
Distinct Gut Microbial Enterotypes and Functional Dynamics in Wild Striped Field Mice (Apodemus agrarius) across Diverse Populations

Abstract
1. Introduction
2. Materials and Methods
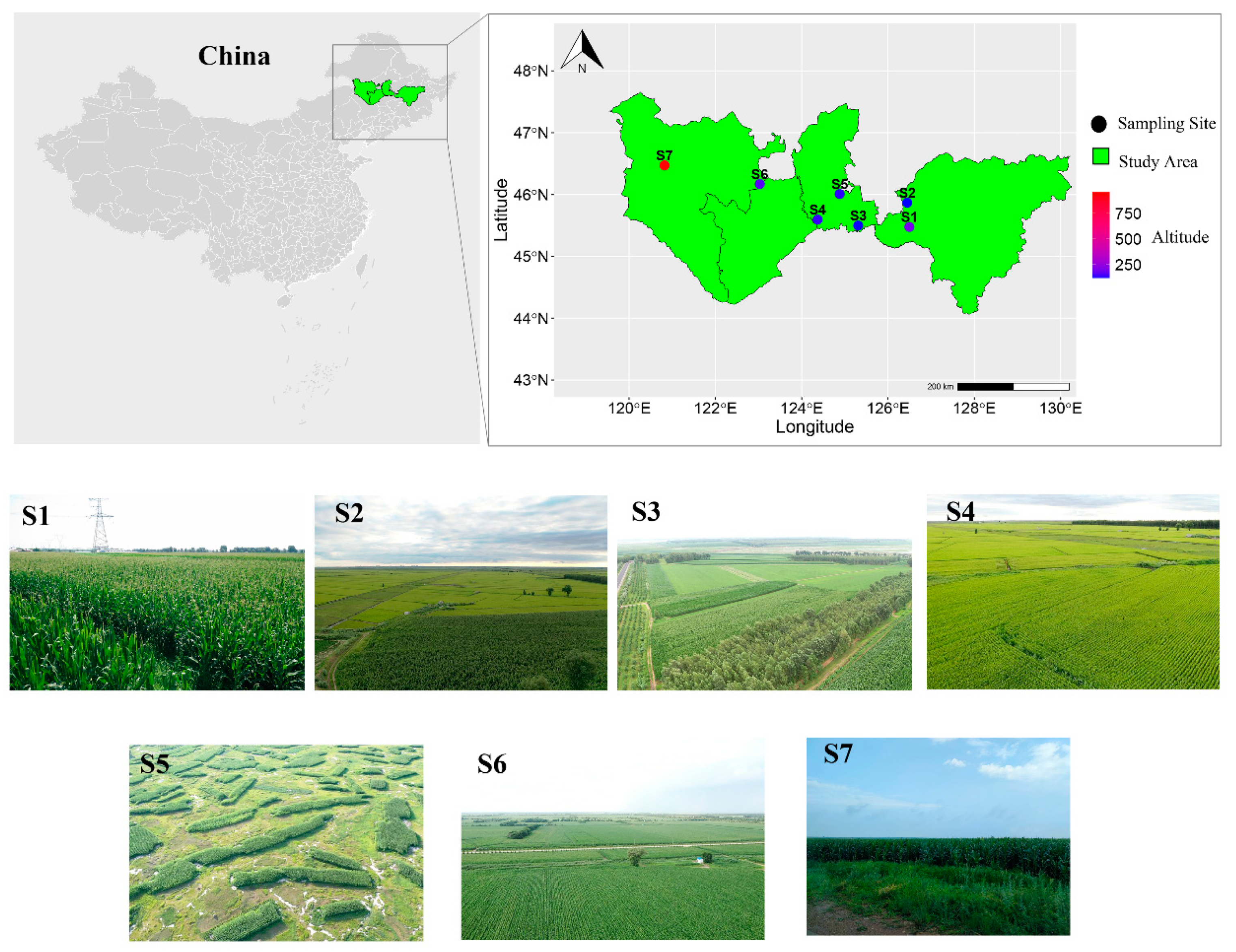
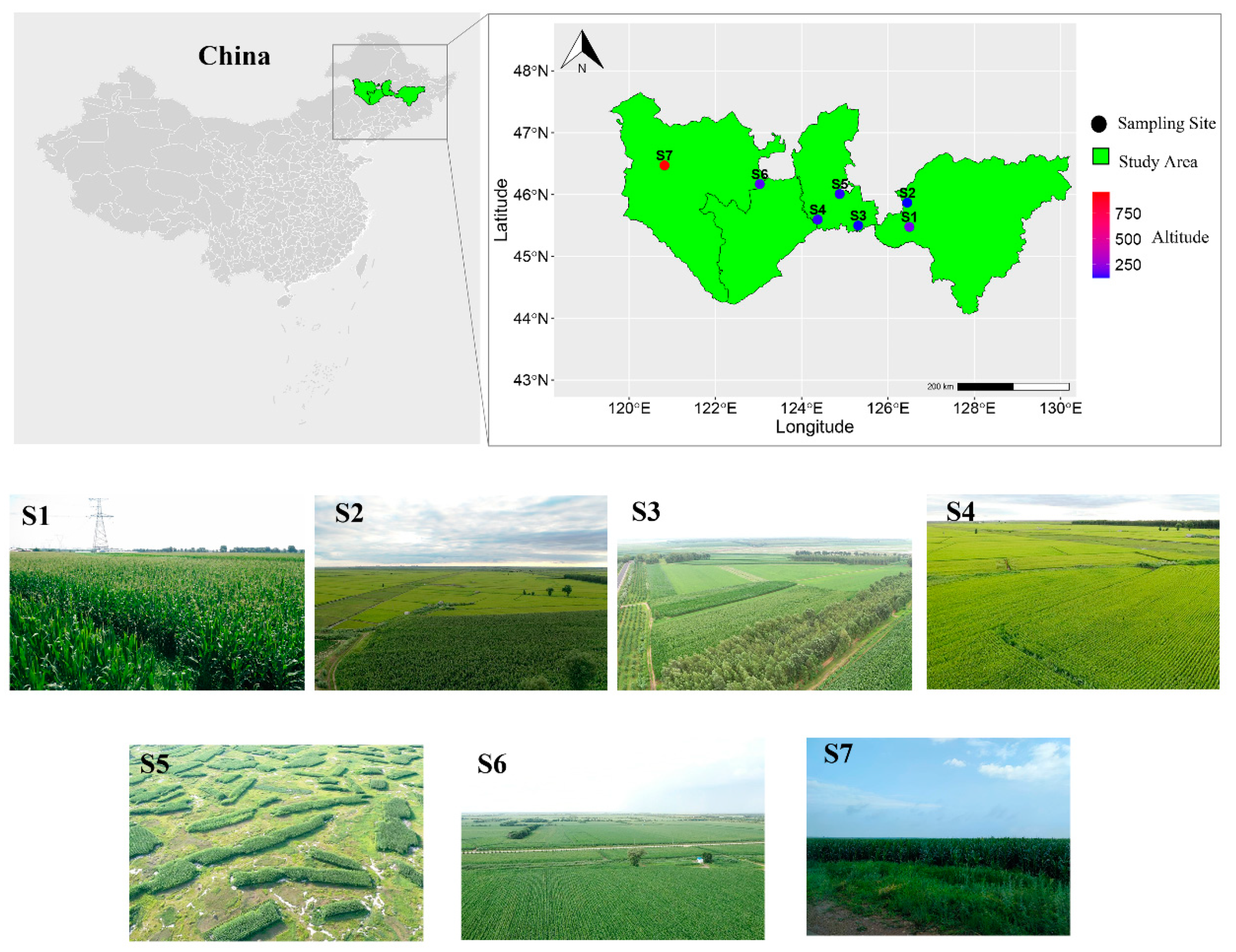
2.1. Sample Collection
2.2. DNA Extraction and Sequencing
2.3. Bioinformatics Analysis
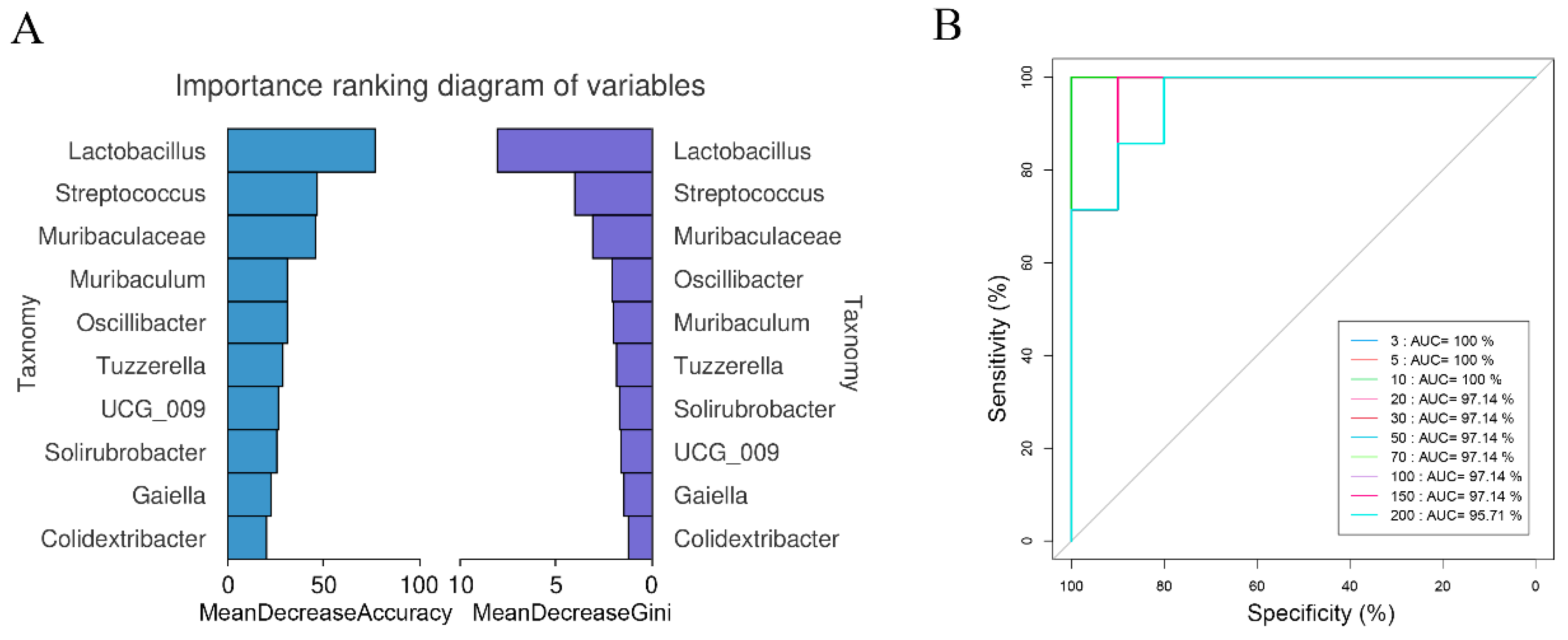
2.4. Random Forest Classifier Models
2.5. Metagenome Prediction
2.6. Ecological Network Analysis
2.7. Bacterial Community Assembly Analyses
2.8. Statistical Analyses
2.9. Data Availability
3. Results
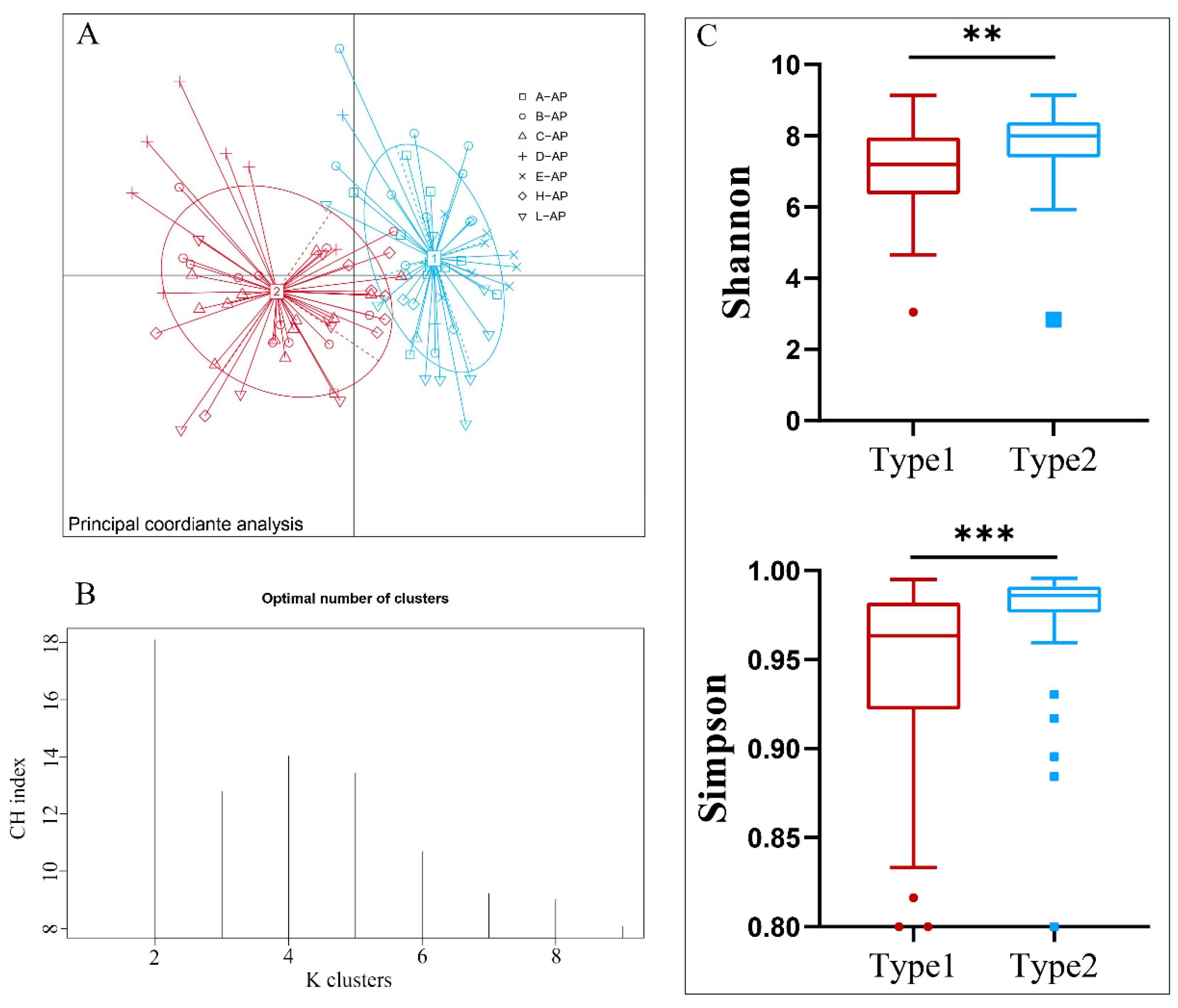
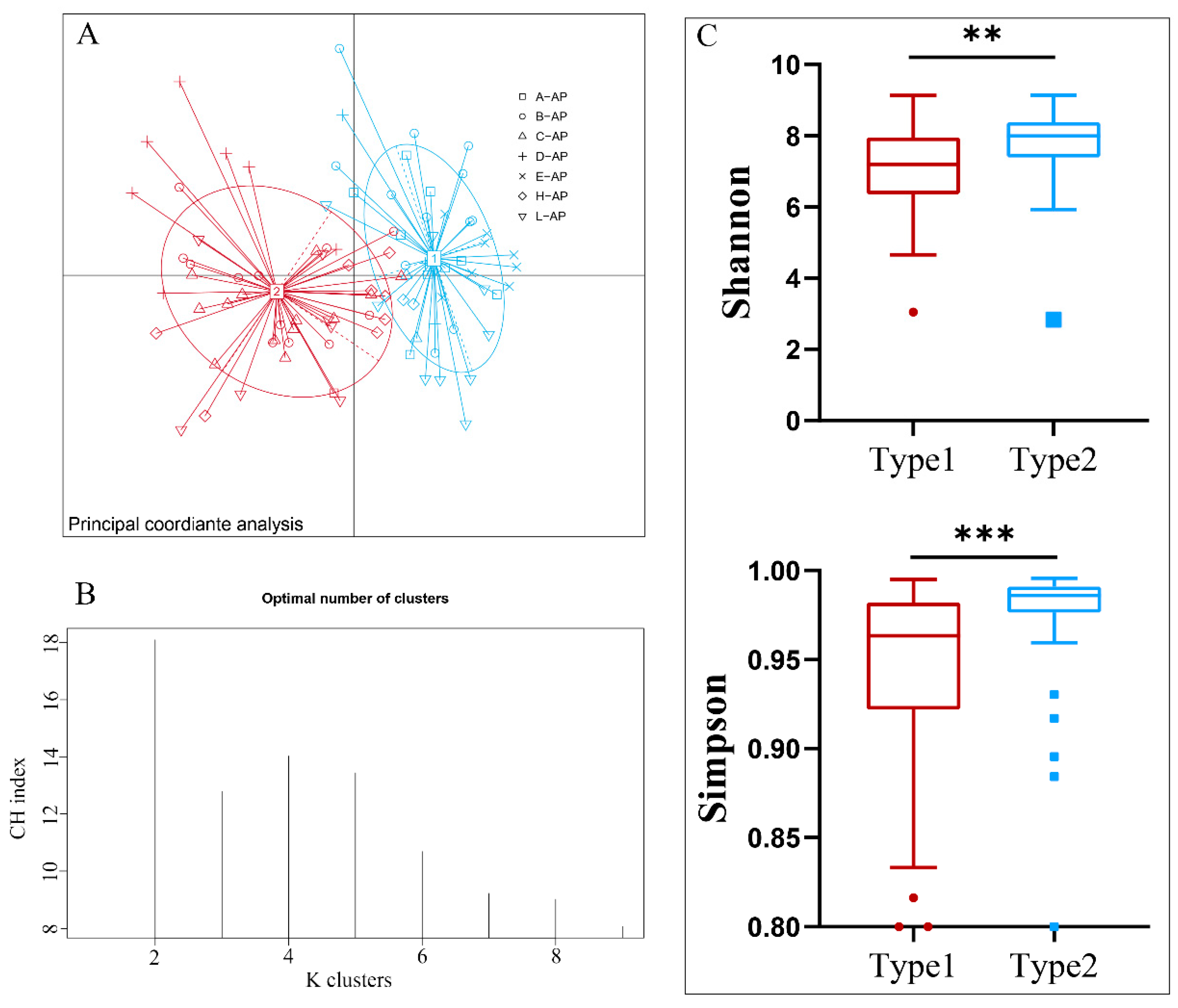
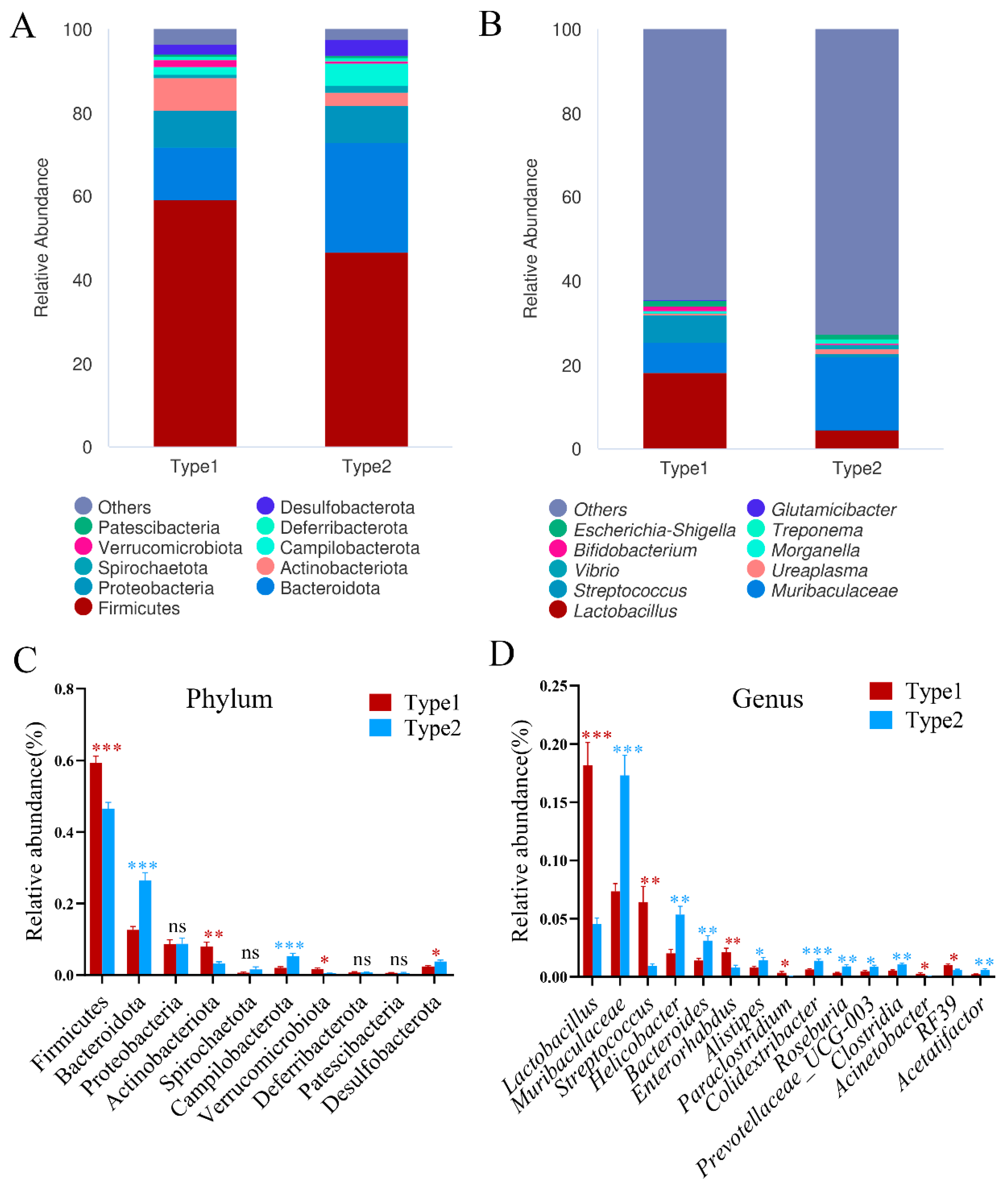
3.1. Identification and Diversity of Enterotypes in A. agrarius
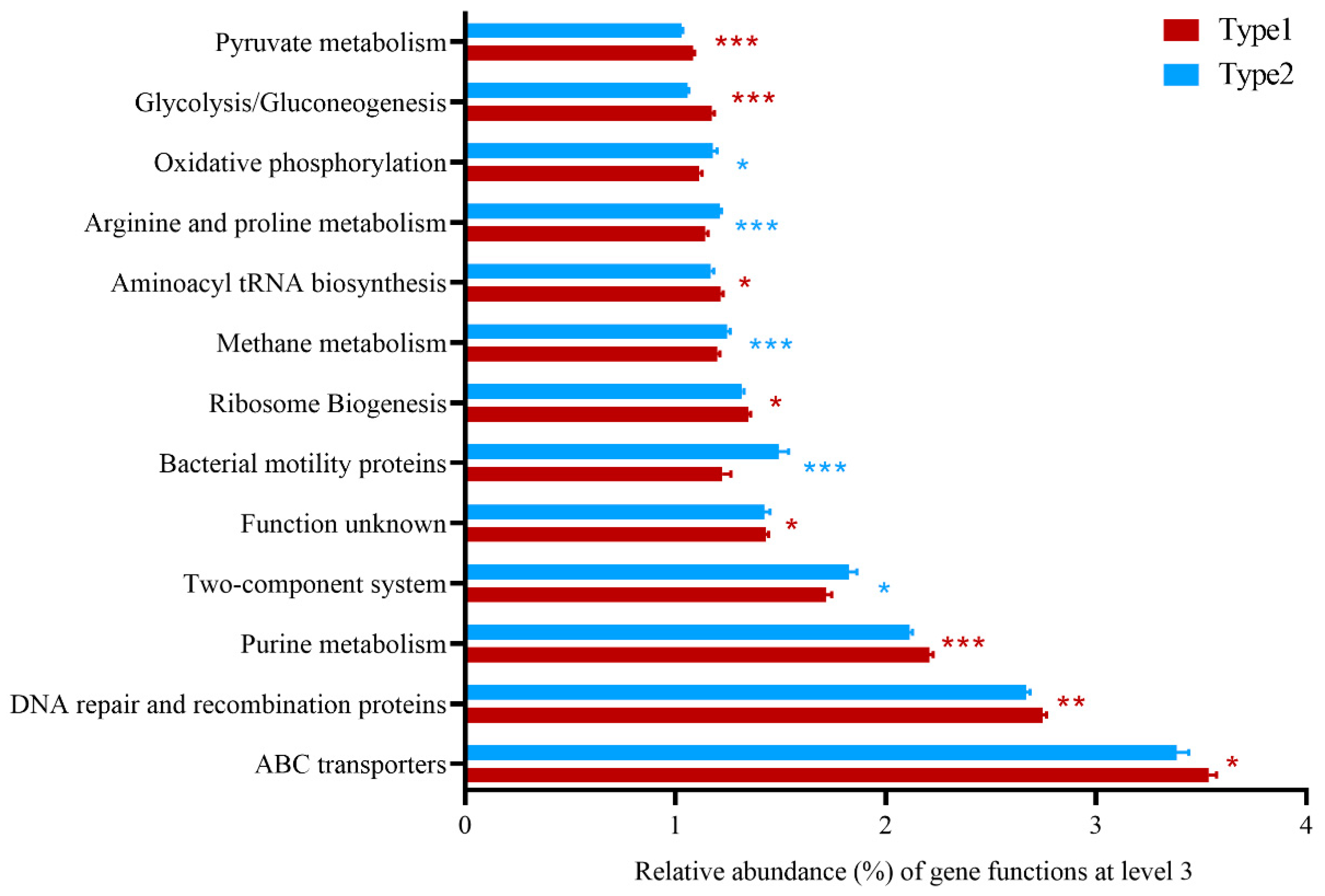
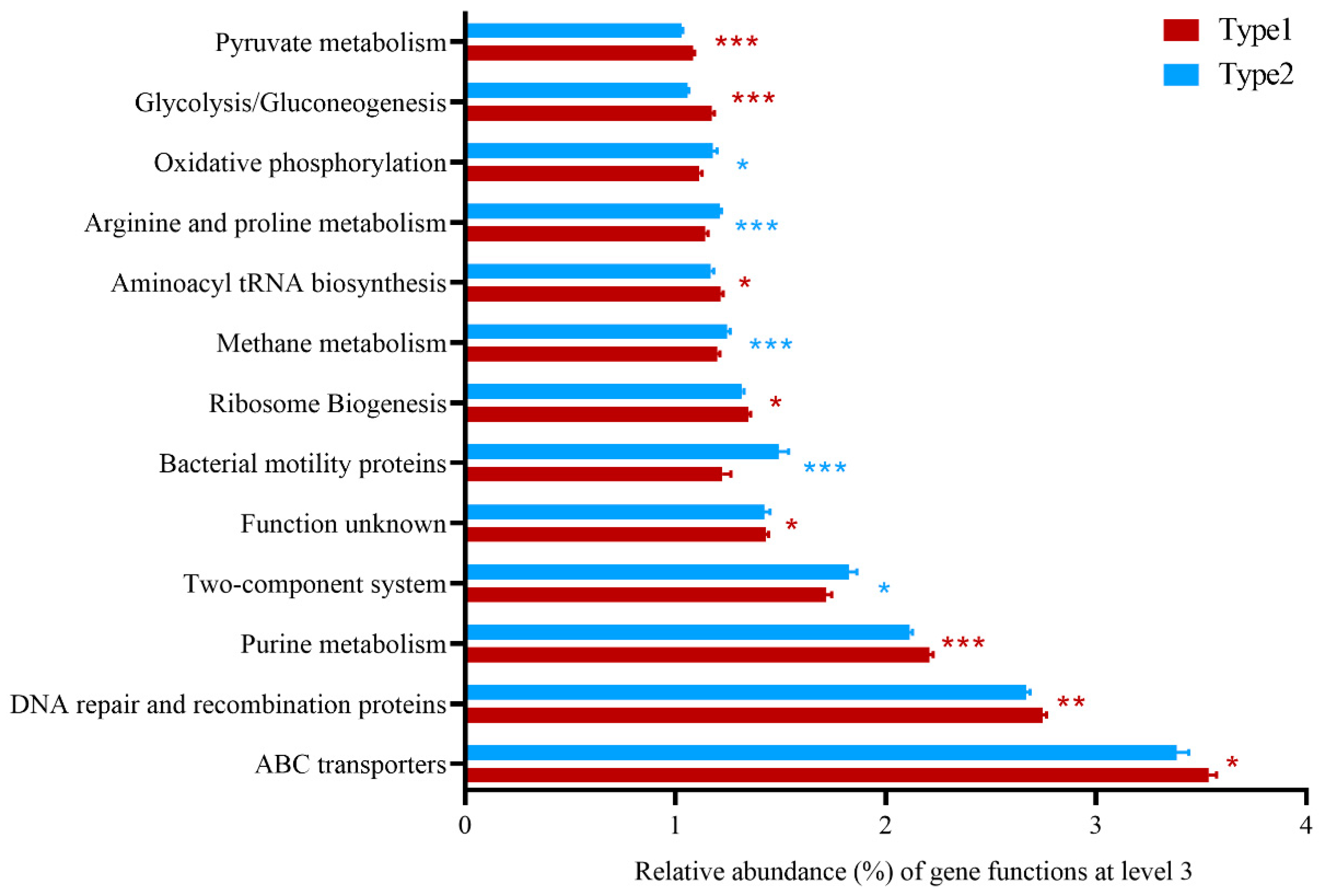
3.2. Differences of Predicted Gene Functions between Enterotypes
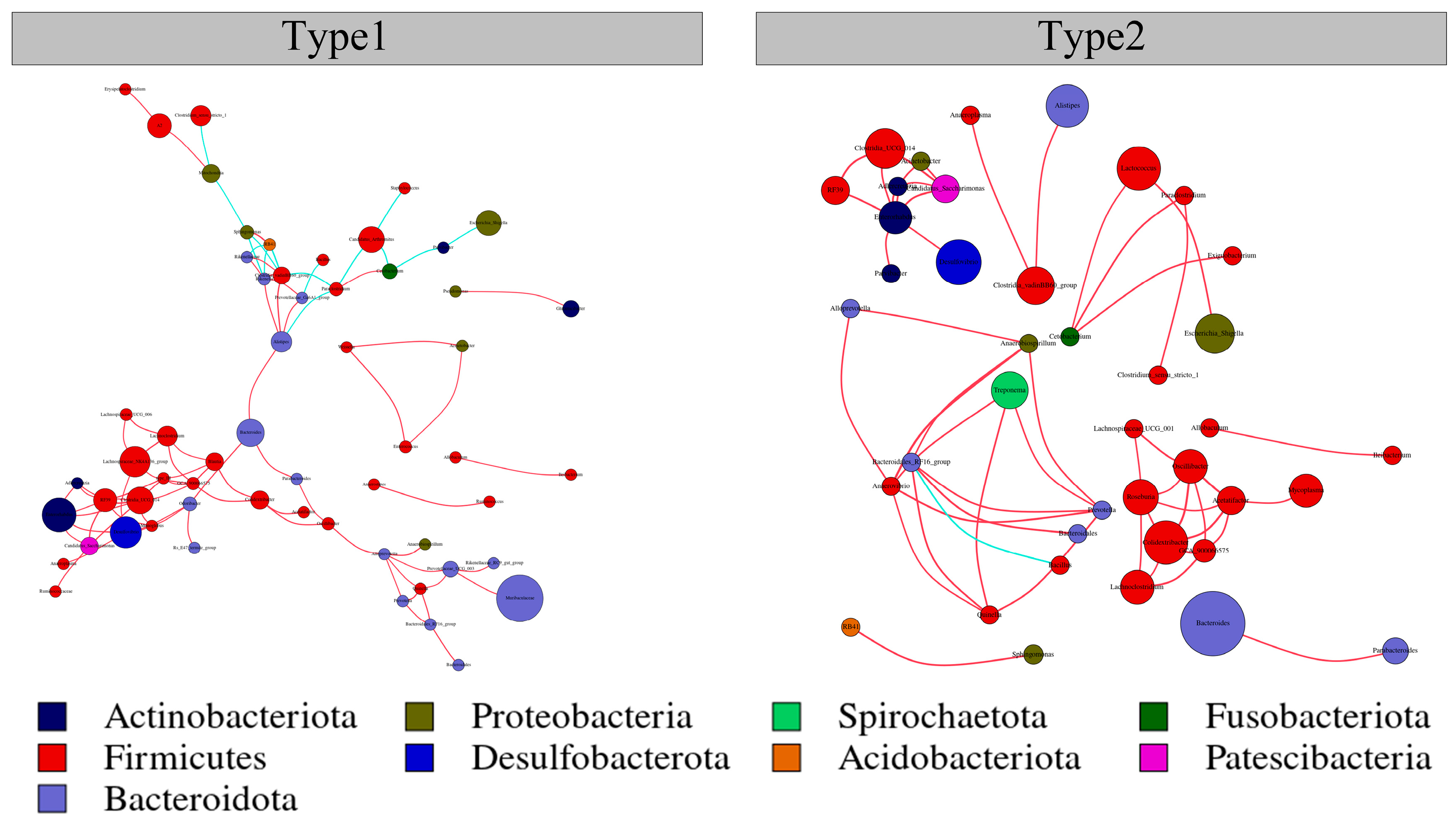
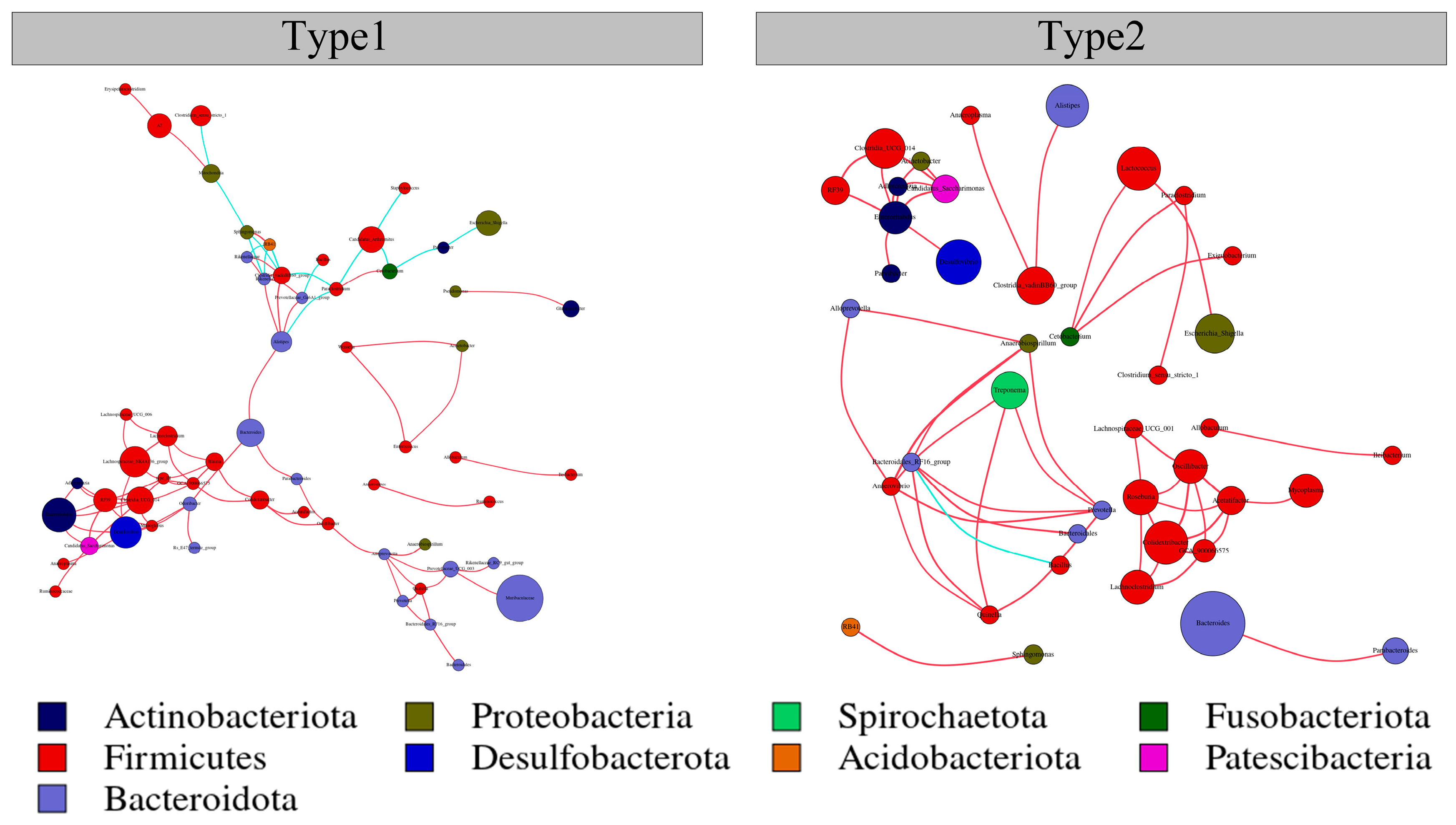
3.3. Network Interactions of the Two Enterotypes
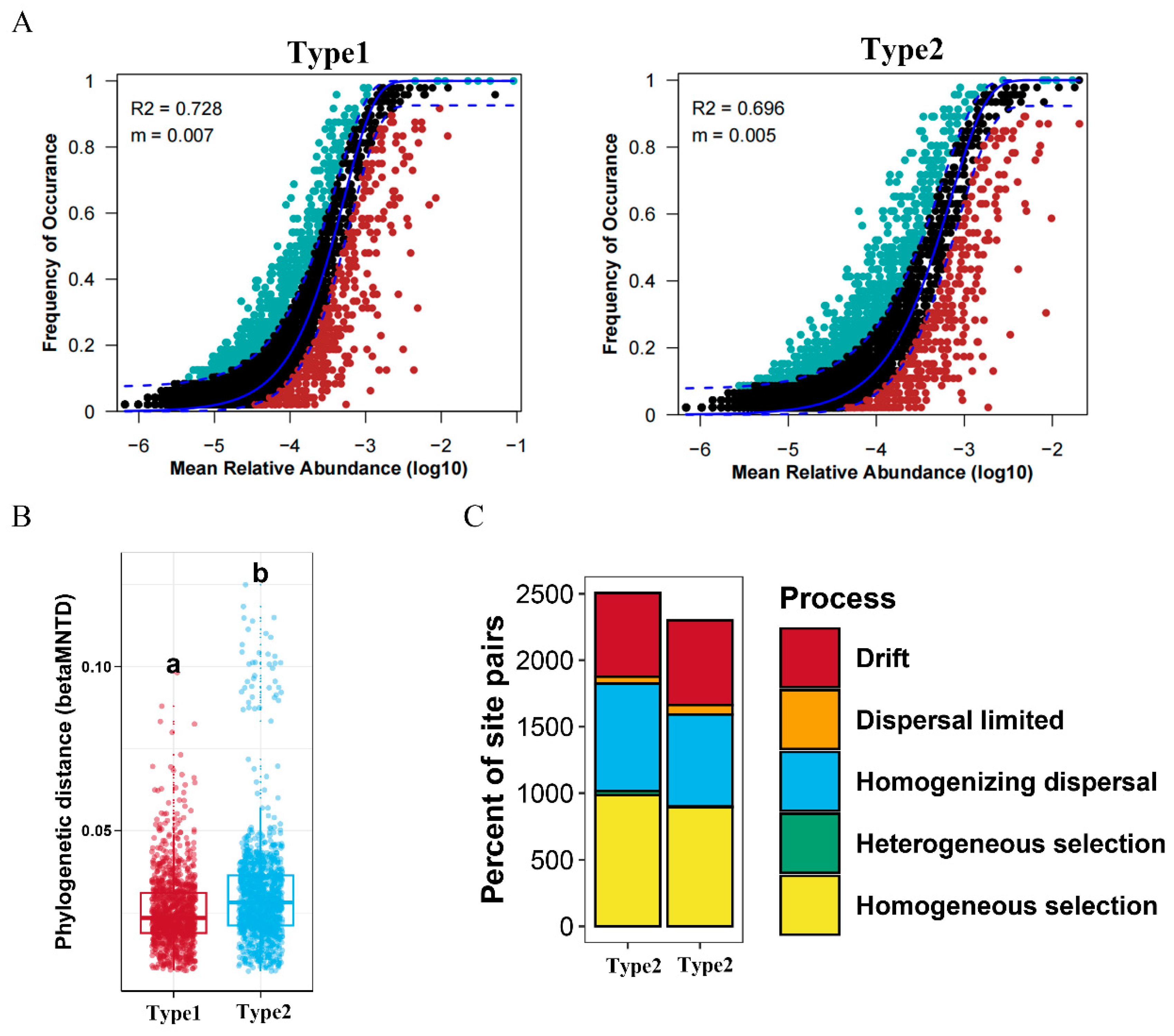
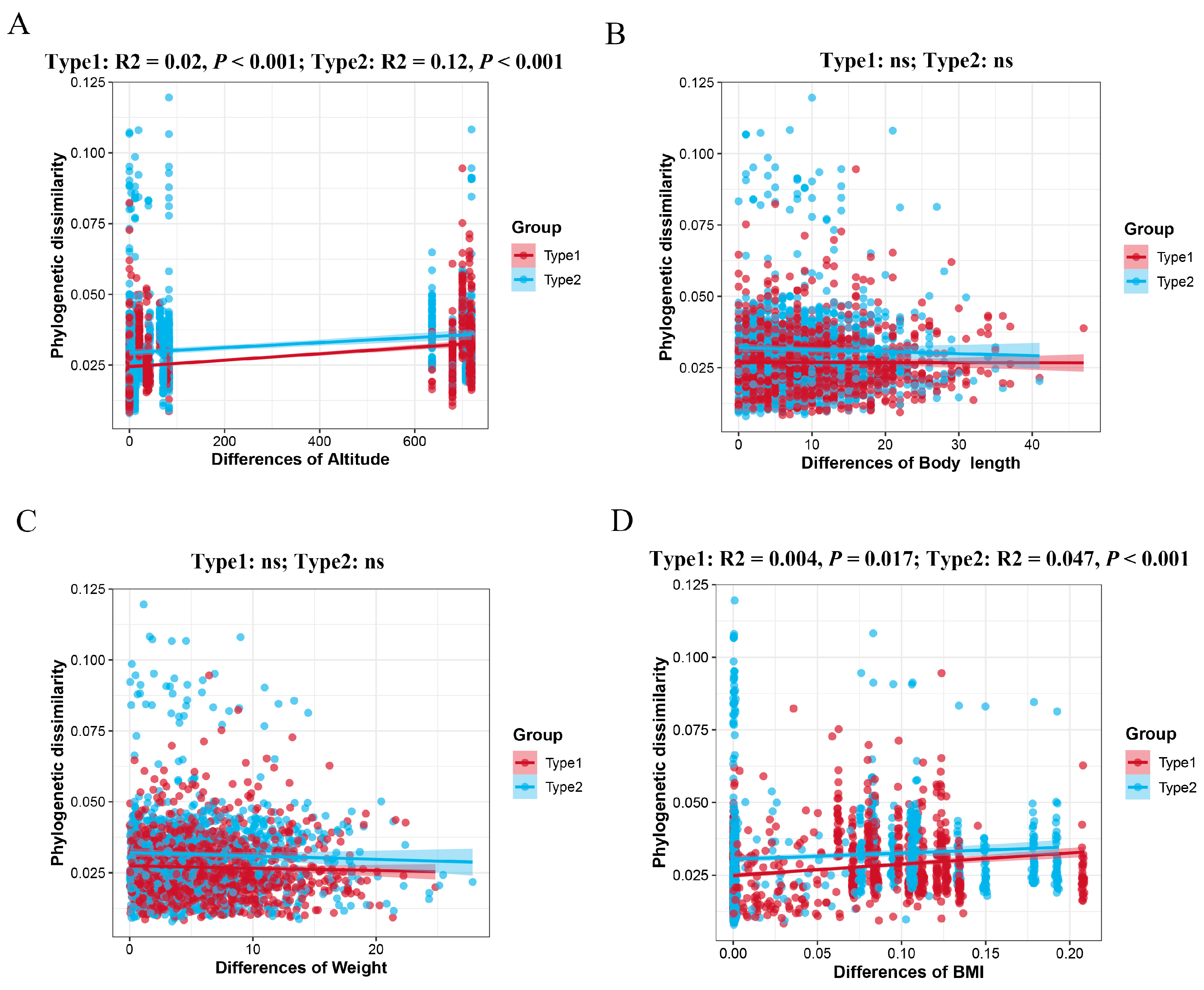
3.4. Community Assembly Process in the Two Enterotypes of A. agrarius
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cheng, M.Y.; Ning, K. Stereotypes About Enterotype: The Old and New Ideas. Genom. Proteom. Bioinform. 2019, 17, 4–12. [Google Scholar] [CrossRef]
- Gorvitovskaia, A.; Holmes, S.P.; Huse, S.M. Interpreting and as biomarkers of diet and lifestyle. Microbiome 2016, 4, 507–525. [Google Scholar] [CrossRef]
- Arumugam, M.; Raes, J.; Pelletier, E.; Le Paslier, D.; Yamada, T.; Mende, D.R.; Fernandes, G.R.; Tap, J.; Bruls, T.; Batto, J.M.; et al. Enterotypes of the human gut microbiome. Nature 2011, 473, 174–180. [Google Scholar] [CrossRef]
- Christensen, L.; Sorensen, C.V.; Wohlk, F.U.; Kjolbaek, L.; Astrup, A.; Sanz, Y.; Hjorth, M.F.; Benitez-Paez, A. Microbial enterotypes beyond genus level: Bacteroides species as a predictive biomarker for weight change upon controlled intervention with arabinoxylan oligosaccharides in overweight subjects. Gut Microbes 2020, 12, 1847627. [Google Scholar] [CrossRef]
- Cuevas-Sierra, A.; Riezu-Boj, J.I.; Guruceaga, E.; Martínez, J.A.; Milagro, F.I. Prevotella enterotype is associated with higher body mass index, HOMA level, and fiber intake. Ann. Nutr. Metab. 2019, 75, 1–75. [Google Scholar] [CrossRef]
- Jamieson, P.E.; Smart, E.B.; Bouranis, J.A.; Choi, J.; Danczak, R.E.; Wong, C.P.; Paraiso, I.L.; Maier, C.S.; Ho, E.; Sharpton, T.J.; et al. Gut enterotype-dependent modulation of gut microbiota and their metabolism in response to xanthohumol supplementation in healthy adults. Gut Microbes 2024, 16, 2315633. [Google Scholar] [CrossRef]
- Hicks, A.L.; Lee, K.J.; Couto-Rodriguez, M.; Patel, J.; Sinha, R.; Guo, C.; Olson, S.H.; Seimon, A.; Seimon, T.A.; Ondzie, A.U.; et al. Gut microbiomes of wild great apes fluctuate seasonally in response to diet. Nat. Commun. 2018, 9, 1786. [Google Scholar] [CrossRef]
- Rodríguez-Daza, M.C.; Roquim, M.; Dudonné, S.; Pilon, G.; Levy, E.; Marette, A.; Roy, D.; Desjardins, Y. Berry Polyphenols and Fibers Modulate Distinct Microbial Metabolic Functions and Gut Microbiota Enterotype-Like Clustering in Obese Mice. Front. Microbiol. 2020, 11, 2032. [Google Scholar] [CrossRef]
- Wang, J.; Linnenbrink, M.; Kunzel, S.; Fernandes, R.; Nadeau, M.J.; Rosenstiel, P.; Baines, J.F. Dietary history contributes to enterotype-like clustering and functional metagenomic content in the intestinal microbiome of wild mice. Proc. Natl. Acad. Sci. USA 2014, 111, E2703–E2710. [Google Scholar] [CrossRef]
- Haddad, E.N.; Sugino, K.Y.; Tucker, R.M.; Comstock, S.S. Gut enterotypes are stable during Bifidobacterium and Lactobacillus probiotic supplementation. J. Food Sci. 2020, 85, 1596–1604. [Google Scholar] [CrossRef]
- Ke, S.L.; Fang, S.M.; He, M.Z.; Huang, X.C.; Yang, H.; Yang, B.; Chen, C.Y.; Huang, L.S. Age-based dynamic changes of phylogenetic composition and interaction networks of health pig gut microbiome feeding in a uniformed condition. BMC Vet. Res. 2019, 15, 172. [Google Scholar] [CrossRef]
- Xiao, L.W.; Wang, J.F.; Zheng, J.Y.; Li, X.Q.; Zhao, F.Q. Deterministic transition of enterotypes shapes the infant gut microbiome at an early age. Genome Biol. 2021, 22, 243. [Google Scholar] [CrossRef] [PubMed]
- Moeller, A.H.; Degnan, P.H.; Pusey, A.E.; Wilson, M.L.; Hahn, B.H.; Ochman, H. Chimpanzees and humans harbour compositionally similar gut enterotypes. Nat. Commun. 2012, 3, 1179. [Google Scholar] [CrossRef] [PubMed]
- Xu, E.; Yang, H.; Ren, M.M.; Wang, Y.X.; Xiao, M.F.; Tang, Q.S.; Zhu, M.; Xiao, Y.P. Identification of Enterotype and Its Effects on Intestinal Butyrate Production in Pigs. Animals 2021, 11, 730. [Google Scholar] [CrossRef] [PubMed]
- Le Sciellour, M.; Zemb, O.; Hochu, I.; Riquet, J.; Gilbert, H.; Giorgi, M.; Billon, Y.; Gourdine, J.L.; Renaudeau, D. Effect of chronic and acute heat challenges on fecal microbiota composition, production, and thermoregulation traits in growing pigs. J. Anim. Sci. 2019, 97, 3845–3858. [Google Scholar] [CrossRef]
- Couch, C.E.; Stagaman, K.; Spaan, R.S.; Combrink, H.J.; Sharpton, T.J.; Beechler, B.R.; Jolles, A.E. Diet and gut microbiome enterotype are associated at the population level in African buffalo. Nat. Commun. 2021, 12, 2267. [Google Scholar] [CrossRef]
- Hildebrand, F.; Nguyen, T.L.A.; Brinkman, B.; Yunta, R.G.; Cauwe, B.; Vandenabeele, P.; Liston, A.; Raes, J. Inflammation-associated enterotypes, host genotype, cage and inter-individual effects drive gut microbiota variation in common laboratory mice. Genome Biol. 2013, 14, R4. [Google Scholar] [CrossRef] [PubMed]
- Li, J.L.; Powell, J.E.; Guo, J.; Evans, J.D.; Wu, J.; Williams, P.; Lin, Q.H.; Moran, N.A.; Zhang, Z.G. Two gut community enterotypes recur in diverse bumblebee species. Curr. Biol. 2015, 25, R652–R653. [Google Scholar] [CrossRef] [PubMed]
- Martinson, V.G.; Douglas, A.E.; Jaenike, J. Community structure of the gut microbiota in sympatric species of wild Drosophila. Ecol. Lett. 2017, 20, 629–639. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.J.; Zhang, L.Z.; Ren, S.E.; Zhao, Y.Q.; Zhang, Y.M. Temporal and geographic distribution of gut microbial enterotypes associated with host thermogenesis characteristics in plateau pikas. Microbiol. Spectr. 2023, 11, e00020-23. [Google Scholar] [CrossRef]
- Fan, C.; Zhang, L.Z.; Fu, H.B.; Liu, C.F.; Li, W.J.; Cheng, Q.; Zhang, H.; Jia, S.A.; Zhang, Y.M. Enterotypes of the Gut Microbial Community and Their Response to Plant Secondary Compounds in Plateau Pikas. Microorganisms 2020, 8, 1311. [Google Scholar] [CrossRef]
- Yu, Q.; Li, G.; Li, H. Two community types occur in gut microbiota of large-sample wild plateau pikas (Ochotona curzoniae). Integr. Zool. 2022, 17, 366–378. [Google Scholar] [CrossRef]
- Costello, E.K.; Stagaman, K.; Dethlefsen, L.; Bohannan, B.J.M.; Relman, D.A. The Application of Ecological Theory Toward an Understanding of the Human Microbiome. Science 2012, 336, 1255–1262. [Google Scholar] [CrossRef]
- Greene, L.K.; Williams, C.V.; Junge, R.E.; Mahefarisoa, K.L.; Rajaonarivelo, T.; Rakotondrainibe, H.; O’Connell, T.M.; Drea, C.M. A role for gut microbiota in host niche differentiation. ISME J. 2020, 14, 1675–1687. [Google Scholar] [CrossRef]
- Liao, J.Q.; Cao, X.F.; Zhao, L.; Wang, J.; Gao, Z.; Wang, M.C.; Huang, Y. The importance of neutral and niche processes for bacterial community assembly differs between habitat generalists and specialists. Fems Microbiol. Ecol. 2016, 92, fiw174. [Google Scholar] [CrossRef] [PubMed]
- Burns, A.R.; Stephens, W.Z.; Stagaman, K.; Wong, S.; Rawls, J.F.; Guillemin, K.; Bohannan, B.J.M. Contribution of neutral processes to the assembly of gut microbial communities in the zebrafish over host development. ISME J. 2016, 10, 655–664. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.; Lacey, E.A.; Bach, B.H.; Bi, K.; Conroy, C.J.; Suvorov, A.; Bowie, R.C.K. Gut microbial diversity across a contact zone for California voles: Implications for lineage divergence of hosts and mitonuclear mismatch in the assembly of the mammalian gut microbiome. Mol. Ecol. 2020, 29, 1873–1889. [Google Scholar] [CrossRef] [PubMed]
- Moeller, A.H.; Suzuki, T.A.; Lin, D.; Lacey, E.A.; Wasser, S.K.; Nachman, M.W. Dispersal limitation promotes the diversification of the mammalian gut microbiota. Proc. Natl. Acad. Sci. USA 2017, 114, 13768–13773. [Google Scholar] [CrossRef]
- Stegen, J.C.; Lin, X.; Fredrickson, J.K.; Konopka, A.E. Estimating and mapping ecological processes influencing microbial community assembly. Front. Microbiol. 2015, 6, 370. [Google Scholar] [CrossRef]
- Zhou, J.; Deng, Y.; Zhang, P.; Xue, K.; Liang, Y.; Van Nostrand, J.D.; Yang, Y.; He, Z.; Wu, L.; Stahl, D.A.; et al. Stochasticity, succession, and environmental perturbations in a fluidic ecosystem. Proc. Natl. Acad. Sci. USA 2014, 111, E836–E845. [Google Scholar] [CrossRef]
- Zhu, Y.X.; Chang, Y.W.; Wen, T.; Yang, R.; Wang, Y.C.; Wang, X.Y.; Lu, M.X.; Du, Y.Z. Species Identity Dominates over Environment in Driving Bacterial Community Assembly in Wild Invasive Leaf Miners. Microbiol. Spectr. 2022, 10, e00266-22. [Google Scholar] [CrossRef]
- Stothart, M.R.; Greuel, R.J.; Gavriliuc, S.; Henry, A.; Wilson, A.J.; McLoughlin, P.D.; Poissant, J. Bacterial dispersal and drift drive microbiome diversity patterns within a population of feral hindgut fermenters. Mol. Ecol. 2021, 30, 555–571. [Google Scholar] [CrossRef]
- Kim, C.M.; Yi, Y.H.; Yu, D.H.; Lee, M.J.; Cho, M.R.; Desai, A.R.; Shringi, S.; Klein, T.A.; Kim, H.C.; Song, J.W.; et al. Tik-borne-borne rickettsial pathogens in ticks and small mammals in Korea. Appl. Environ. Microb. 2006, 72, 5766–5776. [Google Scholar] [CrossRef]
- Jeske, K.; Herzig-Straschil, B.; Raileanu, C.; Kunec, D.; Tauchmann, O.; Emirhar, D.; Schmidt, S.; Trimpert, J.; Silaghi, C.; Heckel, G.; et al. Zoonotic pathogen screening of striped field mice (Apodemus agrarius) from Austria. Transbound. Emerg. Dis. 2022, 69, 886–890. [Google Scholar] [CrossRef]
- Wu, Y.; Zhou, T.; Gu, C.; Yin, B.; Yang, S.; Zhang, Y.; Wu, R.; Wei, W. Geographical distribution and species variation of gut microbiota in small rodents from the agro-pastoral transition ecotone in northern China. Ecol. Evol. 2024, 14, e11084. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Pena, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef]
- Magoc, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef]
- Haas, B.J.; Gevers, D.; Earl, A.M.; Feldgarden, M.; Ward, D.V.; Giannoukos, G.; Ciulla, D.; Tabbaa, D.; Highlander, S.K.; Sodergren, E.; et al. Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Res. 2011, 21, 494–504. [Google Scholar] [CrossRef]
- Marizzoni, M.; Gurry, T.; Provasi, S.; Greub, G.; Lopizzo, N.; Ribaldi, F.; Festari, C.; Mazzelli, M.; Mombelli, E.; Salvatore, M.; et al. Comparison of Bioinformatics Pipelines and Operating Systems for the Analyses of 16S rRNA Gene Amplicon Sequences in Human Fecal Samples. Front. Microbiol. 2020, 11, 1262. [Google Scholar] [CrossRef]
- Knights, D.; Costello, E.K.; Knight, R. Supervised classification of human microbiota. Fems Microbiol. Rev. 2011, 35, 343–359. [Google Scholar] [CrossRef]
- Langille, M.G.I.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Thurber, R.L.V.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef]
- Luo, F.; Zhong, J.X.; Yang, Y.F.; Scheuermann, R.H.; Zhou, J.Z. Application of random matrix theory to biological networks. Phys. Lett. A 2006, 357, 420–423. [Google Scholar] [CrossRef]
- Csardi, G.; Nepusz, T. The igraph software package for complex network research. InterJ. Complex Syst. 2006, 1695, 1–9. [Google Scholar]
- Viney, M. The gut microbiota of wild rodents: Challenges and opportunities. Lab. Anim. 2019, 53, 252–258. [Google Scholar] [CrossRef]
- Shave, J.R.; Waterman, J.M. The effects of familiarity and reproductive status on olfactory discrimination by female Cape ground squirrels (Xerus inauris). Behav. Ecol. Sociobiol. 2017, 71, 184. [Google Scholar] [CrossRef]
- Liu, P.Y.; Cheng, A.C.; Huang, S.W.; Lu, H.P.; Oshida, T.; Liu, W.; Yu, H.T. Body-size Scaling is Related to Gut Microbial Diversity, Metabolism and Dietary Niche of Arboreal Folivorous Flying Squirrels. Sci. Rep. 2020, 10, 7809. [Google Scholar] [CrossRef]
- Ley, R.E.; Hamady, M.; Lozupone, C.; Turnbaugh, P.J.; Ramey, R.R.; Bircher, J.S.; Schlegel, M.L.; Tucker, T.A.; Schrenzel, M.D.; Knight, R.; et al. Evolution of mammals and their gut microbes. Science 2008, 320, 1647–1651. [Google Scholar] [CrossRef]
- Muegge, B.D.; Kuczynski, J.; Knights, D.; Clemente, J.C.; Gonzalez, A.; Fontana, L.; Henrissat, B.; Knight, R.; Gordon, J.I. Diet Drives Convergence in Gut Microbiome Functions Across Mammalian Phylogeny and Within Humans. Science 2011, 332, 970–974. [Google Scholar] [CrossRef]
- Deru, V.; Bouquet, A.; Zemb, O.; Blanchet, B.; De Almeida, M.L.; Cauquil, L.; Carillier-Jacquin, C.; Gilbert, H. Genetic relationships between efficiency traits and gut microbiota traits in growing pigs being fed with a conventional or a high-fiber diet. J. Anim. Sci. 2022, 100, skac183. [Google Scholar] [CrossRef]
- Wu, X.Y.; Wei, Q.G.; Wang, X.B.; Shang, Y.Q.; Zhang, H.H. Evolutionary and dietary relationships of wild mammals based on the gut microbiome. Gene 2022, 808, 145999. [Google Scholar] [CrossRef]
- Spor, A.; Koren, O.; Ley, R. Unravelling the effects of the environment and host genotype on the gut microbiome. Nat. Rev. Microbiol. 2011, 9, 279–290. [Google Scholar] [CrossRef]
- Walters, A.D.; Arp, A.; Cerbie, G.M.; Trujillo, D.A.; Kiss, A.J.; Berg, D.J. Phylogenetic relationship and habitat both impact the gut microbiome in two microendemic gastropods. J. Mollus Stud. 2022, 88, eyac002. [Google Scholar] [CrossRef]
- Theilmann, M.C.; Goh, Y.J.; Nielsen, K.F.; Klaenhammer, T.R.; Barrangou, R.; Abou Hachem, M. Lactobacillus acidophilus Metabolizes Dietary Plant Glucosides and Externalizes Their Bioactive Phytochemicals. mBio 2017, 8, e01421-17. [Google Scholar] [CrossRef]
- Lagkouvardos, I.; Lesker, T.R.; Hitch, T.C.A.; Galvez, E.J.C.; Smit, N.; Neuhaus, K.; Wang, J.; Baines, J.F.; Abt, B.; Stecher, B.; et al. Sequence and cultivation study of Muribaculaceae reveals novel species, host preference, and functional potential of this yet undescribed family. Microbiome 2019, 7, 28. [Google Scholar] [CrossRef]
- Scott, K.P.; Gratz, S.W.; Sheridan, P.O.; Flint, H.J.; Duncan, S.H. The influence of diet on the gut microbiota. Pharmacol. Res. 2013, 69, 52–60. [Google Scholar] [CrossRef]
- Norouzi-Beirami, M.H.; Marashi, S.A.; Banaei-Moghaddam, A.M.; Kavousi, K. Beyond Taxonomic Analysis of Microbiomes: A Functional Approach for Revisiting Microbiome Changes in Colorectal Cancer. Front. Microbiol. 2020, 10, 3117. [Google Scholar] [CrossRef]
- Berry, D.; Widder, S. Deciphering microbial interactions and detecting keystone species with co-occurrence networks. Front. Microbiol. 2014, 5, 219. [Google Scholar] [CrossRef]
- Nemergut, D.R.; Schmidt, S.K.; Fukami, T.; O’Neill, S.P.; Bilinski, T.M.; Stanish, L.F.; Knelman, J.E.; Darcy, J.L.; Lynch, R.C.; Wickey, P.; et al. Patterns and processes of microbial community assembly. Microbiol. Mol. Biol. Rev. 2013, 77, 342–356. [Google Scholar] [CrossRef]
- Linnenbrink, M.; Wang, J.; Hardouin, E.A.; Kunzel, S.; Metzler, D.; Baines, J.F. The role of biogeography in shaping diversity of the intestinal microbiota in house mice. Mol. Ecol. 2013, 22, 1904–1916. [Google Scholar] [CrossRef]
- Suzuki, T.A.; Martins, F.M.; Phifer-Rixey, M.; Nachman, M.W. The gut microbiota and Bergmann’s rule in wild house mice. Mol. Ecol. 2020, 29, 2300–2311. [Google Scholar] [CrossRef]
- Goertz, S.; de Menezes, A.B.; Birtles, R.J.; Fenn, J.; Lowe, A.E.; MacColl, A.D.C.; Poulin, B.; Young, S.; Bradley, J.E.; Taylor, C.H. Geographical location influences the composition of the gut microbiota in wild house mice (Mus musculus domesticus) at a fine spatial scale. PLoS ONE 2019, 14, e0222501. [Google Scholar] [CrossRef]
- Suzuki, T.A.; Martins, F.M.; Nachman, M.W. Altitudinal variation of the gut microbiota in wild house mice. Mol. Ecol. 2019, 28, 2378–2390. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, C.; Li, G.; Yi, X. The influence of species identity and geographic locations on gut microbiota of small rodents. Front. Microbiol. 2022, 13, 983660. [Google Scholar] [CrossRef]
- Weinstein, S.B.; Martinez-Mota, R.; Stapleton, T.E.; Klure, D.M.; Greenhalgh, R.; Orr, T.J.; Dale, C.; Kohl, K.D.; Dearing, M.D. Microbiome stability and structure is governed by host phylogeny over diet and geography in woodrats (Neotoma spp.). Proc. Natl. Acad. Sci. USA, 2021; 118, e2108787118. [Google Scholar] [CrossRef]
- Zhao, J.S.; Yao, Y.F.; Li, D.Y.; Xu, H.M.; Wu, J.Y.; Wen, A.X.; Xie, M.; Ni, Q.Y.; Zhang, M.W.; Peng, G.N.; et al. Characterization of the Gut Microbiota in Six Geographical Populations of Chinese Rhesus Macaques (Macaca mulatta), Implying an Adaptation to High-Altitude Environment. Microb. Ecol. 2018, 76, 565–577. [Google Scholar] [CrossRef]
- Bezek, K.; Petelin, A.; Praznikar, J.; Nova, E.; Redondo, N.; Marcos, A.; Praznikar, Z.J. Obesity Measures and Dietary Parameters as Predictors of Gut Microbiota Phyla in Healthy Individuals. Nutrients 2020, 12, 2695. [Google Scholar] [CrossRef]
- Nakajima, A.; Sasaki, T.; Itoh, K.; Kitahara, T.; Takema, Y.; Hiramatsu, K.; Ishikawa, D.; Shibuya, T.; Kobayashi, O.; Osada, T.; et al. A Soluble Fiber Diet Increases Bacteroides fragilis Group Abundance and Immunoglobulin A Production in the Gut. Appl. Environ. Microb. 2020, 86, e00405-20. [Google Scholar] [CrossRef]
- Holmes, E.; Nicholson, J. Variation in gut microbiota strongly influences individual rodent phenotypes. Toxicol. Sci. 2005, 87, 1–2. [Google Scholar] [CrossRef]
- Bo, T.B.; Zhang, X.Y.; Wen, J.; Deng, K.; Qin, X.W.; Wang, D.H. The microbiota-gut-brain interaction in regulating host metabolic adaptation to cold in male Brandt’s voles (Lasiopodomys brandtii). ISME J. 2019, 13, 3037–3053. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
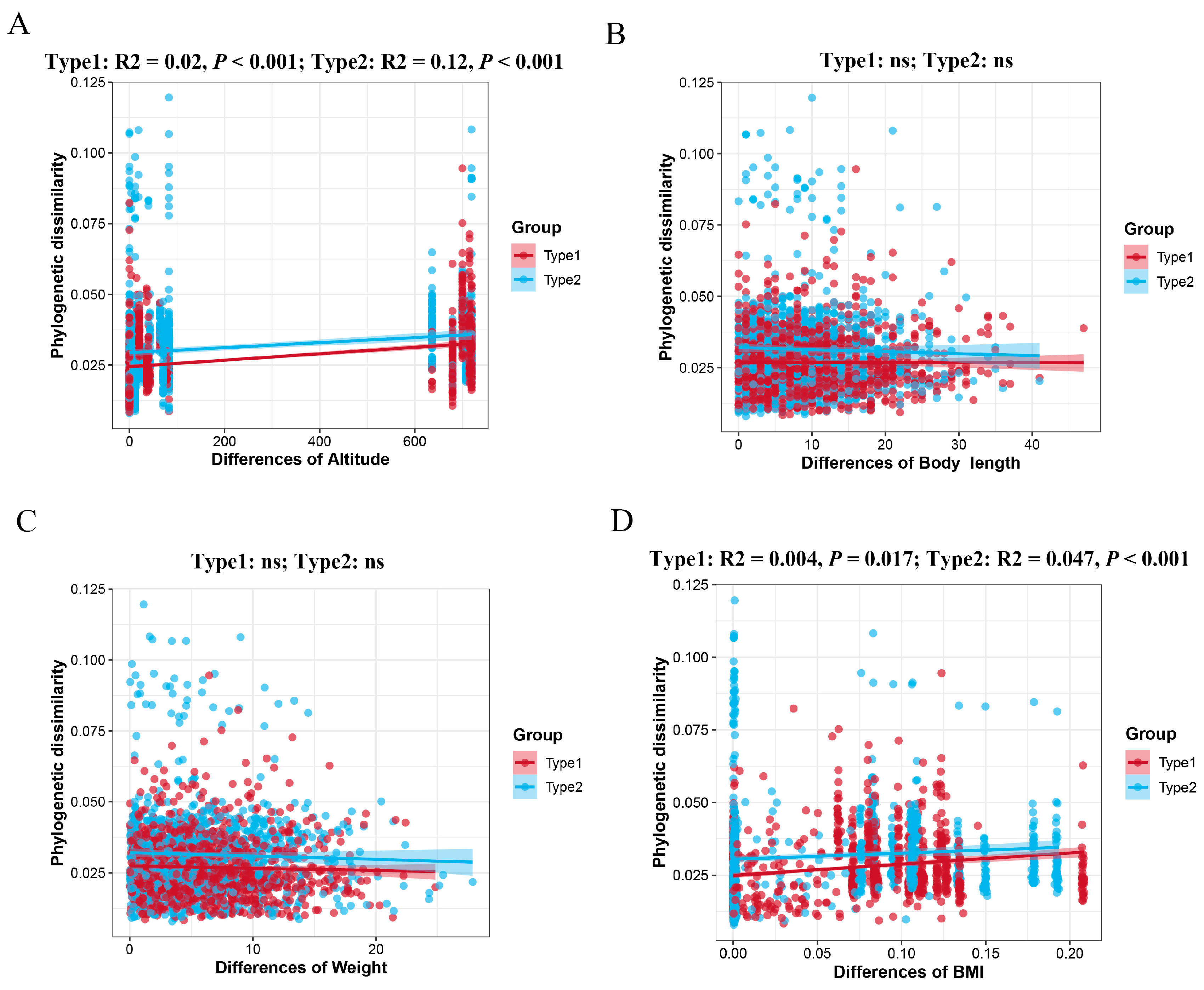{kind=link}
| Network Diameter | Modularity | Clustering Coefficient | Graph Density | Average Degree | Average Path Length | |
|---|---|---|---|---|---|---|
| Type1 | 12 | 0.706 | 0.392 | 0.018 | 3.469 | 5.109 |
| Type2 | 4 | 0.768 | 0.489 | 0.010 | 2.061 | 1.673 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, Y.; Zhou, T.; Yang, S.; Yin, B.; Wu, R.; Wei, W. Distinct Gut Microbial Enterotypes and Functional Dynamics in Wild Striped Field Mice (Apodemus agrarius) across Diverse Populations. Microorganisms 2024, 12, 671. https://doi.org/10.3390/microorganisms12040671
Wu Y, Zhou T, Yang S, Yin B, Wu R, Wei W. Distinct Gut Microbial Enterotypes and Functional Dynamics in Wild Striped Field Mice (Apodemus agrarius) across Diverse Populations. Microorganisms. 2024; 12(4):671. https://doi.org/10.3390/microorganisms12040671
Chicago/Turabian StyleWu, Yongzhen, Taoxiu Zhou, Shengmei Yang, Baofa Yin, Ruiyong Wu, and Wanhong Wei. 2024. "Distinct Gut Microbial Enterotypes and Functional Dynamics in Wild Striped Field Mice (Apodemus agrarius) across Diverse Populations" Microorganisms 12, no. 4: 671. https://doi.org/10.3390/microorganisms12040671
APA StyleWu, Y., Zhou, T., Yang, S., Yin, B., Wu, R., & Wei, W. (2024). Distinct Gut Microbial Enterotypes and Functional Dynamics in Wild Striped Field Mice (Apodemus agrarius) across Diverse Populations. Microorganisms, 12(4), 671. https://doi.org/10.3390/microorganisms12040671