
Lnc-RPS6P3 Inhibits Influenza A Virus Replication and Attenuates the Inhibitory Effect of NS1 on Innate Immune Response


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Line and Viruses
2.2. Plasmids and Antibodies
2.3. Cell Transfection and Lentiviral Production
2.4. Immunoblotting
2.5. RNA Isolation and Quantitative RT-PCR (RT-qPCR)
2.6. Luciferase Reporter Assay
2.7. Co-Immunoprecipitation (Co-IP) and RNA Immunoprecipitation (RIP)
2.8. RNA Pulldown Assay and Mass Spectrometry Analysis
2.9. Plaque Assay
2.10. RNA Fluorescence In Situ Hybridization (FISH)
2.11. Cell Fractionation
2.12. Statistical Analysis
3. Results
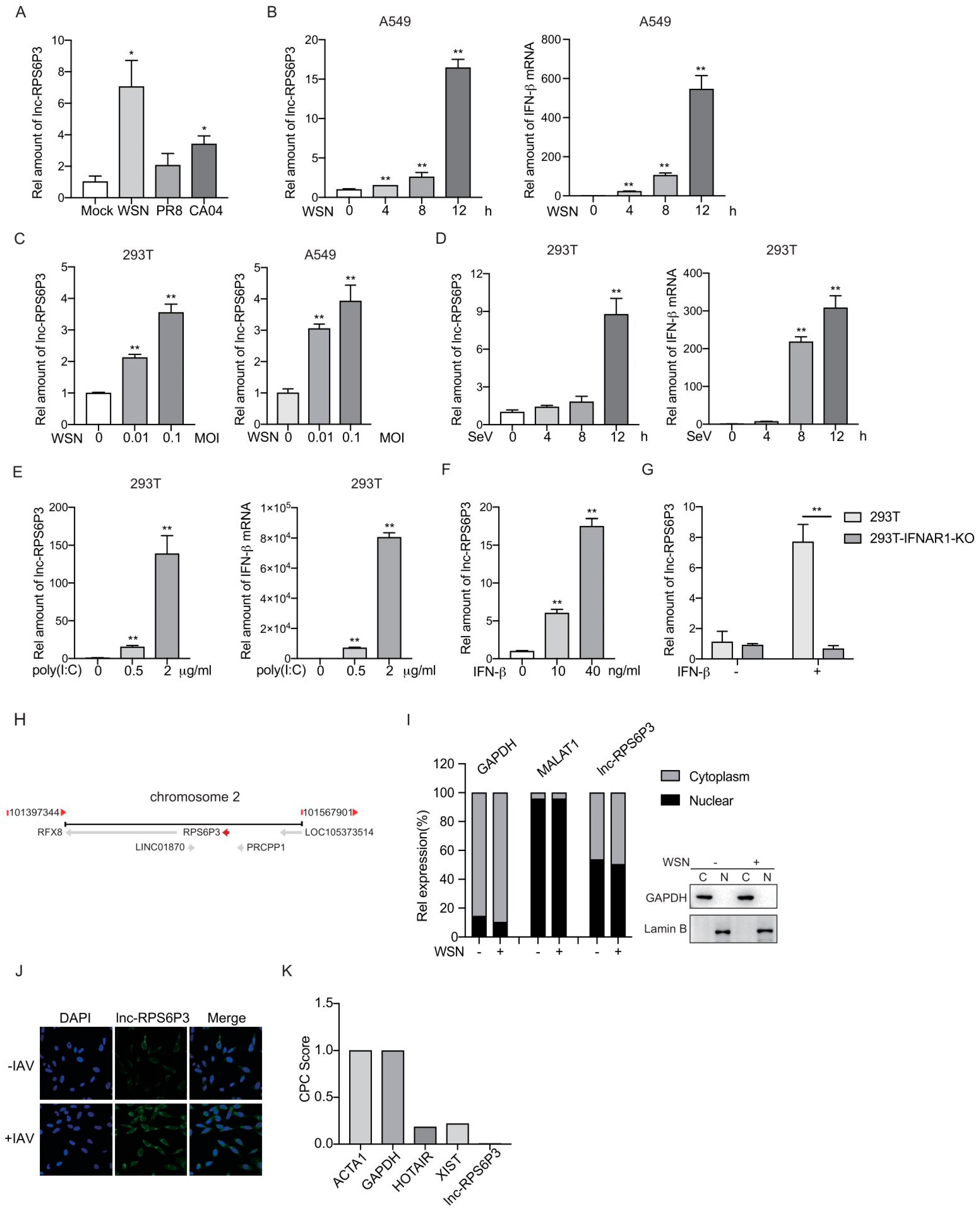
3.1. Lnc-RPS6P3 Is Induced upon IAV Infection and IFN-β Treatment
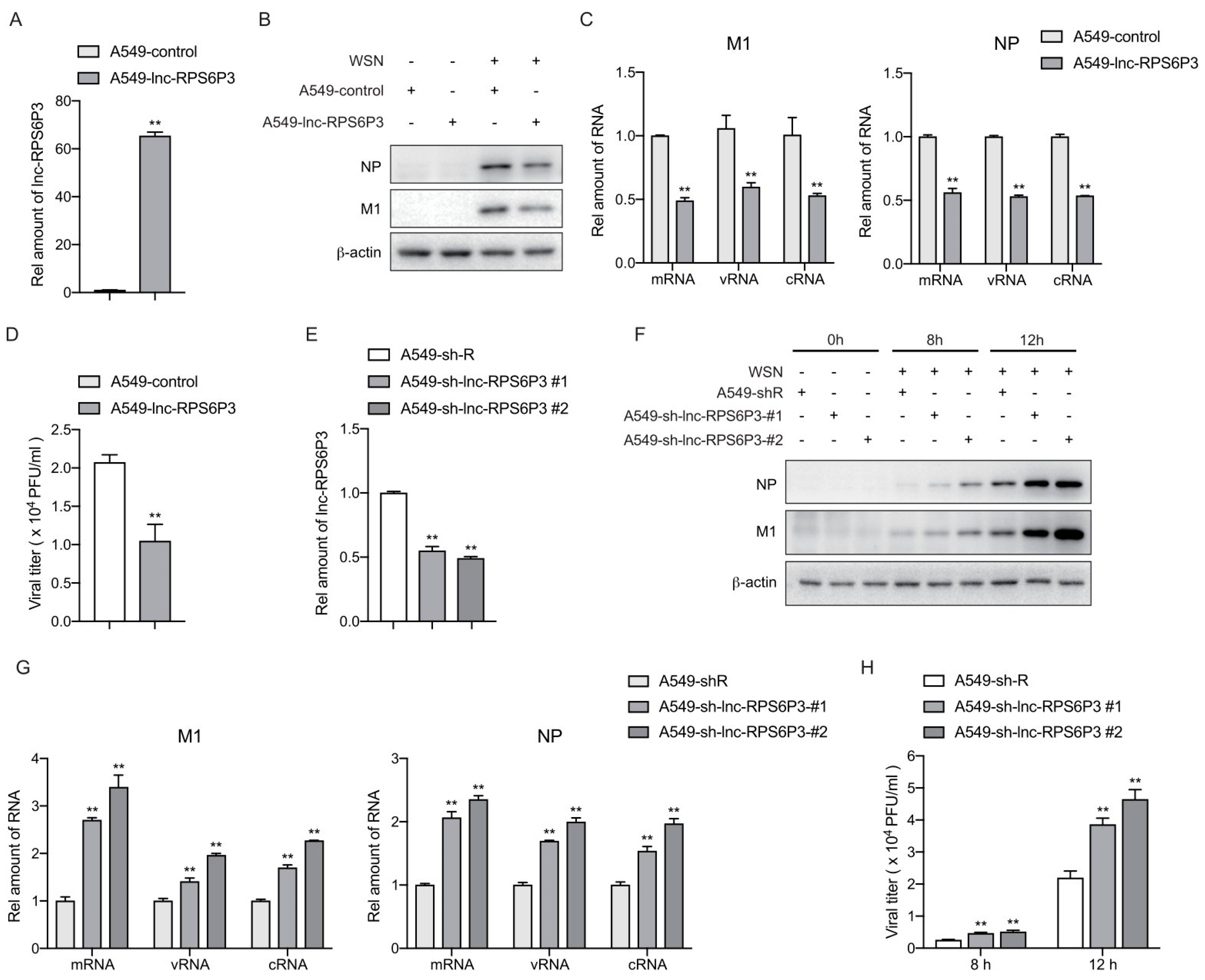
3.2. Lnc-RPS6P3 Inhibits the Replication of Influenza A Virus
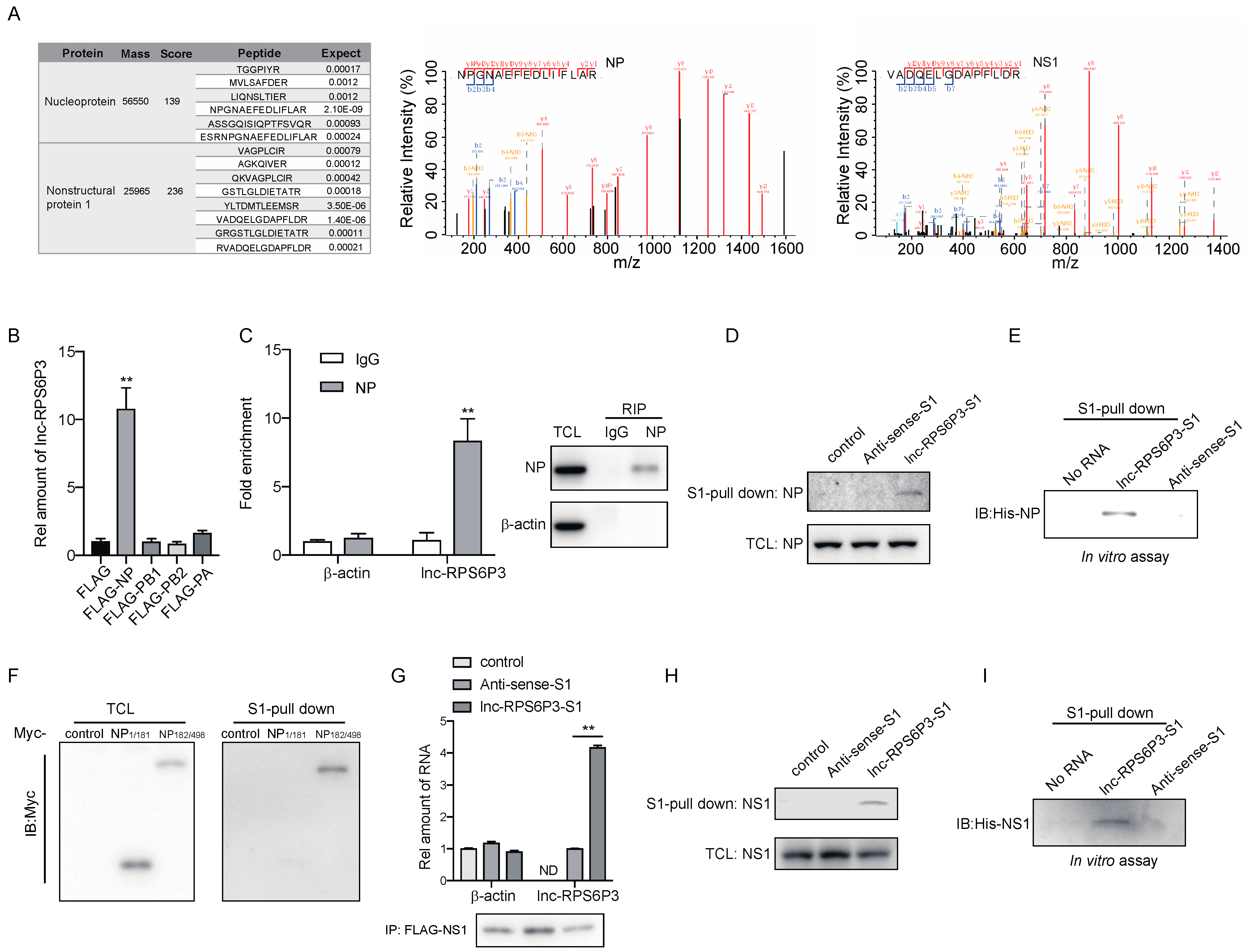
3.3. Lnc-RPS6P3 Interacts with NP and NS1 Proteins
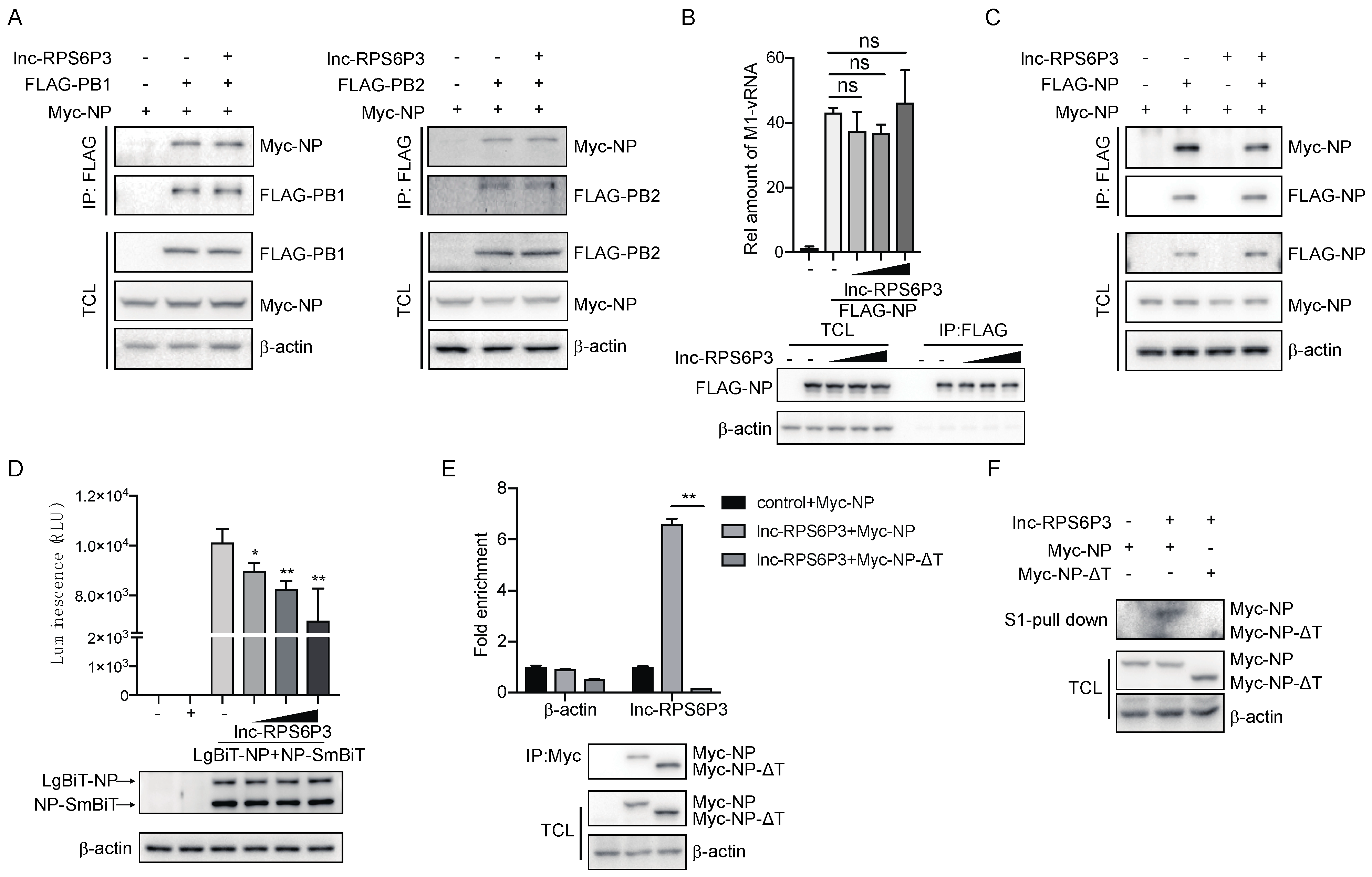
3.4. Lnc-RPS6P3 Blocks vRNP Activity of Influenza A Virus by Interfering with NP Oligomerization
3.5. Lnc-RPS6P3 Reduces the Inhibitory Effect of NS1 on IFN-β Activation by Decreasing the Interaction of NS1 with RIG-I and TRIM25
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Krammer, F.; Smith, G.J.D.; Fouchier, R.A.M.; Peiris, M.; Kedzierska, K.; Doherty, P.C.; Palese, P.; Shaw, M.L.; Treanor, J.; Webster, R.G. Influenza. Nat. Rev. Dis. Primers 2018, 4, 3. [Google Scholar] [CrossRef] [PubMed]
- Han, A.X.; de Jong, S.P.J.; Russell, C.A. Co-Evolution of Immunity and Seasonal Influenza Viruses. Nat. Rev. Microbiol. 2023, 21, 805–817. [Google Scholar] [CrossRef] [PubMed]
- Resa-Infante, P.; Jorba, N.; Coloma, R.; Ortin, J. The Influenza Virus RNA Synthesis Machine. RNA Biol. 2011, 8, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Reich, S.; Guilligay, D.; Pflug, A.; Malet, H.; Berger, I.; Crépin, T.; Hart, D.; Lunardi, T.; Nanao, M.; Ruigrok, R.W.H. Structural Insight into Cap-Snatching and RNA Synthesis by Influenza Polymerase. Nature 2014, 516, 361–366. [Google Scholar] [CrossRef] [PubMed]
- Te Velthuis, A.J.W.; Fodor, E. Influenza Virus RNA Polymerase: Insights into the Mechanisms of Viral RNA Synthesis. Nature Rev. Microbiol. 2016, 14, 479–493. [Google Scholar] [CrossRef] [PubMed]
- Lo, C.-Y.; Tang, Y.-S.; Shaw, P.-C. Structure and Function of Influenza Virus Ribonucleoprotein. In Virus Protein and Nucleoprotein Complexes; Subcellular Biochemistry; Springer: Singapore, 2018; Volume 88, pp. 95–128. [Google Scholar]
- Lee, N.; Le Sage, V.; Nanni, A.V.; Snyder, D.J.; Cooper, V.S.; Lakdawala, S.S. Genome-Wide Analysis of Influenza Viral RNA and Nucleoprotein Association. Nucleic Acids Res. 2017, 45, 8968–8977. [Google Scholar] [CrossRef] [PubMed]
- Chan, W.-H.; Ng, A.K.-L.; Robb, N.C.; Lam, M.K.-H.; Chan, P.K.-S.; Au, S.W.-N.; Wang, J.-H.; Fodor, E.; Shaw, P.-C. Functional Analysis of the Influenza Virus H5N1 Nucleoprotein Tail Loop Reveals Amino Acids That Are Crucial for Oligomerization and Ribonucleoprotein Activities. J. Virol. 2010, 84, 7337–7345. [Google Scholar] [CrossRef] [PubMed]
- Turrell, L.; Lyall, J.W.; Tiley, L.S.; Fodor, E.; Vreede, F.T. The Role and Assembly Mechanism of Nucleoprotein in Influenza A Virus Ribonucleoprotein Complexes. Nat. Commun. 2013, 4, 1591. [Google Scholar] [CrossRef] [PubMed]
- Geiss, G.K.; Salvatore, M.; Tumpey, T.M.; Carter, V.S.; Wang, X.; Basler, C.F.; Taubenberger, J.K.; Bumgarner, R.E.; Palese, P.; Katze, M.G. Cellular Transcriptional Profiling in Influenza A Virus-Infected Lung Epithelial Cells: The Role of the Nonstructural NS1 Protein in the Evasion of the Host Innate Defense and Its Potential Contribution to Pandemic Influenza. Proc. Natl. Acad. Sci. USA 2002, 99, 10736–10741. [Google Scholar] [CrossRef]
- Krug, R.M. Functions of the Influenza A Virus NS1 Protein in Antiviral Defense. Curr. Opin. Virol. 2015, 12, 1–6. [Google Scholar] [CrossRef]
- Lamotte, L.-A.; Tafforeau, L. How Influenza A Virus NS1 Deals with the Ubiquitin System to Evade Innate Immunity. Viruses 2021, 13, 2309. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Chen, L.; Zeng, H.; Gomez, J.A.; Plowden, J.; Fujita, T.; Katz, J.M.; Donis, R.O.; Sambhara, S. NS1 Protein of Influenza A Virus Inhibits the Function of Intracytoplasmic Pathogen Sensor, RIG-I. Am. J. Respir. Cell Mol. Biol. 2007, 36, 263–269. [Google Scholar] [CrossRef]
- Mibayashi, M.; Martínez-Sobrido, L.; Loo, Y.M.; Cárdenas, W.B.; Gale, M., Jr.; García-Sastre, A. Inhibition of Retinoic Acid-Inducible Gene I-Mediated Induction of Beta Interferon by the NS1 Protein of Influenza A Virus. J. Virol. 2007, 81, 514–524. [Google Scholar] [CrossRef] [PubMed]
- Opitz, B.; Rejaibi, A.; Dauber, B.; Eckhard, J.; Vinzing, M.; Schmeck, B.; Hippenstiel, S.; Suttorp, N.; Wolff, T. IFNβ Induction by Influenza A Virus Is Mediated by RIG-I Which Is Regulated by the Viral NS1 Protein. Cell. Microbiol. 2007, 9, 930–938. [Google Scholar] [CrossRef] [PubMed]
- Gack, M.U.; Albrecht, R.A.; Urano, T.; Inn, K.-S.; Huang, I.-C.; Carnero, E.; Farzan, M.; Inoue, S.; Jung, J.U.; García-Sastre, A. Influenza A Virus NS1 Targets the Ubiquitin Ligase TRIM25 to Evade Recognition by the Host Viral RNA Sensor RIG-I. Cell Host Microbe 2009, 5, 439–449. [Google Scholar] [CrossRef]
- Koliopoulos, M.G.; Lethier, M.; Van Der Veen, A.G.; Haubrich, K.; Hennig, J.; Kowalinski, E.; Stevens, R.V.; Martin, S.R.; Reis, E.; Sousa, C.; et al. Molecular Mechanism of Influenza A NS1-Mediated TRIM25 Recognition and Inhibition. Nat. Commun. 2018, 9, 1820. [Google Scholar] [CrossRef] [PubMed]
- Nagata, K.; Kawaguchi, A.; Naito, T. Host Factors for Replication and Transcription of the Influenza Virus Genome. Rev. Med. Virol. 2008, 18, 247–260. [Google Scholar] [CrossRef] [PubMed]
- König, R.; Stertz, S.; Zhou, Y.; Inoue, A.; Hoffmann, H.-H.; Bhattacharyya, S.; Alamares, J.G.; Tscherne, D.M.; Ortigoza, M.B.; Liang, Y. Human Host Factors Required for Influenza Virus Replication. Nature 2010, 463, 813–817. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Zhang, B.; Deng, L.; Liang, B.; Ping, J. Virus-Host Interaction Networks as New Antiviral Drug Targets for IAV and SARS-CoV-2. Emerg. Microbes Infect. 2022, 11, 1371–1389. [Google Scholar] [CrossRef]
- Sun, X.; Zeng, H.; Kumar, A.; Belser, J.A.; Maines, T.R.; Tumpey, T.M. Constitutively Expressed IFITM3 Protein in Human Endothelial Cells Poses an Early Infection Block to Human Influenza Viruses. J. Virol. 2016, 90, 11157–11167. [Google Scholar] [CrossRef]
- Deeg, C.; Mutz, P.; Kallfass, C.; Nürnberger, C.; Soubies, S.; Staeheli, P. 64: Transgenic Mice Carrying Interferon-Regulated Human MxA Locus Are Highly Resistant to Avian but Not Human Influenza A Viruses. Cytokine 2013, 63, 258. [Google Scholar] [CrossRef]
- Deeg, C.M.; Hassan, E.; Mutz, P.; Rheinemann, L.; Götz, V.; Magar, L.; Schilling, M.; Kallfass, C.; Nürnberger, C.; Soubies, S. In Vivo Evasion of MxA by Avian Influenza Viruses Requires Human Signature in the Viral Nucleoprotein. J. Exp. Med. 2017, 214, 1239–1248. [Google Scholar] [CrossRef]
- Kulkarni, V.; Jayakumar, S.; Mohan, M.; Kulkarni, S. Aid or Antagonize: Nuclear Long Noncoding RNAs Regulate Host Responses and Outcomes of Viral Infections. Cells 2023, 12, 987. [Google Scholar] [CrossRef] [PubMed]
- Maarouf, M.; Chen, B.; Chen, Y.; Wang, X.; Rai, K.R.; Zhao, Z.; Liu, S.; Li, Y.; Xiao, M.; Chen, J.-L. Identification of lncRNA-155 Encoded by MIR155HG as a Novel Regulator of Innate Immunity against Influenza A Virus Infection. Cell. Microbiol. 2019, 21, e13036. [Google Scholar] [CrossRef] [PubMed]
- Chai, W.; Li, J.; Shangguan, Q.; Liu, Q.; Li, X.; Qi, D.; Tong, X.; Liu, W.; Ye, X. Lnc-ISG20 Inhibits Influenza A Virus Replication by Enhancing ISG20 Expression. J. Virol. 2018, 92, e00539-18. [Google Scholar] [CrossRef]
- Min, J.; Li, Y.; Li, X.; Wang, M.; Li, H.; Bi, Y.; Xu, P.; Liu, W.; Ye, X.; Li, J. The circRNA circVAMP3 Restricts Influenza A Virus Replication by Interfering with NP and NS1 Proteins. PLoS Pathog. 2023, 19, e1011577. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, J.; Zhu, X.; Chen, Y.; Wei, H.; Chen, Q.; Chi, X.; Qi, B.; Zhang, L.; Zhao, Y.; Gao, G.F. NRAV, a Long Noncoding RNA, Modulates Antiviral Responses through Suppression of Interferon-Stimulated Gene Transcription. Cell Host Microbe 2014, 16, 616–626. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Guo, G.; Lu, M.; Chai, W.; Li, Y.; Tong, X.; Li, J.; Jia, X.; Liu, W.; Qi, D. Long Noncoding RNA Lnc-MxA Inhibits Beta Interferon Transcription by Forming RNA-DNA Triplexes at Its Promoter. J. Virol. 2019, 93, e00786-19. [Google Scholar] [CrossRef]
- Kang, Y.-J.; Yang, D.-C.; Kong, L.; Hou, M.; Meng, Y.-Q.; Wei, L.; Gao, G. CPC2: A Fast and Accurate Coding Potential Calculator Based on Sequence Intrinsic Features. Nucleic Acids Res. 2017, 45, W12–W16. [Google Scholar] [CrossRef]
- Dixon, A.S.; Schwinn, M.K.; Hall, M.P.; Zimmerman, K.; Otto, P.; Lubben, T.H.; Butler, B.L.; Binkowski, B.F.; Machleidt, T.; Kirkland, T.A. NanoLuc Complementation Reporter Optimized for Accurate Measurement of Protein Interactions in Cells. ACS Chem. Biol. 2016, 11, 400–408. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, Y.; Li, Q.; Zhao, J.; Yi, D.; Ding, J.; Zhao, F.; Hu, S.; Zhou, J.; Deng, T. Influenza Virus Exploits an Interferon-Independent lncRNA to Preserve Viral RNA Synthesis through Stabilizing Viral RNA Polymerase PB1. Cell Rep. 2019, 27, 3295–3304.e4. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, Y.; Zhou, R.; Zhao, J.; Zhang, Y.; Yi, D.; Li, Q.; Zhou, J.; Guo, F.; Liang, C. Host Long Noncoding RNA lncRNA-PAAN Regulates the Replication of Influenza A Virus. Viruses 2018, 10, 330. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, P.; Zhang, Y.; Xu, J.; Li, Z.; Li, Z.; Zhou, Z.; Liu, L.; Cao, X. Decreased Expression of the Host Long-Noncoding RNA-GM Facilitates Viral Escape by Inhibiting the Kinase Activity TBK1 via S-Glutathionylation. Immunity 2020, 53, 1168–1181.e7. [Google Scholar] [CrossRef]
- Munger, J.; Bennett, B.D.; Parikh, A.; Feng, X.-J.; McArdle, J.; Rabitz, H.A.; Shenk, T.; Rabinowitz, J.D. Systems-Level Metabolic Flux Profiling Identifies Fatty Acid Synthesis as a Target for Antiviral Therapy. Nat. Biotechnol. 2008, 26, 1179–1186. [Google Scholar] [CrossRef] [PubMed]
- Bedi, S.; Ono, A. Friend or Foe: The Role of the Cytoskeleton in Influenza A Virus Assembly. Viruses 2019, 11, 46. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Wang, Z.-G.; Ma, A.-X.; Liu, S.-L.; Pang, D.-W. Uncovering the F-Actin-Based Nuclear Egress Mechanism of Newly Synthesized Influenza A Virus Ribonucleoprotein Complexes by Single-Particle Tracking. Anal. Chem. 2022, 94, 5624–5633. [Google Scholar] [CrossRef]
- Banerjee, I.; Miyake, Y.; Nobs, S.P.; Schneider, C.; Horvath, P.; Kopf, M.; Matthias, P.; Helenius, A.; Yamauchi, Y. Influenza A Virus Uses the Aggresome Processing Machinery for Host Cell Entry. Science 2014, 346, 473–477. [Google Scholar] [CrossRef]
- Zhang, F.; Li, Y.; Gan, L.; Tong, X.; Qi, D.; Wang, Q.; Ye, X. HBx-Upregulated MAFG-AS1 Promotes Cell Proliferation and Migration of Hepatoma Cells by Enhancing MAFG Expression and Stabilizing Nonmuscle Myosin IIA. FASEB J. 2021, 35, e21529. [Google Scholar] [CrossRef] [PubMed]
- Shang, W.; Gao, Y.; Tang, Z.; Zhang, Y.; Yang, R. The Pseudogene Olfr29-Ps1 Promotes the Suppressive Function and Differentiation of Monocytic MDSCs. Cancer Immunol. Res. 2019, 7, 813–827. [Google Scholar] [CrossRef]
- Wang, J.; He, X.; Yao, Q.; Wang, C.; Lu, X.; Wang, R.; Miao, D. LncRNA PTTG3P Promotes Tumorigenesis and Metastasis of NSCLC by Binding with ILF3 to Maintain mRNA Stability and Form a Positive Feedback Loop with E2F1. Int. J. Biol. Sci. 2023, 19, 4291–4310. [Google Scholar] [CrossRef]
- Kovalenko, T.F.; Yadav, B.; Anufrieva, K.S.; Larionova, T.D.; Aksinina, T.E.; Latyshev, Y.A.; Bastola, S.; Shakhparonov, M.I.; Pandey, A.K.; Pavlyukov, M.S. PTEN Regulates Expression of Its Pseudogene in Glioblastoma Cells in DNA Methylation-Dependent Manner. Biochimie 2023, 219, 74–83. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Zhu, X.; Wen, W.; Yuan, J.; Hu, Y.; Chen, J.; An, S.; Dong, X.; Lin, C.; Yu, J.; et al. Dengue Virus Subverts Host Innate Immunity by Targeting Adaptor Protein MAVS. J. Virol. 2016, 90, 7219–7230. [Google Scholar] [CrossRef] [PubMed]
- Harman, A.N.; Nasr, N.; Feetham, A.; Galoyan, A.; Alshehri, A.A.; Rambukwelle, D.; Botting, R.A.; Hiener, B.M.; Diefenbach, E.; Diefenbach, R.J.; et al. HIV Blocks Interferon Induction in Human Dendritic Cells and Macrophages by Dysregulation of TBK1. J. Virol. 2015, 89, 6575–6584. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, M.; Yao, X.; Tong, X.; Qi, D.; Ye, X. Lnc-RPS6P3 Inhibits Influenza A Virus Replication and Attenuates the Inhibitory Effect of NS1 on Innate Immune Response. Microorganisms 2024, 12, 654. https://doi.org/10.3390/microorganisms12040654
Wang M, Yao X, Tong X, Qi D, Ye X. Lnc-RPS6P3 Inhibits Influenza A Virus Replication and Attenuates the Inhibitory Effect of NS1 on Innate Immune Response. Microorganisms. 2024; 12(4):654. https://doi.org/10.3390/microorganisms12040654
Chicago/Turabian StyleWang, Mingge, Xinli Yao, Xiaomei Tong, Dandan Qi, and Xin Ye. 2024. "Lnc-RPS6P3 Inhibits Influenza A Virus Replication and Attenuates the Inhibitory Effect of NS1 on Innate Immune Response" Microorganisms 12, no. 4: 654. https://doi.org/10.3390/microorganisms12040654
APA StyleWang, M., Yao, X., Tong, X., Qi, D., & Ye, X. (2024). Lnc-RPS6P3 Inhibits Influenza A Virus Replication and Attenuates the Inhibitory Effect of NS1 on Innate Immune Response. Microorganisms, 12(4), 654. https://doi.org/10.3390/microorganisms12040654