
Genomoviruses in Liver Samples of Molossus molossus Bats


 , , , , , , ,
, , , , , , ,  and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Processing of Samples
2.3. Nucleic Acid Extraction (DNA/RNA)
2.4. Preparation of Libraries for the Illumina Platform
2.5. Bioinformatics Analysis
2.6. Genome Annotation
2.7. Genetic Distance
2.8. Phylogenetic Analysis
3. Results
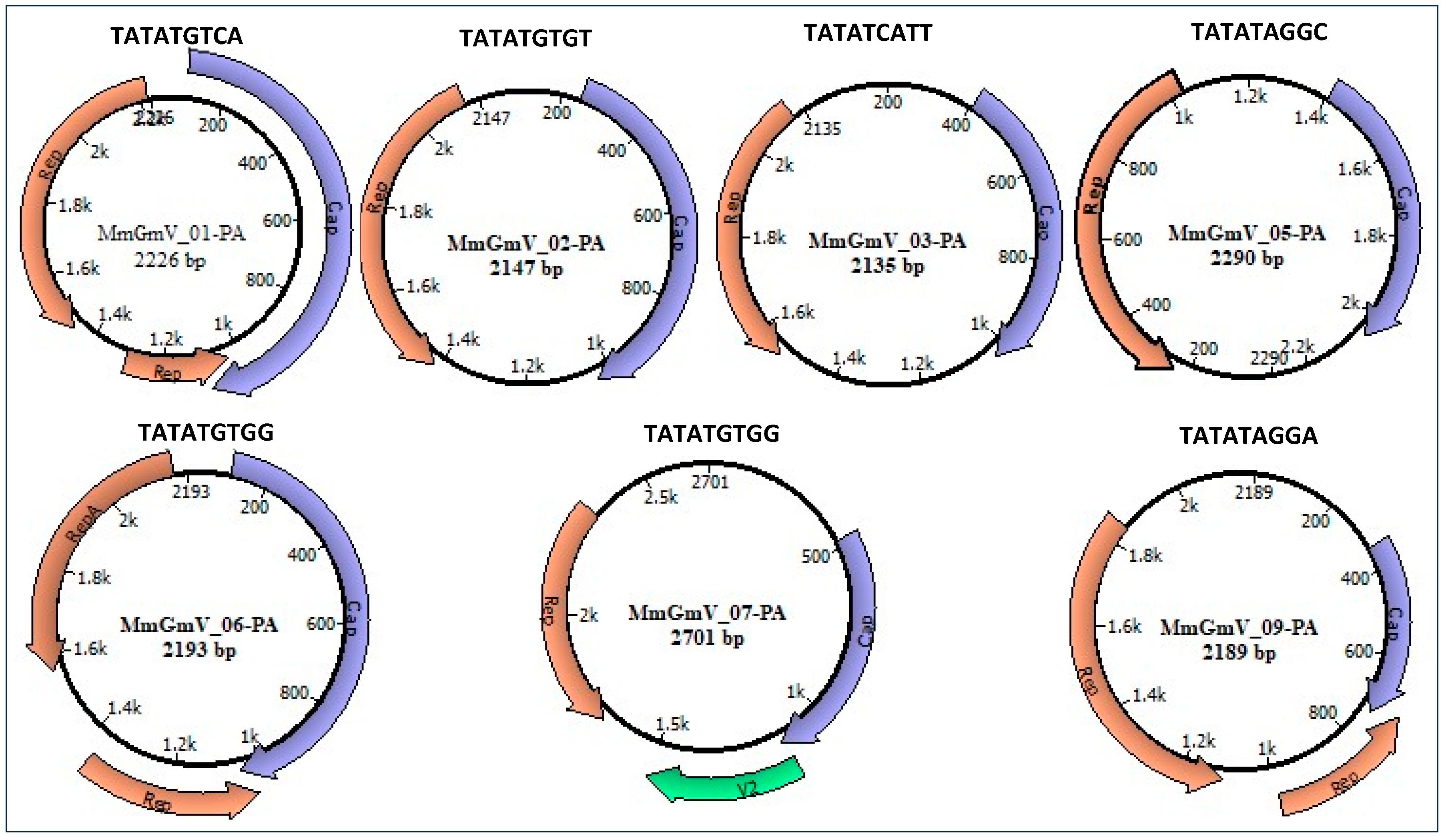
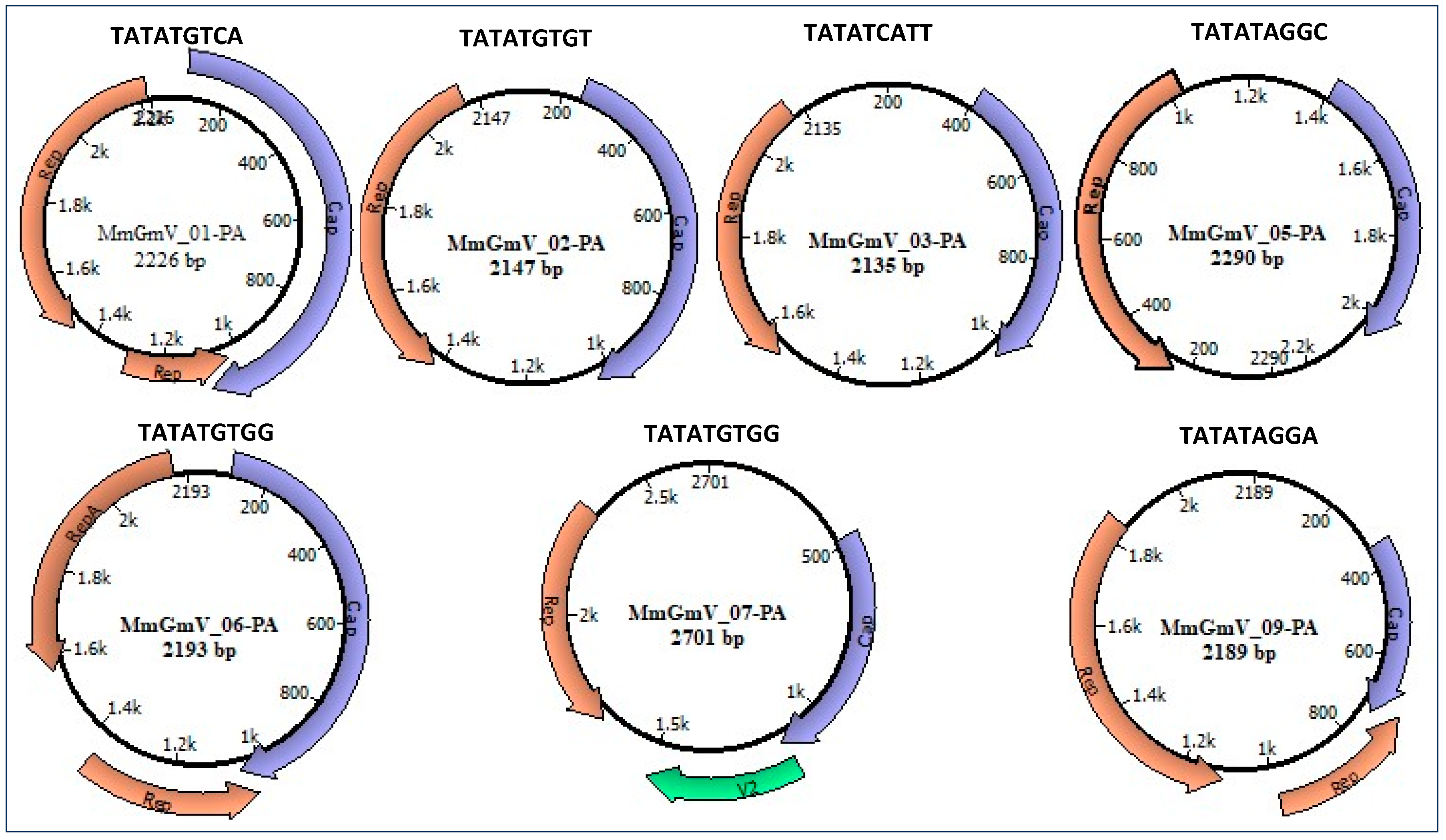
3.1. Identification of Genomoviridae
3.2. Genome Characterization
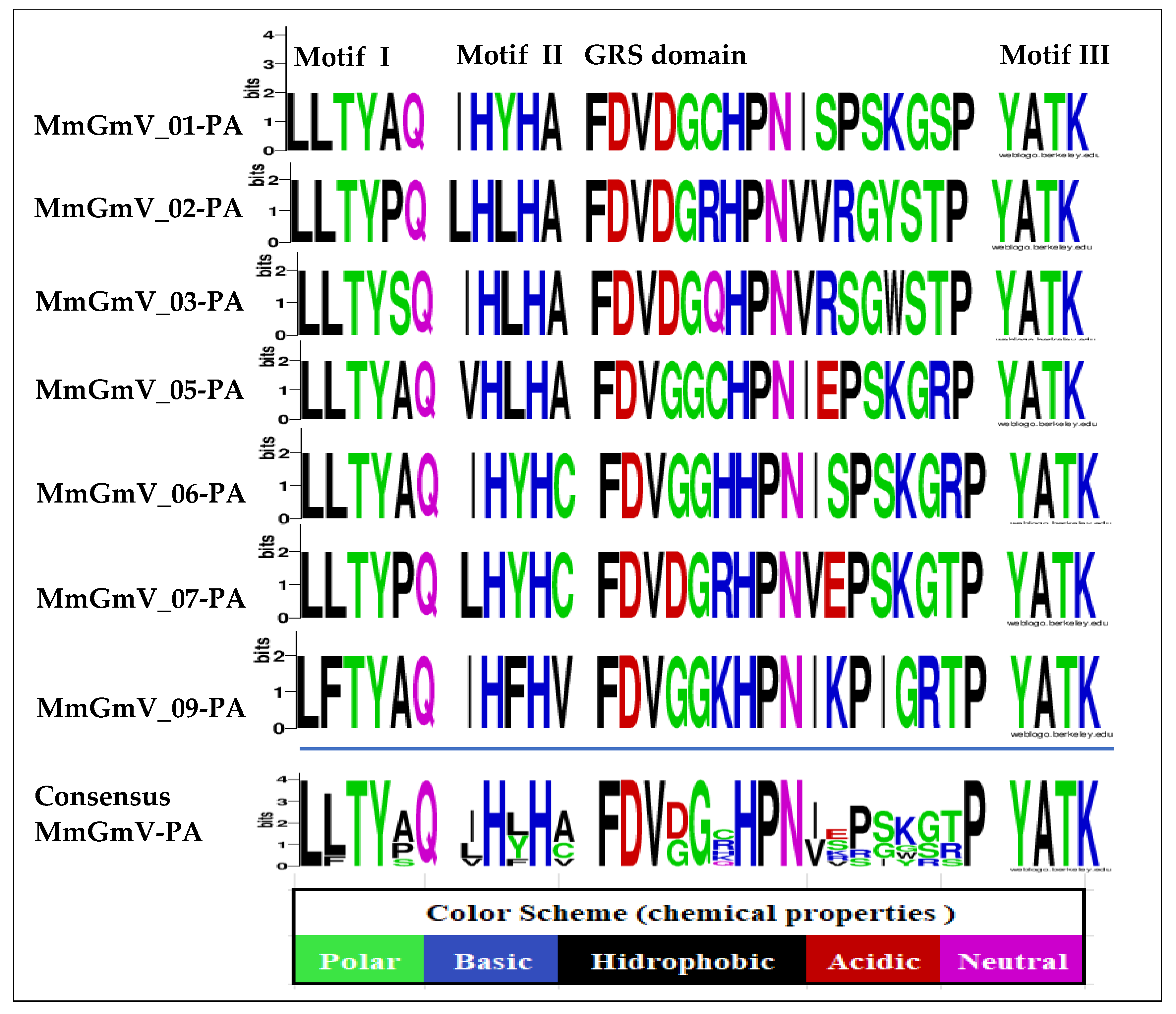
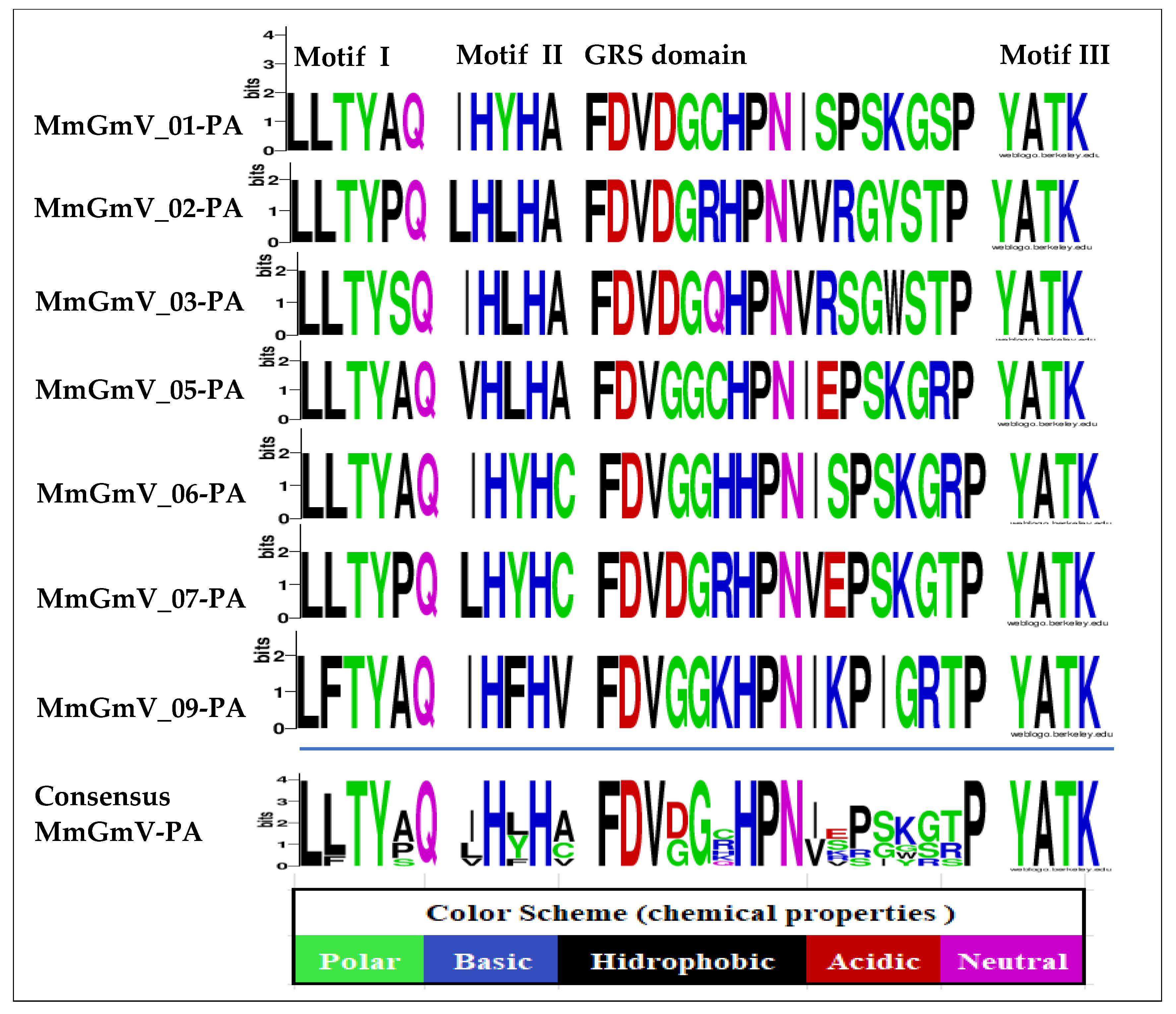
3.3. Analysis of Conserved Motifs
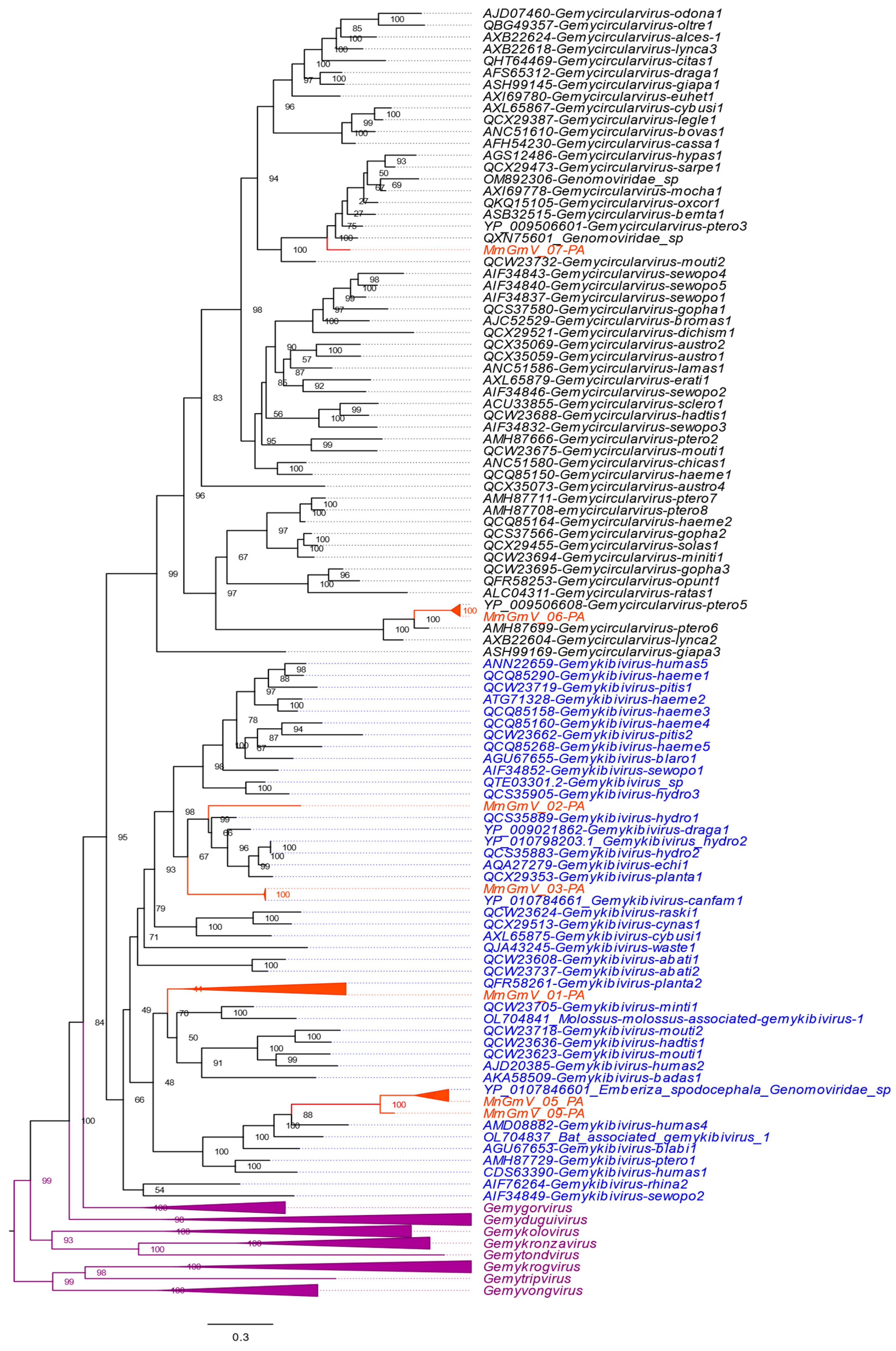
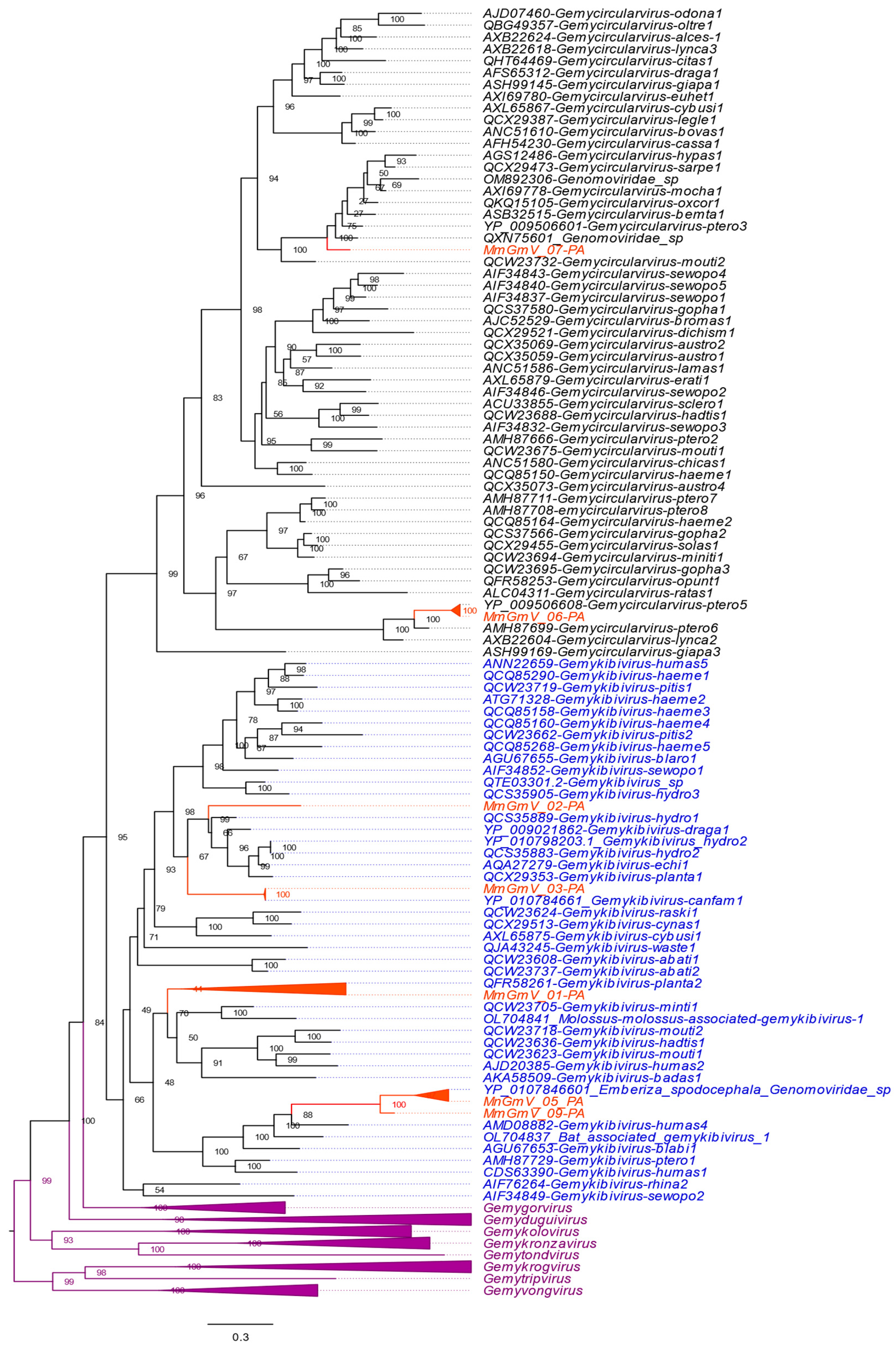
3.4. Genetic Distances and Phylogenetic Inferences
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Roes, F.L. On the Evolution of Virulent Zoonotic Viruses in Bats. Biol. Theory 2020, 15, 223–225. [Google Scholar] [CrossRef]
- Wang, L.-F.; Anderson, D.E. Viruses in bats and potential spillover to animals and humans. Curr. Opin. Virol. 2019, 34, 79–89. [Google Scholar] [CrossRef]
- Kirtipal, N.; Bharadwaj, S.; Kang, S.G. From SARS to SARS-CoV-2, insights on structure, pathogenicity and immunity aspects of pandemic human coronaviruses. Infect. Genet. Evol. 2020, 85, 104502. [Google Scholar] [CrossRef]
- Epstein, J.H.; Anthony, S.J.; Islam, A.; Kilpatrick, A.M.; Khan, S.A.; Balkey, M.D.; Ross, N.; Smith, I.; Zambrana-Torrelio, C.; Tao, Y.; et al. Nipah virus dynamics in bats and implications for spillover to humans. Proc. Natl. Acad. Sci. USA 2020, 117, 29190–29201. [Google Scholar] [CrossRef]
- Khusro, A.; Aarti, C.; Pliego, A.B.; Cipriano-Salazar, M. Hendra Virus Infection in Horses: A Review on Emerging Mystery Paramyxovirus. J. Equine Vet. Sci. 2020, 91, 103149. [Google Scholar] [CrossRef]
- Samies, N.L.; James, S.H. Prevention and treatment of neonatal herpes simplex virus infection. Antivir. Res. 2020, 176, 104721. [Google Scholar] [CrossRef]
- Leguia, M.; Vila-Sanjurjo, A.; Chain, P.S.G.; Berry, I.M.; Jarman, R.G.; Pollett, S. Precision Medicine and Precision Public Health in the Era of Pathogen Next-Generation Sequencing. J. Infect. Dis. 2020, 221, S289–S291. [Google Scholar] [CrossRef]
- Somasekar, S.; Lee, D.; Rule, J.; Naccache, S.N.; Stone, M.; Busch, M.P.; Sanders, C.; Lee, W.M.; Chiu, C.Y. Viral Surveillance in Serum Samples from Patients with Acute Liver Failure By Metagenomic Next-Generation Sequencing. Clin. Infect. Dis. 2017, 65, 1477–1485. [Google Scholar] [CrossRef]
- Zhong, Y.; Xu, F.; Wu, J.; Schubert, J.; Li, M.M. Application of Next Generation Sequencing in Laboratory Medicine. Ann. Lab. Med. 2021, 41, 25–43. [Google Scholar] [CrossRef]
- Bolatti, E.M.; Viarengo, G.; Zorec, T.M.; Cerri, A.; Montani, M.E.; Hosnjak, L.; Casal, P.E.; Bortolotto, E.; Di Domenica, V.; Chouhy, D.; et al. Viral Metagenomic Data Analyses of Five New World Bat Species from Argentina: Identification of 35 Novel DNA Viruses. Microorganisms 2022, 10, 266. [Google Scholar] [CrossRef]
- Han, H.; Wen, H.; Zhao, L.; Liu, J.; Luo, L.; Zhou, C.; Qin, X.; Zhu, Y.; Liu, M.; Qi, R.; et al. Novel coronaviruses, astroviruses, adenoviruses and circoviruses in insectivorous bats from northern China. Zoonoses Public. Health 2017, 64, 636–646. [Google Scholar] [CrossRef]
- Wang, J.; Li, Y.; He, X.; Ma, J.; Hong, W.; Hu, F.; Zhao, L.; Li, Q.; Zhang, J.; Zhang, C.; et al. Gemykibivirus Genome in Lower Respiratory Tract of Elderly Woman With Unexplained Acute Respiratory Distress Syndrome. Clin. Infect. Dis. 2019, 69, 861–864. [Google Scholar] [CrossRef]
- Lamberto, I.; Gunst, K.; Müller, H.; Zur Hausen, H.; de Villiers, E.-M. Mycovirus-like DNA virus sequences from cattle serum and human brain and serum samples from multiple sclerosis patients. Genome Announc. 2014, 2, e00848-14. [Google Scholar] [CrossRef]
- Bezerra, R.S.; Bitencourt, H.T.; Covas, D.T.; Kashima, S.; Slavov, S.N. Metagenomic identification of human Gemykibivirus-2 (HuGkV-2) in parenterally infected blood donors from the Brazilian Amazon. Int. J. Infect. Dis. 2020, 98, 249–251. [Google Scholar] [CrossRef]
- Bolatti, E.M.; Zorec, T.M.; Montani, M.E.; Hošnjak, L.; Chouhy, D.; Viarengo, G.; Casal, P.E.; Barquez, R.M.; Poljak, M.; Giri, A.A. A Preliminary Study of the Virome of the South American Free-Tailed Bats (Tadarida brasiliensis) and Identification of Two Novel Mammalian Viruses. Viruses 2020, 12, 422. [Google Scholar] [CrossRef]
- Orton, J.P.; Morales, M.; Fontenele, R.S.; Schmidlin, K.; Kraberger, S.; Leavitt, D.J.; Webster, T.H.; Wilson, M.A.; Kusumi, K.; Dolby, G.A.; et al. Virus Discovery in Desert Tortoise Fecal Samples: Novel Circular Single-Stranded DNA Viruses. Viruses 2020, 12, 143. [Google Scholar] [CrossRef]
- Kaszab, E.; Lengyel, G.; Marton, S.; Dán, Á.; Bányai, K.; Fehér, E. Occurrence and genetic diversity of CRESS DNA viruses in wild birds: A Hungarian study. Sci. Rep. 2020, 10, 7036. [Google Scholar] [CrossRef]
- Rosario, K.; Dayaram, A.; Marinov, M.; Ware, J.; Kraberger, S.; Stainton, D.; Breitbart, M.; Varsani, A. Diverse circular ssDNA viruses discovered in dragonflies (Odonata: Epiprocta). J. Gen. Virol. 2012, 93 Pt 12, 2668–2681. [Google Scholar] [CrossRef]
- Dayaram, A.; Potter, K.A.; Pailes, R.; Marinov, M.; Rosenstein, D.D.; Varsani, A. Identification of diverse circular single-stranded DNA viruses in adult dragonflies and damselflies (Insecta: Odonata) of Arizona and Oklahoma, USA. Infect. Genet. Evol. 2015, 30, 278–287. [Google Scholar] [CrossRef]
- Du, Z.; Tang, Y.; Zhang, S.; She, X.; Lan, G.; Varsani, A.; He, Z. Identification and molecular characterization of a single-stranded circular DNA virus with similarities to Sclerotinia sclerotiorum hypovirulence-associated DNA virus 1. Arch. Virol. 2014, 159, 1527–1531. [Google Scholar] [CrossRef]
- Kinsella, C.M.; Bart, A.; Deijs, M.; Broekhuizen, P.; Kaczorowska, J.; Jebbink, M.F.; van Gool, T.; Cotten, M.; van der Hoek, L. Entamoeba and Giardia parasites implicated as hosts of CRESS viruses. Nat. Commun. 2020, 11, 4620. [Google Scholar] [CrossRef]
- Makoa-Meng, M.; Semmar, R.; Antezack, A.; Penant, G.; La Scola, B.; Monnet-Corti, V.; Colson, P. Correlation of Redondovirus and Entamoeba gingivalis Detections in the Human Oral Cavity Suggests That This Amoeba Is Possibly the Redondovirus Host. Int. J. Mol. Sci. 2023, 24, 6303. [Google Scholar] [CrossRef]
- Assis, M.R.d.S.; Vieira, C.B.; Fioretti, J.M.; Rocha, M.S.; de Almeida, P.I.N.; Miagostovich, M.P.; Fumian, T.M. Detection and Molecular Characterization of Gemycircularvirus from Environmental Samples in Brazil. Food Environ. Virol. 2016, 8, 305–309. [Google Scholar] [CrossRef]
- Siddell, S.G.; Walker, P.J.; Lefkowitz, E.J.; Mushegian, A.R.; Dutilh, B.E.; Harrach, B.; Harrison, R.L.; Junglen, S.; Knowles, N.J.; Kropinski, A.M.; et al. Binomial nomenclature for virus species: A consultation. Arch. Virol. 2020, 165, 519–525. [Google Scholar] [CrossRef]
- Varsani, A.; Krupovic, M. Family Genomoviridae: 2021 taxonomy update. Arch. Virol. 2021, 166, 2911–2926. [Google Scholar] [CrossRef]
- Simmonds, P.; Adams, M.J.; Benkő, M.; Breitbart, M.; Brister, J.R.; Carstens, E.B.; Davison, A.J.; Delwart, E.; Gorbalenya, A.E.; Harrach, B.; et al. Consensus statement: Virus taxonomy in the age of metagenomics. Nat. Rev. Microbiol. 2017, 15, 161–168. [Google Scholar] [CrossRef]
- Kazlauskas, D.; Varsani, A.; Krupovic, M. Pervasive Chimerism in the Replication-Associated Proteins of Uncultured Single-Stranded DNA Viruses. Viruses 2018, 10, 187. [Google Scholar] [CrossRef]
- Varsani, A.; Krupovic, M. Sequence-based taxonomic framework for the classification of uncultured single-stranded DNA viruses of the family Genomoviridae. Virus Evol. 2017, 3, vew037. [Google Scholar] [CrossRef]
- Gorbalenya, A.E.; Koonin, E.V.; Wolf, Y.I. A new superfamily of putative NTP-binding domains encoded by genomes of small DNA and RNA viruses. FEBS Lett. 1990, 262, 145–148. [Google Scholar] [CrossRef]
- Koonin, E.V.; Ilyina, T.V. Geminivirus replication proteins are related to prokaryotic plasmid rolling circle DNA replication initiator proteins. J. Gen. Virol. 1992, 73 Pt 10, 2763–2766. [Google Scholar] [CrossRef]
- Nash, T.E.; Dallas, M.B.; Reyes, M.I.; Buhrman, G.K.; Ascencio-Ibañez, J.T.; Hanley-Bowdoin, L. Functional analysis of a novel motif conserved across geminivirus Rep proteins. J. Virol. 2011, 85, 1182–1192. [Google Scholar] [CrossRef] [PubMed]
- Vega-Rocha, S.; Byeon, I.-J.L.; Gronenborn, B.; Gronenborn, A.M.; Campos-Olivas, R. Solution structure, divalent metal and DNA binding of the endonuclease domain from the replication initiation protein from porcine circovirus 2. J. Mol. Biol. 2007, 367, 473–487. [Google Scholar] [CrossRef] [PubMed]
- Rosario, K.; Duffy, S.; Breitbart, M. A field guide to eukaryotic circular single-stranded DNA viruses: Insights gained from metagenomics. Arch. Virol. 2012, 157, 1851–1871. [Google Scholar] [CrossRef] [PubMed]
- Deltoro, D.; Ortiz, D.; Ordyan, M.; Sippy, J.; Oh, C.-S.; Keller, N.; Feiss, M.; Catalano, C.E.; Smith, D.E. Walker-A Motif Acts to Coordinate ATP Hydrolysis with Motor Output in Viral DNA Packaging. J. Mol. Biol. 2016, 428, 2709–2729. [Google Scholar] [CrossRef]
- Kanade, M.; Chakraborty, S.; Shelke, S.S.; Gayathri, P. A Distinct Motif in a Prokaryotic Small Ras-Like GTPase Highlights Unifying Features of Walker B Motifs in P-Loop NTPases. J. Mol. Biol. 2020, 432, 5544–5564. [Google Scholar] [CrossRef] [PubMed]
- Krupovic, M.; Ghabrial, S.A.; Jiang, D.; Varsani, A. Genomoviridae: A new family of widespread single-stranded DNA viruses. Arch. Virol. 2016, 161, 2633–2643. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Wang, S.; Zhang, L.; Qiu, D.; Zhou, X.; Guo, L. A tripartite ssDNA mycovirus from a plant pathogenic fungus is infectious as cloned DNA and purified virions. Sci. Adv. 2020, 6, eaay9634. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Huson, D.H.; Xie, C. A poor man’s BLASTX--high-throughput metagenomic protein database search using PAUDA. Bioinformatics 2014, 30, 38–39. [Google Scholar] [CrossRef] [PubMed]
- Pirooznia, M.; Perkins, E.J.; Deng, Y. Batch Blast Extractor: An automated blastx parser application. BMC Genom. 2008, 9 (Suppl. S2), S10. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Muhire, B.M.; Varsani, A.; Martin, D.P. SDT: A virus classification tool based on pairwise sequence alignment and identity calculation. PLoS ONE 2014, 9, e108277. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: A multiple sequence alignment method with reduced time and space complexity. BMC Bioinform. 2004, 5, 113. [Google Scholar] [CrossRef] [PubMed]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [PubMed]
- de Rezende, R.R.; Mar, T.B.; Páez, L.M.C.; Silva Xavier, A.D.; Xavier, C.A.D.; Navas-Castillo, J.; Zerbini, F.M.; Alfenas-Zerbini, P. Complete genome sequences of two gemycircularviruses associated with non-cultivated plants in Brazil. Arch. Virol. 2018, 163, 3163–3166. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Wu, H.; Sun, G.; Yang, S.; Shen, Q.; Wang, X.; Zhang, W. Identification of diverse novel genomoviruses in gut of wild birds. Biosaf. Health 2021, 3, 136–141. [Google Scholar] [CrossRef]
- Krupovic, M. Networks of evolutionary interactions underlying the polyphyletic origin of ssDNA viruses. Curr. Opin. Virol. 2013, 3, 578–586. [Google Scholar] [CrossRef] [PubMed]
- Campos-Olivas, R.; Louis, J.M.; Clerot, D.; Gronenborn, B.; Gronenborn, A.M. The structure of a replication initiator unites diverse aspects of nucleic acid metabolism. Proc. Natl. Acad. Sci. USA 2002, 99, 10310–10315. [Google Scholar] [CrossRef]
- Steinfeldt, T.; Finsterbusch, T.; Mankertz, A. Demonstration of nicking/joining activity at the origin of DNA replication associated with the rep and rep’ proteins of porcine circovirus type 1. J. Virol. 2006, 80, 6225–6234. [Google Scholar] [CrossRef]
- Nakasu, E.Y.T.; Melo, F.L.; Michereff-Filho, M.; Nagata, T.; Ribeiro, B.M.; Ribeiro, S.G.; Lacorte, C. Discovery of two small circular ssDNA viruses associated with the whitefly Bemisia tabaci. Arch. Virol. 2017, 162, 2835–2838. [Google Scholar] [CrossRef] [PubMed]
- Kemenesi, G.; Kurucz, K.; Zana, B.; Földes, F.; Urbán, P.; Vlaschenko, A.; Kravchenko, K.; Budinski, I.; Szodoray-Parádi, F.; Bücs, S.; et al. Diverse replication-associated protein encoding circular DNA viruses in guano samples of Central-Eastern European bats. Arch. Virol. 2018, 163, 671–678. [Google Scholar] [CrossRef] [PubMed]
- Cibulski, S.P.; Lima, F.E.d.S.; Teixeira, T.F.; Varela, A.P.M.; Scheffer, C.M.; Mayer, F.Q.; Witt, A.A.; Roehe, P.M. Detection of multiple viruses in oropharyngeal samples from Brazilian free-tailed bats (Tadarida brasiliensis) using viral metagenomics. Arch. Virol. 2021, 166, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Kirk, M.D.; Pires, S.M.; Black, R.E.; Caipo, M.; Crump, J.A.; Devleesschauwer, B.; Döpfer, D.; Fazil, A.; Fischer-Walker, C.L.; Hald, T.; et al. World Health Organization Estimates of the Global and Regional Disease Burden of 22 Foodborne Bacterial, Protozoal, and Viral Diseases, 2010: A Data Synthesis. PLoS Med. 2015, 12, e1001921. [Google Scholar]
- Jones, S.; Baizan-Edge, A.; MacFarlane, S.; Torrance, L. Viral Diagnostics in Plants Using Next Generation Sequencing: Computational Analysis in Practice. Front. Plant Sci. 2017, 8, 1770. [Google Scholar] [CrossRef]
- Ghurye, J.S.; Cepeda-Espinoza, V.; Pop, M. Metagenomic Assembly: Overview, Challenges and Applications. Yale J. Biol. Med. 2016, 89, 353–362. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
| ContigID (Genome Size in Base Pairs) | BLAST Search Results | ||||||
|---|---|---|---|---|---|---|---|
| Genbank Best-Hit | Coverage | E-Values | Identity | ||||
| BLASTN | BLASTX | BLASTN | BLASTX | BLASTN | BLASTX | ||
| Genomovirus01 (2226) | MT205711.1 | 11% | 26% | 1 × 10−15 | 2 × 10−64 | 74.03% | 56.90% |
| Genomovirus02 (2147) | NC_076359.1 | 45% | 45% | 1 × 10−125 | 3 × 10−114 | 81.05% | 63.75% |
| Genomovirus03 (2135) | NC_075339.1 | 99% | 51% | 0.0 | 0.0 | 98.71% | 86.70% |
| Genomovirus05 (2290) | MW182919.1 | 41% | 45% | 0.0 | 2 × 10−148 | 88.89% | 66.65% |
| Genomovirus06 (2193) | NC_038488.1 | 58% | 27% | 0.0 | 2 × 10−135 | 93.70% | 98.00% |
| Genomovirus07 (2701) | MW678943.1 | 43% | 44% | 0.0 | 0.0 | 87.01% | 69.85% |
| Genomovirus09 (2189) | NC_038497.1 | 42% | 26% | 0.0 | 3 × 10−98 | 88.50% | 78.65% |
| Sample | Size (Base Pairs) | Best-Hit GenbankID | Coverage | E-Values | Identity | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Rep | Cap | Rep | Cap | Rep | Cap | Rep | Cap | Rep | Cap | |
| Genomovirus01 | 257 | 329 | QJB18714.1 | UBJ26138.1 | 73% | 91% | 4 × 10−74 | 1 × 10−71 | 60.11% | 43.61% |
| Genomovirus02 | 235 | 295 | QNI80852.1 | QJB18679.1 | 91% | 87% | 1 × 10−111 | 7 × 10−77 | 71.11% | 46.72% |
| Genomovirus03 | 194 | 267 | YP_010784661.1 | UBJ26188.1 | 100% | 100% | 1 × 10−139 | 0.0 | 99.48% | 87.75% |
| Genomovirus05 | 266 | 292 | QTE03605.1 | YP_009021851.1 | 74% | 74% | 5 × 10−124 | 2 × 10−87 | 87.31% | 59.63% |
| Genomovirus06 | 200 | 311 | YP_009506608.1 | YP_010798116.1 | 100% | 100% | 2 × 10−144 | 3 × 10−133 | 98% | 68.25% |
| Genomovirus07 | 215 | 310 | QTZ83241.1 | QXN75602.1 | 93% | 100% | 1 × 10−128 | 0.0 | 88.61% | 82.90% |
| Genomovirus09 | 248 | 130 | QTE03605.1 | QXN75548.1 | 79% | 90% | 1 × 10−108 | 2 × 10−44 | 79.29% | 58.97% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Couto, R.d.S.; Abreu, W.U.; Rodrigues, L.R.R.; Marinho, L.F.; Morais, V.d.S.; Villanova, F.; Pandey, R.P.; Deng, X.; Delwart, E.; da Costa, A.C.; et al. Genomoviruses in Liver Samples of Molossus molossus Bats. Microorganisms 2024, 12, 688. https://doi.org/10.3390/microorganisms12040688
Couto RdS, Abreu WU, Rodrigues LRR, Marinho LF, Morais VdS, Villanova F, Pandey RP, Deng X, Delwart E, da Costa AC, et al. Genomoviruses in Liver Samples of Molossus molossus Bats. Microorganisms. 2024; 12(4):688. https://doi.org/10.3390/microorganisms12040688
Chicago/Turabian StyleCouto, Roseane da Silva, Wandercleyson Uchôa Abreu, Luís Reginaldo Ribeiro Rodrigues, Luis Fernando Marinho, Vanessa dos Santos Morais, Fabiola Villanova, Ramendra Pati Pandey, Xutao Deng, Eric Delwart, Antonio Charlys da Costa, and et al. 2024. "Genomoviruses in Liver Samples of Molossus molossus Bats" Microorganisms 12, no. 4: 688. https://doi.org/10.3390/microorganisms12040688
APA StyleCouto, R. d. S., Abreu, W. U., Rodrigues, L. R. R., Marinho, L. F., Morais, V. d. S., Villanova, F., Pandey, R. P., Deng, X., Delwart, E., da Costa, A. C., & Leal, E. (2024). Genomoviruses in Liver Samples of Molossus molossus Bats. Microorganisms, 12(4), 688. https://doi.org/10.3390/microorganisms12040688