
Multi-Omics Approaches Provide New Insights into the Identification of Putative Fungal Effectors from Valsa mali


 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Material and Methods
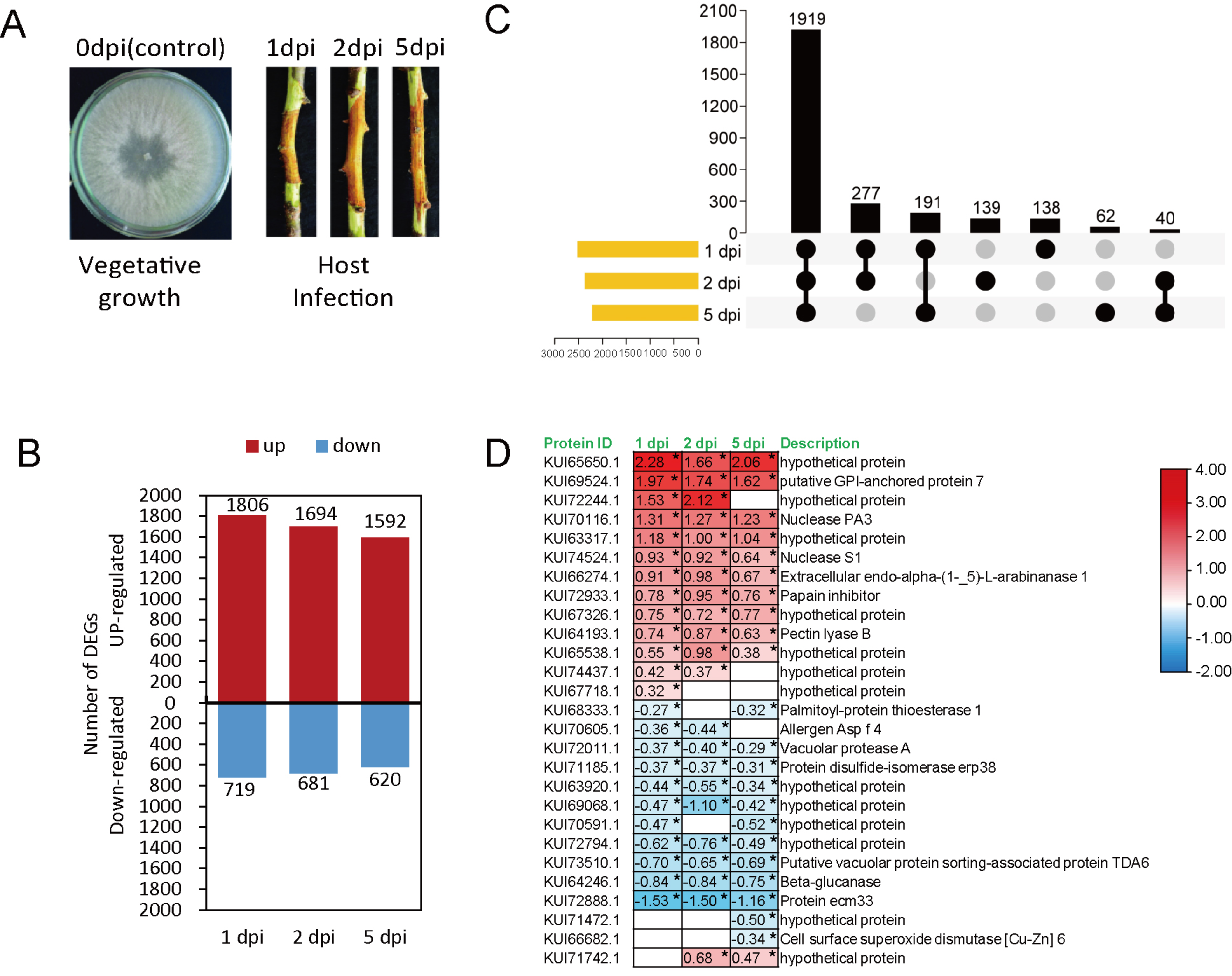
2.1. Fungal Growth and Infection
2.2. Genome-Wide Identification of Effector Candidates from V. mali
2.3. Transcriptome Sequencing and Data Analysis
2.4. Proteome Sequencing and Data Analysis
2.5. Plant Growth, Vector Construction
2.6. In Planta Expression and Symptom Analysis
2.7. Validation of the SP Secretory Activities of Candidate Effectors
2.8. Statistical Analysis
3. Results
3.1. Genome-Wide Identification of Candidate Effector Proteins from V. mali
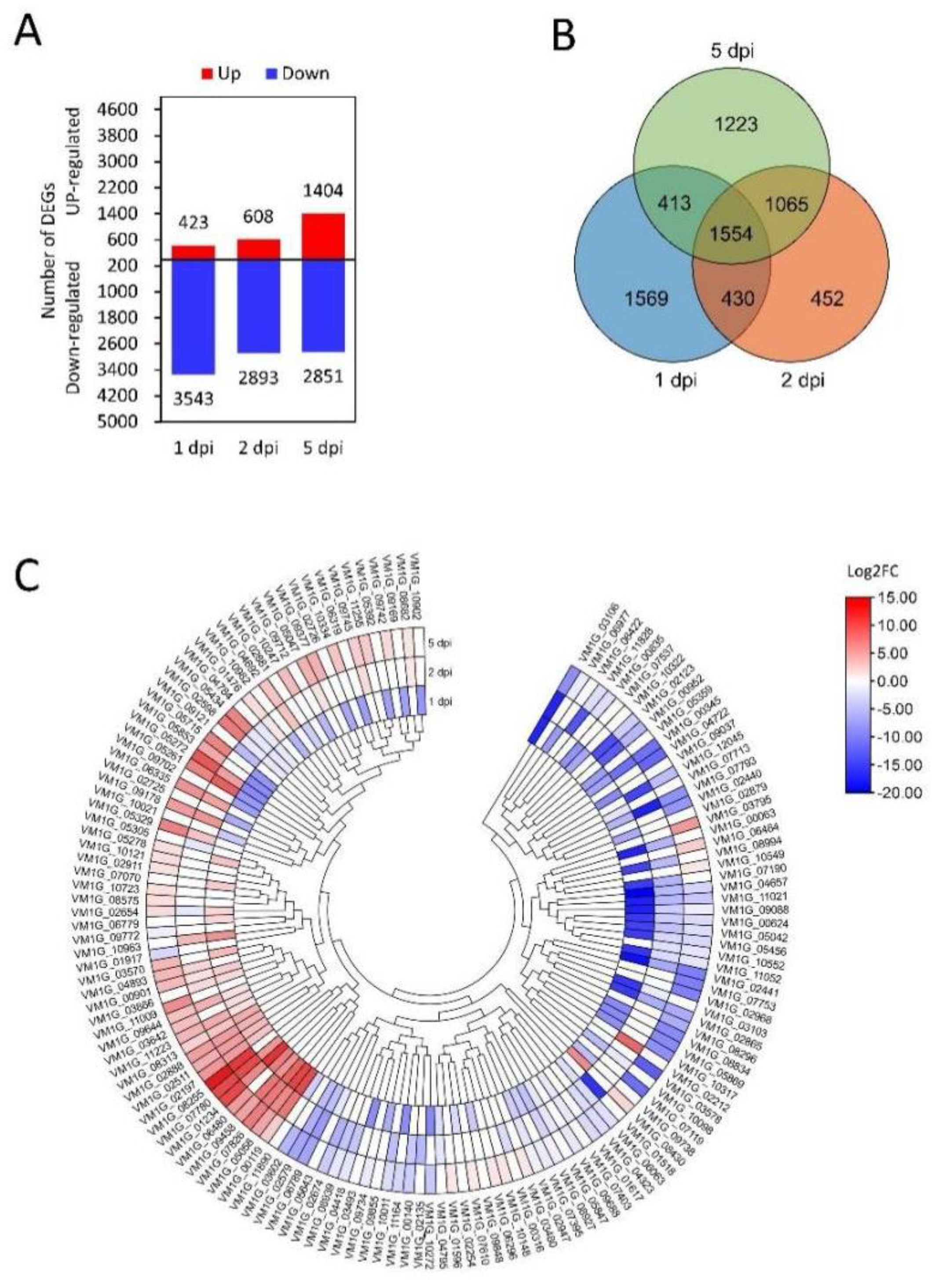
3.2. Transcriptional Analysis of Effector Candidates
3.3. Proteomic Analysis of Effector Candidates
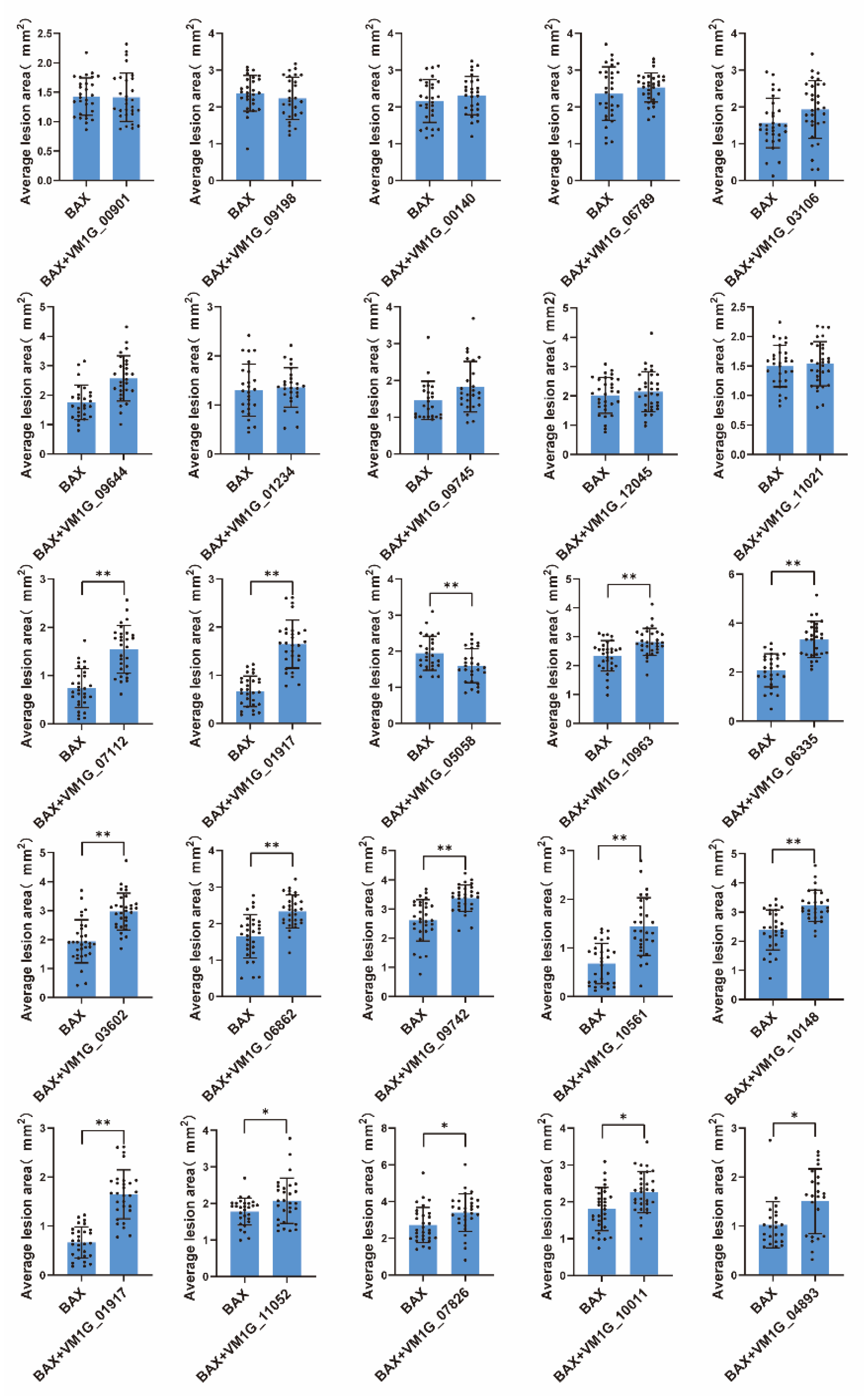
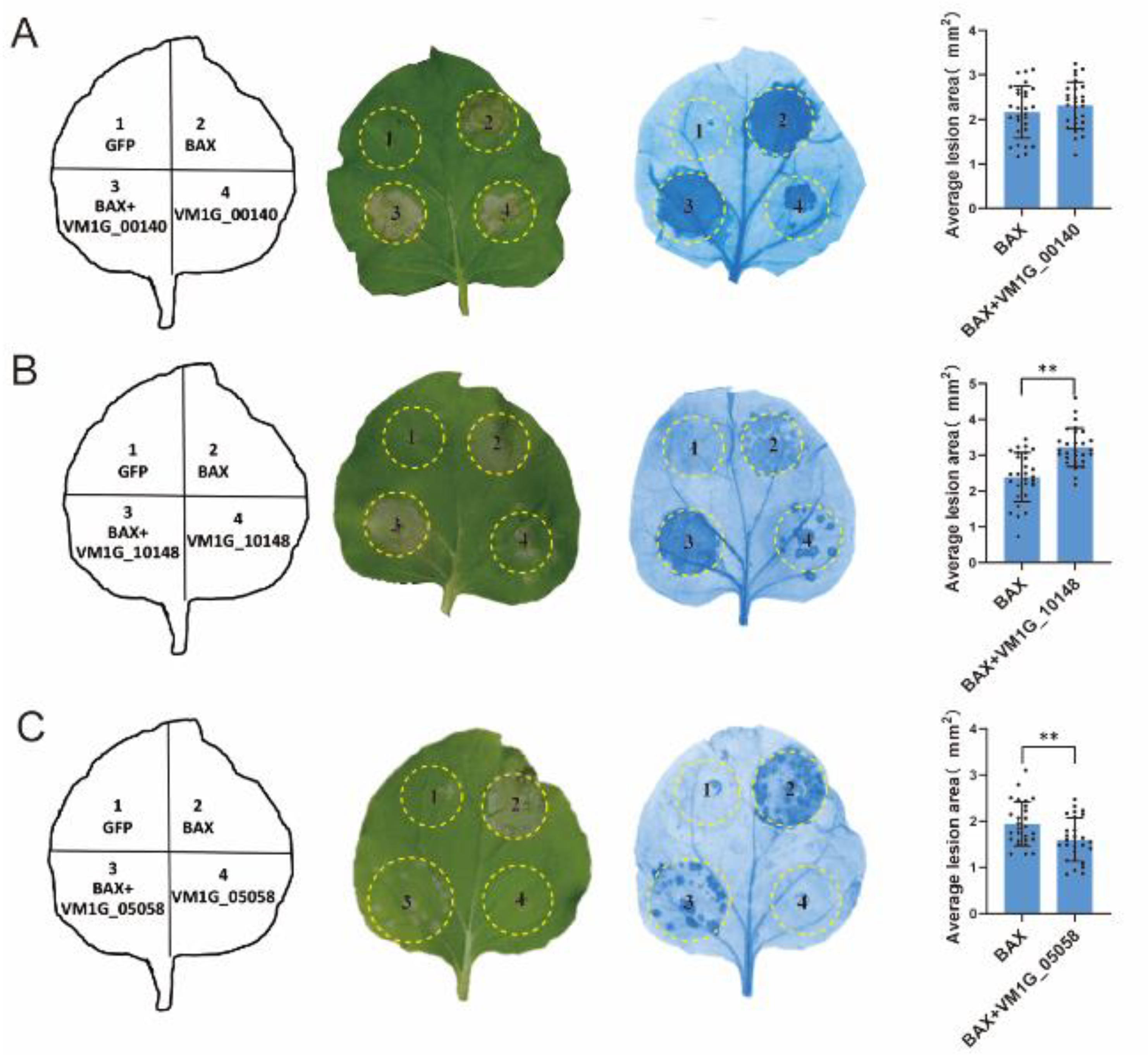
3.4. Pathogenicity Analysis of Selected Effector Candidates
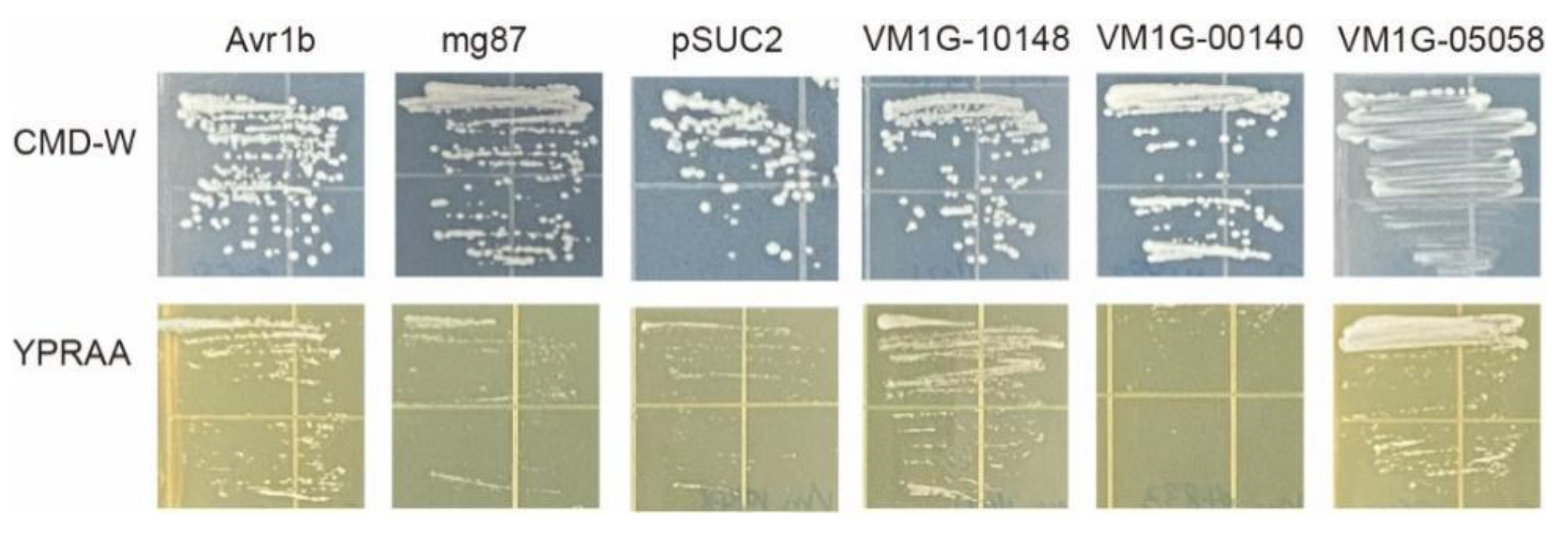
3.5. Secretion Analysis of Selected Effector Candidates
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Boller, T.; He, S.Y. Innate Immunity in Plants: An Arms Race Between Pattern Recognition Receptors in Plants and Effectors in Microbial Pathogens. Science 2009, 324, 742–744. [Google Scholar] [CrossRef] [PubMed]
- de Wit, P.J.G.M.; Mehrabi, R.; van den Burg, H.A.; Stergiopoulos, I. Fungal effector proteins: Past, present and future. Mol. Plant Pathol. 2009, 10, 735–747. [Google Scholar] [CrossRef] [PubMed]
- Giraldo, M.C.; Valent, B. Filamentous plant pathogen effectors in action. Nat. Rev. Microbiol. 2013, 11, 800–814. [Google Scholar] [CrossRef] [PubMed]
- Wawra, S.; Belmonte, R.; Lobach, L.; Saraiva, M.; Willems, A.; van West, P. Secretion, delivery and function of oomycete effector proteins. Curr. Opin. Microbiol. 2012, 15, 685–691. [Google Scholar] [CrossRef] [PubMed]
- Djamei, A.; Schipper, K.; Rabe, F.; Ghosh, A.; Vincon, V.; Kahnt, J.; Osorio, S.; Tohge, T.; Fernie, A.R.; Feussner, I.; et al. Metabolic priming by a secreted fungal effector. Nature 2011, 478, 395–398. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Song, T.; Zhang, X.; Yuan, H.; Su, L.; Li, W.; Xu, J.; Liu, S.; Chen, L.; Chen, T.; et al. Unconventionally secreted effectors of two filamentous pathogens target plant salicylate biosynthesis. Nat. Commun. 2014, 5, 4686. [Google Scholar] [CrossRef] [PubMed]
- Plett, J.M.; Daguerre, Y.; Wittulsky, S.; Vayssieres, A.; Deveau, A.; Melton, S.J.; Kohler, A.; Morrell-Falvey, J.L.; Brun, A.; Veneault-Fourrey, C.; et al. Effector MiSSP7 of the mutualistic fungus Laccaria bicolor stabilizes the Populus JAZ6 protein and represses jasmonic acid (JA) responsive genes. Proc. Natl. Acad. Sci. USA 2014, 111, 8299–8304. [Google Scholar] [CrossRef]
- Okmen, B.; Doehlemann, G. Inside plant: Biotrophic strategies to modulate host immunity and metabolism. Curr. Opin. Plant Biol. 2014, 20, 19–25. [Google Scholar] [CrossRef]
- Kang, H.; Chen, X.; Kemppainen, M.; Pardo, A.G.; Veneault-Fourrey, C.; Kohler, A.; Martin, F.M. The small secreted effector protein MiSSP7.6 of Laccaria bicoloris required for the establishment of ectomycorrhizal symbiosis. Environ. Microbiol. 2020, 22, 1435–1446. [Google Scholar] [CrossRef]
- Sperschneider, J.; Dodds, P.N. EffectorP 3.0: Prediction of apoplastic and cytoplasmic effectors in fungi and oomycetes. Mol. Plant-Microbe Interact. 2021, 32, 146–156. [Google Scholar] [CrossRef]
- Teufel, F.; Almagro Armenteros, J.J.; Johansen, A.R.; Gislason, M.H.; Pihl, S.I.; Tsirigos, K.D.; Winther, O.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 6.0 predicts all five types of signal peptides using protein language models. Nat. Biotechnol. 2022, 40, 1023–1025. [Google Scholar] [CrossRef]
- Carreón-Anguiano, K.G.; Islas-Flores, I.; Vega-Arreguín, J.; Sáenz-Carbonell, L.; Canto-Canché, B. EffHunter: A Tool for Prediction of Effector Protein Candidates in Fungal Proteomic Databases. Biomolecules 2020, 10, 712. [Google Scholar] [CrossRef] [PubMed]
- Seong, K.; Krasileva, K.V. Prediction of effector protein structures from fungal phytopathogens enables evolutionary analyses. Nat. Microbiol. 2023, 8, 174–187. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wei, J.; Huang, L.; Kang, Z. Re-evaluation of pathogens causing Valsa canker on apple in China. Mycologia 2011, 103, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Suzaki, K.Y.K.; Ito, T. Pathogenicity to Apple Branch and Phloridzin Degrading Activity of Valsa ceratosperma Isolated from Some Broad Leaf Trees Including Apple Tree. Annu. Rep. Soc. Plant Prot. N. Jpn. 1997, 48, 145–147. [Google Scholar]
- Yin, Z.; Liu, H.; Li, Z.; Ke, X.; Dou, D.; Gao, X.; Song, N.; Dai, Q.; Wu, Y.; Xu, J.R.; et al. Genome sequence of Valsa canker pathogens uncovers a potential adaptation of colonization of woody bark. New Phytol. 2015, 208, 1202–1216. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Yin, Z.; Fan, Y.; Xu, M.; Kang, Z.; Huang, L. Candidate effector proteins of the necrotrophic apple canker pathogen Valsa mali can suppress BAX-induced PCD. Front. Plant Sci. 2015, 6, 579. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wang, S.; Gong, W.; Lv, L.; Xu, L.; Nie, J.; Huang, L. Valsa mali secretes an effector protein VmEP1 to target a K homology domain-containing protein for virulence in apple. Mol. Plant Pathol. 2022, 23, 1577–1591. [Google Scholar] [CrossRef] [PubMed]
- Che, R.; Liu, C.; Wang, Q.; Tu, W.; Wang, P.; Li, C.; Gong, X.; Mao, K.; Feng, H.; Huang, L.; et al. The Valsa Mali effector Vm1G-1794 protects the aggregated MdEF-Tu from autophagic degradation to promote infection in apple. Autophagy 2022, 19, 1745–1763. [Google Scholar] [CrossRef]
- Liu, X.; Li, X.; Bozorov, T.A.; Ma, R.; Ma, J.; Zhang, Y.; Yang, H.; Li, L.; Zhang, D.; Kim, M.S. Characterization and pathogenicity of six Cytospora strains causing stem canker of wild apple in the Tianshan Forest, China. For. Pathol. 2020, 50, e12587. [Google Scholar] [CrossRef]
- Liu, X.; Li, X.; Wen, X.; Zhang, Y.; Ding, Y.; Zhang, Y.; Gao, B.; Zhang, D. PacBio full-length transcriptome of wild apple (Malus sieversii) provides insights into canker disease dynamic response. BMC Genom. 2021, 22, 52. [Google Scholar] [CrossRef]
- Lacomme, C.; Cruz, S.S. Bax-induced cell death in tobacco is similar to the hypersensitive response. Proc. Natl. Acad. Sci. USA 1999, 96, 7956–7961. [Google Scholar] [CrossRef] [PubMed]
- Dou, D.; Kale, S.D.; Wang, X.; Chen, Y.; Wang, Q.; Wang, X.; Jiang, R.H.; Arredondo, F.D.; Anderson, R.G.; Thakur, P.B.; et al. Conserved C-terminal motifs required for avirulence and suppression of cell death by Phytophthora sojae effector Avr1b. Plant Cell 2008, 20, 1118–1133. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.K.; Young, C.; Lee, M.; Oliva, R.; Bozkurt, T.O.; Cano, L.M.; Win, J.; Bos, J.I.; Liu, H.Y.; van Damme, M.; et al. In planta expression screens of Phytophthora infestans RXLR effectors reveal diverse phenotypes, including activation of the Solanum bulbocastanum disease resistance protein Rpi-blb2. Plant Cell 2009, 21, 2928–2947. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.; Wu, L.; Xue, Q.; Zhang, Q.; Zou, H. Ralstonia solanacearum type III effector RipAA targets chloroplastic AtpB to modulate an incompatible interaction on Nicotiana benthamiana. Front. Microbiol. 2023, 14, 1179824. [Google Scholar] [CrossRef] [PubMed]
- Sperschneider, J.; Williams, A.H.; Hane, J.K.; Singh, K.B.; Taylor, J.M. Evaluation of Secretion Prediction Highlights Differing Approaches Needed for Oomycete and Fungal Effectors. Front. Plant Sci. 2015, 6, 1168. [Google Scholar] [CrossRef]
- Shao, D.D.; Smith, D.L.; Kabbage, M.; Roth, M.G. Effectors of Plant Necrotrophic Fungi. Front. Plant Sci. 2021, 12, 687713. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Kelley, B.S.; Damasceno, C.M.; St John, B.; Kim, B.S.; Kim, B.D.; Rose, J.K. A Functional Screen to Characterize the Secretomes of Eukaryotic Pathogens and Their Hosts In Planta. Mol. Plant. Microbe. Interact. 2006, 19, 1368–1377. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Li, H.; Zhou, Y.; Bao, Y.; Duan, Z.; Wang, C.; Powell, C.A.; Chen, B.; Zhang, M.; Yao, W. Predication of the Effector Proteins Secreted by Fusarium sacchari Using Genomic Analysis and Heterogenous Expression. J. Fungi 2022, 8, 59. [Google Scholar] [CrossRef] [PubMed]
- Mentges, M.; Glasenapp, A.; Boenisch, M.; Malz, S.; Henrissat, B.; Frandsen, R.J.N.; Guldener, U.; Munsterkotter, M.; Bormann, J.; Lebrun, M.H.; et al. Infection cushions of Fusarium graminearum are fungal arsenals for wheat infection. Mol. Plant Pathol. 2020, 21, 1070–1087. [Google Scholar] [CrossRef]
- Jones, D.A.B.; Bertazzoni, S.; Turo, C.J.; Syme, R.A.; Hane, J.K. Bioinformatic prediction of plant–pathogenicity effector proteins of fungi. Curr. Opin. Microbiol. 2018, 46, 43–49. [Google Scholar] [CrossRef]
- Kubicek, C.P.; Starr, T.L.; Glass, N.L. Plant Cell Wall–Degrading Enzymes and Their Secretion in Plant-Pathogenic Fungi. Annu. Rev. Phytopathol. 2014, 52, 427–451. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Xie, S.; Zhao, Y.; Meng, X.; Song, L.; Feng, H.; Huang, L. Hce2 domain-containing effectors contribute to the full virulence of Valsa mali in a redundant manner. Mol. Plant Pathol. 2019, 20, 843–856. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Feng, H.; Zhao, Y.; Song, L.; Gao, C.; Xu, X.; Huang, L. Valsa mali Pathogenic Effector VmPxE1 Contributes to Full Virulence and Interacts With the Host Peroxidase MdAPX1 as a Potential Target. Front. Microbiol. 2018, 9, 821. [Google Scholar] [CrossRef]
- Torto, T.A.; Li, S.; Styer, A.; Huitema, E.; Testa, A.; Gow, N.A.; van West, P.; Kamoun, S. EST mining and functional expression assays identify extracellular effector proteins from the plant pathogen Phytophthora. Genome Res. 2003, 13, 1675–1685. [Google Scholar] [CrossRef] [PubMed]
- Kamoun, S. A catalogue of the effector secretome of plant pathogenic oomycetes. Annu. Rev. Phytopathol. 2006, 44, 41–60. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Chen, Y.; Jian, H.; Yang, D.; Dai, Y.; Pan, L.; Shi, F.; Yang, S.; Liu, Q. Large-Scale Identification and Characterization of Heterodera avenae Putative Effectors Suppressing or Inducing Cell Death in Nicotiana benthamiana. Front. Plant Sci. 2018, 8, 2062. [Google Scholar] [CrossRef]
- Vleeshouwers, V.G.; Oliver, R.P. Effectors as tools in disease resistance breeding against biotrophic, hemibiotrophic, and necrotrophic plant pathogens. Mol. Plant Microbe Interact. 2014, 27, 196–206. [Google Scholar] [CrossRef] [PubMed]
- Uhse, S.; Djamei, A. Effectors of plant-colonizing fungi and beyond. PLoS Pathog. 2018, 14, e1006992. [Google Scholar] [CrossRef] [PubMed]
- Stergiopoulos, I.; de Wit, P.J. Fungal effector proteins. Annu. Rev. Phytopathol. 2009, 47, 233–263. [Google Scholar] [CrossRef]
- Heijne, G.v. Life and death of a signal peptide. Nature 1989, 396, 111–113. [Google Scholar] [CrossRef]
- Niu, X.; Yang, G.; Lin, H.; Liu, Y.; Li, P.; Zheng, A. A Novel, Small Cysteine-Rich Effector, RsSCR10 in Rhizoctonia solani Is Sufficient to Trigger Plant Cell Death. Front. Microbiol. 2021, 12, 684923. [Google Scholar] [CrossRef] [PubMed]
- Shen, D.; Wang, Y.; Chen, X.; Srivastava, V.; Toffolatti, S.L. Editorial: Advances in multi-omics study of filamentous plant pathogens. Front. Microbiol. 2022, 13, 998501. [Google Scholar] [CrossRef] [PubMed]
- Halaouli, S.; Asther, M.; Sigoillot, J.C.; Hamdi, M.; Lomascolo, A. Fungal tyrosinases: New prospects in molecular characteristics, bioengineering and biotechnological applications. J. Appl. Microbiol. 2006, 100, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Zhang, X.; Zhang, Y.; Yi, G.; Xie, J.; Viljoen, A.; Wang, W.; Mostert, D.; Fu, G.; Peng, C.; et al. FocECM33, a GPI-anchored protein, regulates vegetative growth and virulence in Fusarium oxysporum f. sp. cubense tropical race 4. Fungal Biol. 2022, 126, 213–223. [Google Scholar] [CrossRef]
- Chang, P.-K.; Zhang, Q.; Scharfenstein, L.; Mack, B.; Yoshimi, A.; Miyazawa, K.; Abe, K. Aspergillus flavus GPI-anchored protein-encoding ecm33 has a role in growth, development, aflatoxin biosynthesis, and maize infection. Appl. Microbiol. Biotechnol. 2018, 102, 5209–5220. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kahar, G.; Haxim, Y.; Waheed, A.; Bozorov, T.A.; Liu, X.; Wen, X.; Zhao, M.; Zhang, D. Multi-Omics Approaches Provide New Insights into the Identification of Putative Fungal Effectors from Valsa mali. Microorganisms 2024, 12, 655. https://doi.org/10.3390/microorganisms12040655
Kahar G, Haxim Y, Waheed A, Bozorov TA, Liu X, Wen X, Zhao M, Zhang D. Multi-Omics Approaches Provide New Insights into the Identification of Putative Fungal Effectors from Valsa mali. Microorganisms. 2024; 12(4):655. https://doi.org/10.3390/microorganisms12040655
Chicago/Turabian StyleKahar, Gulnaz, Yakupjan Haxim, Abdul Waheed, Tohir A. Bozorov, Xiaojie Liu, Xuejing Wen, Mingqi Zhao, and Daoyuan Zhang. 2024. "Multi-Omics Approaches Provide New Insights into the Identification of Putative Fungal Effectors from Valsa mali" Microorganisms 12, no. 4: 655. https://doi.org/10.3390/microorganisms12040655
APA StyleKahar, G., Haxim, Y., Waheed, A., Bozorov, T. A., Liu, X., Wen, X., Zhao, M., & Zhang, D. (2024). Multi-Omics Approaches Provide New Insights into the Identification of Putative Fungal Effectors from Valsa mali. Microorganisms, 12(4), 655. https://doi.org/10.3390/microorganisms12040655