar] [CrossRef] [PubMed]
- Llarena, A.K.; Aspholm, M.; O’Sullivan, K.; Wêgrzyn, G.; Lindbäck, T. Replication Region Analysis Reveals Non-lambdoid Shiga Toxin Converting Bacteriophages. Front. Microbiol. 2021, 12, 640945. [Google Scholar] [CrossRef] [PubMed]
- Gottesman, M.E.; Weisberg, R.A. Little lambda, who made thee? Microbiol. Mol. Biol. Rev. 2004, 68, 796–813. [Google Scholar] [CrossRef] [PubMed]
- van Belkum, A.; Scherer, S.; van Alphen, L.; Verbrugh, H. Short-sequence DNA repeats in prokaryotic genomes. Microbiol. Mol. Biol. Rev. MMBR 1998, 62, 275–293. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.; Bibi, E. A single membrane-embedded negative charge is critical for recognizing positively charged drugs by the Escherichia coli multidrug resistance protein MdfA. EMBO J. 1999, 18, 822–832. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Lin, R.; Zhou, Z.; Ma, J.; Lin, H.; Zheng, X.; Wang, J.; Wu, J.; Dong, Y.; Jiang, H.; et al. Antimicrobial resistance and genomic characterization of Escherichia coli from pigs and chickens in Zhejiang, China. Front. Microbiol. 2022, 13, 1018682. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.Y.; Lavelle, K.; Huang, A.; Atwill, E.R.; Pitesky, M.; Li, X. Assessment of Prevalence and Diversity of Antimicrobial Resistant Escherichia coli from Retail Meats in Southern California. Antibiotics 2023, 12, 782. [Google Scholar] [CrossRef] [PubMed]
- Awosile, B.; Fritzler, J.; Levent, G.; Rahman, M.K.; Ajulo, S.; Daniel, I.; Tasnim, Y.; Sarkar, S. Genomic Characterization of Fecal Escherichia coli Isolates with Reduced Susceptibility to Beta-Lactam Antimicrobials from Wild Hogs and Coyotes. Pathogens 2023, 12, 929. [Google Scholar] [CrossRef]
- Law, D. Virulence factors of Escherichia coli O157 and other Shiga toxin-producing E. coli. J. Appl. Microbiol. 2000, 88, 729–745. [Google Scholar] [CrossRef] [PubMed]
- Levine, M.M. Escherichia-Coli That Cause Diarrhea-Enterotoxigenic, Enteropathogenic, Enteroinvasive, Enterohemorrhagic, and Enteroadherent. J. Infect. Dis. 1987, 155, 377–389. [Google Scholar] [CrossRef] [PubMed]
- Franzin, F.M.; Sircili, M.P. Locus of enterocyte effacement: A pathogenicity island involved in the virulence of enteropathogenic and enterohemorragic Escherichia coli subjected to a complex network of gene regulation. Biomed. Res. Int. 2015, 2015, 534738. [Google Scholar] [CrossRef]
- McDaniel, T.K.; Jarvis, K.G.; Donnenberg, M.S.; Kaper, J.B. A genetic locus of enterocyte effacement conserved among diverse enterobacterial pathogens. Proc. Natl. Acad. Sci. USA 1995, 92, 1664–1668. [Google Scholar] [CrossRef] [PubMed]
- Sperandio, V.; Kaper, J.B.; Bortolini, M.R.; Neves, B.C.; Keller, R.; Trabulsi, L.R. Characterization of the locus of enterocyte effacement (LEE) in different enteropathogenic Escherichia coli (EPEC) and Shiga-toxin producing Escherichia coli (STEC) serotypes. FEMS Microbiol. Lett. 1998, 164, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Jerse, A.E.; Yu, J.; Tall, B.D.; Kaper, J.B. A genetic locus of enteropathogenic Escherichia coli necessary for the production of attaching and effacing lesions on tissue culture cells. Proc. Natl. Acad. Sci. USA 1990, 87, 7839–7843. [Google Scholar] [CrossRef] [PubMed]
- Bibbal, D.; Loukiadis, E.; Kérourédan, M.; Peytavin de Garam, C.; Ferré, F.; Cartier, P.; Gay, E.; Oswald, E.; Auvray, F.; Brugère, H. Intimin gene (eae) subtype-based real-time PCR strategy for specific detection of Shiga toxin-producing Escherichia coli serotypes O157:H7, O26:H11, O103:H2, O111:H8, and O145:H28 in cattle feces. Appl. Environ. Microbiol. 2014, 80, 1177–1184. [Google Scholar] [CrossRef]
- Oswald, E.; Schmidt, H.; Morabito, S.; Karch, H.; Marchès, O.; Caprioli, A. Typing of intimin genes in human and animal enterohemorrhagic and enteropathogenic Escherichia coli: Characterization of a new intimin variant. Infect. Immun. 2000, 68, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Moon, H.W.; Whipp, S.C.; Argenzio, R.A.; Levine, M.M.; Giannella, R.A. Attaching and effacing activities of rabbit and human enteropathogenic Escherichia coli in pig and rabbit intestines. Infect. Immun. 1983, 41, 1340–1351. [Google Scholar] [CrossRef] [PubMed]
- Kaper, J.B.; Nataro, J.P.; Mobley, H.L. Pathogenic Escherichia coli. Nat. Rev. Microbiol. 2004, 2, 123–140. [Google Scholar] [CrossRef] [PubMed]
- Jores, J.; Rumer, L.; Wieler, L.H. Impact of the locus of enterocyte effacement pathogenicity island on the evolution of pathogenic Escherichia coli. Int. J. Med. Microbiol. 2004, 294, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Bono, J.L.; Weinroth, M.; Clawson, M.; Harhay, G.P.; Eppinger, M.; Harhay, D.; Smith, T.P. Escherichia coli O157:H7 tir 255 T > A allele strains differ in chromosomal and plasmid composition. Front. Microbiol. 2023, 14, 1303387. [Google Scholar] [CrossRef]
- Burland, V.; Shao, Y.; Perna, N.T.; Plunkett, G.; Sofia, H.J.; Blattner, F.R. The complete DNA sequence and analysis of the large virulence plasmid of Escherichia coli O157:H7. Nucleic Acids Res. 1998, 26, 4196–4204. [Google Scholar] [CrossRef] [PubMed]
- Rump, L.V.; Meng, J.; Strain, E.A.; Cao, G.; Allard, M.W.; Gonzalez-Escalona, N. Complete DNA sequence analysis of enterohemorrhagic Escherichia coli plasmid pO157_2 in beta-glucuronidase-positive E. coli O157:H7 reveals a novel evolutionary path. J. Bacteriol. 2012, 194, 3457–3463. [Google Scholar] [CrossRef] [PubMed]
- Brunder, W.; Karch, H.; Schmidt, H. Complete sequence of the large virulence plasmid pSFO157 of the sorbitol-fermenting enterohemorrhagic Escherichia coli O157:H- strain 3072/96. Int. J. Med. Microbiol. 2006, 296, 467–474. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Escalona, N.; Allard, M.A.; Brown, E.W.; Sharma, S.; Hoffmann, M. Nanopore sequencing for fast determination of plasmids, phages, virulence markers, and antimicrobial resistance genes in Shiga toxin-producing Escherichia coli. PLoS ONE 2019, 14, e0220494. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.Y.; La, H.J.; Sheng, H.; Forney, L.J.; Hovde, C.J. Influence of plasmid pO157 on Escherichia coli O157:H7 Sakai biofilm formation. Appl. Environ. Microbiol. 2010, 76, 963–966. [Google Scholar] [CrossRef] [PubMed]
- Johnson, T.J.; Nolan, L.K. Pathogenomics of the virulence plasmids of Escherichia coli. Microbiol. Mol. Biol. Rev. MMBR 2009, 73, 750–774. [Google Scholar] [CrossRef]
- Lim, J.Y.; Sheng, H.; Seo, K.S.; Park, Y.H.; Hovde, C.J. Characterization of an Escherichia coli O157:H7 plasmid O157 deletion mutant and its survival and persistence in cattle. Appl. Environ. Microbiol. 2007, 73, 2037–2047. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, H.; Beutin, L.; Karch, H. Molecular analysis of the plasmid-encoded hemolysin of Escherichia coli O157:H7 strain EDL 933. Infect. Immun. 1995, 63, 1055–1061. [Google Scholar] [CrossRef] [PubMed]
- Bielaszewska, M.; Rüter, C.; Kunsmann, L.; Greune, L.; Bauwens, A.; Zhang, W.; Kuczius, T.; Kim, K.S.; Mellmann, A.; Schmidt, M.A.; et al. Enterohemorrhagic Escherichia coli hemolysin employs outer membrane vesicles to target mitochondria and cause endothelial and epithelial apoptosis. PLoS Pathog. 2013, 9, e1003797. [Google Scholar] [CrossRef] [PubMed]
- Lathem, W.W.; Grys, T.E.; Witowski, S.E.; Torres, A.G.; Kaper, J.B.; Tarr, P.I.; Welch, R.A. StcE, a metalloprotease secreted by Escherichia coli O157:H7, specifically cleaves C1 esterase inhibitor. Mol. Microbiol. 2002, 45, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Serra-Moreno, R.; Jofre, J.; Muniesa, M. The CI repressors of Shiga toxin-converting prophages are involved in coinfection of Escherichia coli strains, which causes a down regulation in the production of Shiga toxin 2. J. Bacteriol. 2008, 190, 4722–4735. [Google Scholar] [CrossRef] [PubMed]
- Donohue-Rolfe, A.; Kondova, I.; Oswald, S.; Hutto, D.; Tzipori, S. Escherichia coli O157:H7 strains that express Shiga toxin (Stx) 2 alone are more neurotropic for gnotobiotic piglets than are isotypes producing only Stx1 or both Stx1 and Stx2. J. Infect. Dis. 2000, 181, 1825–1829. [Google Scholar] [CrossRef]
- Russo, L.M.; Melton-Celsa, A.R.; O’Brien, A.D. Shiga Toxin (Stx) Type 1a Reduces the Oral Toxicity of Stx Type 2a. J. Infect. Dis. 2016, 213, 1271–1279. [Google Scholar] [CrossRef] [PubMed]
- Castro, V.S.; Ortega Polo, R.; Figueiredo, E.E.S.; Bumunange, E.W.; McAllister, T.; King, R.; Conte-Junior, C.A.; Stanford, K. Inconsistent PCR detection of Shiga toxin-producing Escherichia coli: Insights from whole genome sequence analyses. PLoS ONE 2021, 16, e0257168. [Google Scholar] [CrossRef] [PubMed]
- Whitworth, J.H.; Fegan, N.; Keller, J.; Gobius, K.S.; Bono, J.L.; Call, D.R.; Hancock, D.D.; Besser, T.E. International comparison of clinical, bovine, and environmental Escherichia coli O157 isolates on the basis of Shiga toxin-encoding bacteriophage insertion site genotypes. Appl. Environ. Microbiol. 2008, 74, 7447–7450. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Besser, T.E.; Shaikh, N.; Holt, N.J.; Tarr, P.I.; Konkel, M.E.; Malik-Kale, P.; Walsh, C.W.; Whittam, T.S.; Bono, J.L. Greater diversity of Shiga toxin-encoding bacteriophage insertion sites among Escherichia coli O157:H7 isolates from cattle than in those from humans. Appl. Environ. Microbiol. 2007, 73, 671–679. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Russo, L.M.; Melton-Celsa, A.R.; Smith, M.J.; O’Brien, A.D. Comparisons of native Shiga toxins (Stxs) type 1 and 2 with chimeric toxins indicate that the source of the binding subunit dictates degree of toxicity. PLoS ONE 2014, 9, e93463. [Google Scholar] [CrossRef] [PubMed]
- Ostroff, S.M.; Tarr, P.I.; Neill, M.A.; Lewis, J.H.; Hargrett-Bean, N.; Kobayashi, J.M. Toxin genotypes and plasmid profiles as determinants of systemic sequelae in Escherichia coli O157:H7 infections. J. Infect. Dis. 1989, 160, 994–998. [Google Scholar] [CrossRef] [PubMed]
- Petro, C.D.; Trojnar, E.; Sinclair, J.; Liu, Z.M.; Smith, M.; O’Brien, A.D.; Melton-Celsa, A. Shiga Toxin Type 1a (Stx1a) Reduces the Toxicity of the More Potent Stx2a In Vivo and In Vitro. Infect. Immun. 2019, 87, e00787-18. [Google Scholar] [CrossRef] [PubMed]
- Allue-Guardia, A.; Koenig, S.S.K.; Martinez, R.A.; Rodriguez, A.L.; Bosilevac, J.M.; Feng, P.; Eppinger, M. Pathogenomes and variations in Shiga toxin production among geographically distinct clones of Escherichia coli O113:H21. Microb. Genom. 2022, 8, 000796. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, A.L.; Rasko, D.A.; Mobley, H.L. Defining genomic islands and uropathogen-specific genes in uropathogenic Escherichia coli. J. Bacteriol. 2007, 189, 3532–3546. [Google Scholar] [CrossRef] [PubMed]
- Perna, N.T.; Plunkett, G., 3rd; Burland, V.; Mau, B.; Glasner, J.D.; Rose, D.J.; Mayhew, G.F.; Evans, P.S.; Gregor, J.; Kirkpatrick, H.A.; et al. Genome sequence of enterohaemorrhagic Escherichia coli O157:H7. Nature 2001, 409, 529–533. [Google Scholar] [CrossRef] [PubMed]
- Imamovic, L.; Muniesa, M. Characterizing RecA-independent induction of Shiga toxin2-encoding phages by EDTA treatment. PLoS ONE 2012, 7, e32393. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, S.; Muhldorfer, I.; Donohue-Rolfe, A.; Kerenyi, M.; Emody, L.; Alexiev, R.; Nenkov, P.; Hacker, J. Influence of RecA on in vivo virulence and Shiga toxin 2 production in Escherichia coli pathogens. Microb. Pathog. 1999, 27, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Fernández De Henestrosa, A.R.; Ogi, T.; Aoyagi, S.; Chafin, D.; Hayes, J.J.; Ohmori, H.; Woodgate, R. Identification of additional genes belonging to the LexA regulon in Escherichia coli. Mol. Microbiol. 2000, 35, 1560–1572. [Google Scholar] [CrossRef] [PubMed]
- Holmes, A.; Allison, L.; Ward, M.; Dallman, T.J.; Clark, R.; Fawkes, A.; Murphy, L.; Hanson, M. Utility of Whole-Genome Sequencing of Escherichia coli O157 for Outbreak Detection and Epidemiological Surveillance. J. Clin. Microbiol. 2015, 53, 3565–3573. [Google Scholar] [CrossRef] [PubMed]
- Denamur, E.; Clermont, O.; Bonacorsi, S.; Gordon, D. The population genetics of pathogenic Escherichia coli. Nat. Rev. Microbiol. 2021, 19, 37–54. [Google Scholar] [CrossRef] [PubMed]
- Griffing, S.M.; MacCannell, D.R.; Schmidtke, A.J.; Freeman, M.M.; Hyytia-Trees, E.; Gerner-Smidt, P.; Ribot, E.M.; Bono, J.L. Canonical Single Nucleotide Polymorphisms (SNPs) for High-Resolution Subtyping of Shiga-Toxin Producing Escherichia coli (STEC) O157:H7. PLoS ONE 2015, 10, e0131967. [Google Scholar] [CrossRef] [PubMed]
- Karch, H.; Meyer, T.; Rüssmann, H.; Heesemann, J. Frequent loss of Shiga-like toxin genes in clinical isolates of Escherichia coli upon subcultivation. Infect. Immun. 1992, 60, 3464–3467. [Google Scholar] [CrossRef] [PubMed]
- Schutz, K.; Cowley, L.A.; Shaaban, S.; Carroll, A.; McNamara, E.; Gally, D.L.; Godbole, G.; Jenkins, C.; Dallman, T.J. Evolutionary Context of Non-Sorbitol-Fermenting Shiga Toxin-Producing Escherichia coli O55:H7. Emerg. Infect. Dis. 2017, 23, 1966–1973. [Google Scholar] [CrossRef] [PubMed]
- Lainhart, W.; Stolfa, G.; Koudelka, G.B. Shiga toxin as a bacterial defense against a eukaryotic predator, Tetrahymena thermophila. J. Bacteriol. 2009, 191, 5116–5122. [Google Scholar] [CrossRef] [PubMed]
- King, A.J.; Sundaram, S.; Cendoroglo, M.; Acheson, D.W.; Keusch, G.T. Shiga toxin induces superoxide production in polymorphonuclear cells with subsequent impairment of phagocytosis and responsiveness to phorbol esters. J. Infect. Dis. 1999, 179, 503–507. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, K.M.; Levin, B.R. Grazing protozoa and the evolution of the Escherichia coli O157:H7 Shiga toxin-encoding prophage. Proc. Biol. Sci. 2007, 274, 1921–1929. [Google Scholar] [CrossRef] [PubMed]
- Poirier, K.; Faucher, S.P.; Beland, M.; Brousseau, R.; Gannon, V.; Martin, C.; Harel, J.; Daigle, F. Escherichia coli O157:H7 survives within human macrophages: Global gene expression profile and involvement of the Shiga toxins. Infect. Immun. 2008, 76, 4814–4822. [Google Scholar] [CrossRef] [PubMed]
- Eklund, M.; Leino, K.; Siitonen, A. Clinical Escherichia coli strains carrying stx genes: Stx variants and stx-positive virulence profiles. J. Clin. Microbiol. 2002, 40, 4585–4593. [Google Scholar] [CrossRef] [PubMed]
- Los, J.M.; Los, M.; Wegrzyn, A.; Wegrzyn, G. Altruism of Shiga toxin-producing Escherichia coli: Recent hypothesis versus experimental results. Front. Cell. Infect. Microbiol. 2012, 2, 166. [Google Scholar] [CrossRef]
- Los, J.M.; Los, M.; Wegrzyn, G. Bacteriophages carrying Shiga toxin genes: Genomic variations, detection and potential treatment of pathogenic bacteria. Future Microbiol. 2011, 6, 909–924. [Google Scholar] [CrossRef] [PubMed]
- Stolfa, G.; Koudelka, G.B. Entry and killing of Tetrahymena thermophila by bacterially produced Shiga toxin. mBio 2012, 4, e00416-12. [Google Scholar] [CrossRef] [PubMed]
- Veses-Garcia, M.; Liu, X.; Rigden, D.J.; Kenny, J.G.; McCarthy, A.J.; Allison, H.E. Transcriptomic analysis of Shiga-toxigenic bacteriophage carriage reveals a profound regulatory effect on acid resistance in Escherichia coli. Appl. Environ. Microbiol. 2015, 81, 8118–8125. [Google Scholar] [CrossRef] [PubMed]
- Riley, L.M.; Veses-Garcia, M.; Hillman, J.D.; Handfield, M.; McCarthy, A.J.; Allison, H.E. Identification of genes expressed in cultures of E. coli lysogens carrying the Shiga toxin-encoding prophage Phi24B. BMC Microbiol. 2012, 12, 42. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; McAteer, S.P.; Tree, J.J.; Shaw, D.J.; Wolfson, E.B.; Beatson, S.A.; Roe, A.J.; Allison, L.J.; Chase-Topping, M.E.; Mahajan, A.; et al. Lysogeny with Shiga toxin 2-encoding bacteriophages represses type III secretion in enterohemorrhagic Escherichia coli. PLoS Pathog. 2012, 8, e1002672. [Google Scholar] [CrossRef] [PubMed]
- Flockhart, A.F.; Tree, J.J.; Xu, X.; Karpiyevich, M.; McAteer, S.P.; Rosenblum, R.; Shaw, D.J.; Low, C.J.; Best, A.; Gannon, V.; et al. Identification of a novel prophage regulator in Escherichia coli controlling the expression of type III secretion. Mol. Microbiol. 2012, 83, 208–223. [Google Scholar] [CrossRef]
- Berger, P.; Kouzel, I.U.; Berger, M.; Haarmann, N.; Dobrindt, U.; Koudelka, G.B.; Mellmann, A. Carriage of Shiga toxin phage profoundly affects Escherichia coli gene expression and carbon source utilization. BMC Genom. 2019, 20, 504. [Google Scholar] [CrossRef]
- Robinson, C.M.; Sinclair, J.F.; Smith, M.J.; O’Brien, A.D. Shiga toxin of enterohemorrhagic Escherichia coli type O157:H7 promotes intestinal colonization. Proc. Natl. Acad. Sci. USA 2006, 103, 9667–9672. [Google Scholar] [CrossRef] [PubMed]
- Yokoigawa, K.; Takikawa, A.; Okubo, Y.; Umesako, S. Acid tolerance and gad mRNA levels of Escherichia coli O157:H7 grown in foods. Int. J. Food Microbiol. 2003, 82, 203–211. [Google Scholar] [CrossRef]
- Saile, N.; Voigt, A.; Kessler, S.; Stressler, T.; Klumpp, J.; Fischer, L.; Schmidt, H. Escherichia coli O157:H7 Strain EDL933 Harbors Multiple Functional Prophage-Associated Genes Necessary for the Utilization of 5-N-Acetyl-9-O-Acetyl Neuraminic Acid as a Growth Substrate. Appl. Environ. Microbiol. 2016, 82, 5940–5950. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Doria, J.D.; Sperandio, V. Bacteriophage Transcription Factor Cro Regulates Virulence Gene Expression in Enterohemorrhagic Escherichia coli. Cell Host Microbe 2018, 23, 607–617. [Google Scholar] [CrossRef] [PubMed]
- Su, L.K.; Lu, C.P.; Wang, Y.; Cao, D.M.; Sun, J.H.; Yan, Y.X. Lysogenic infection of a Shiga toxin 2-converting bacteriophage changes host gene expression, enhances host acid resistance and motility. Mol. Biol. 2010, 44, 54–66. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).