
Back to the Future: Immune Protection or Enhancement of Future Coronaviruses


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
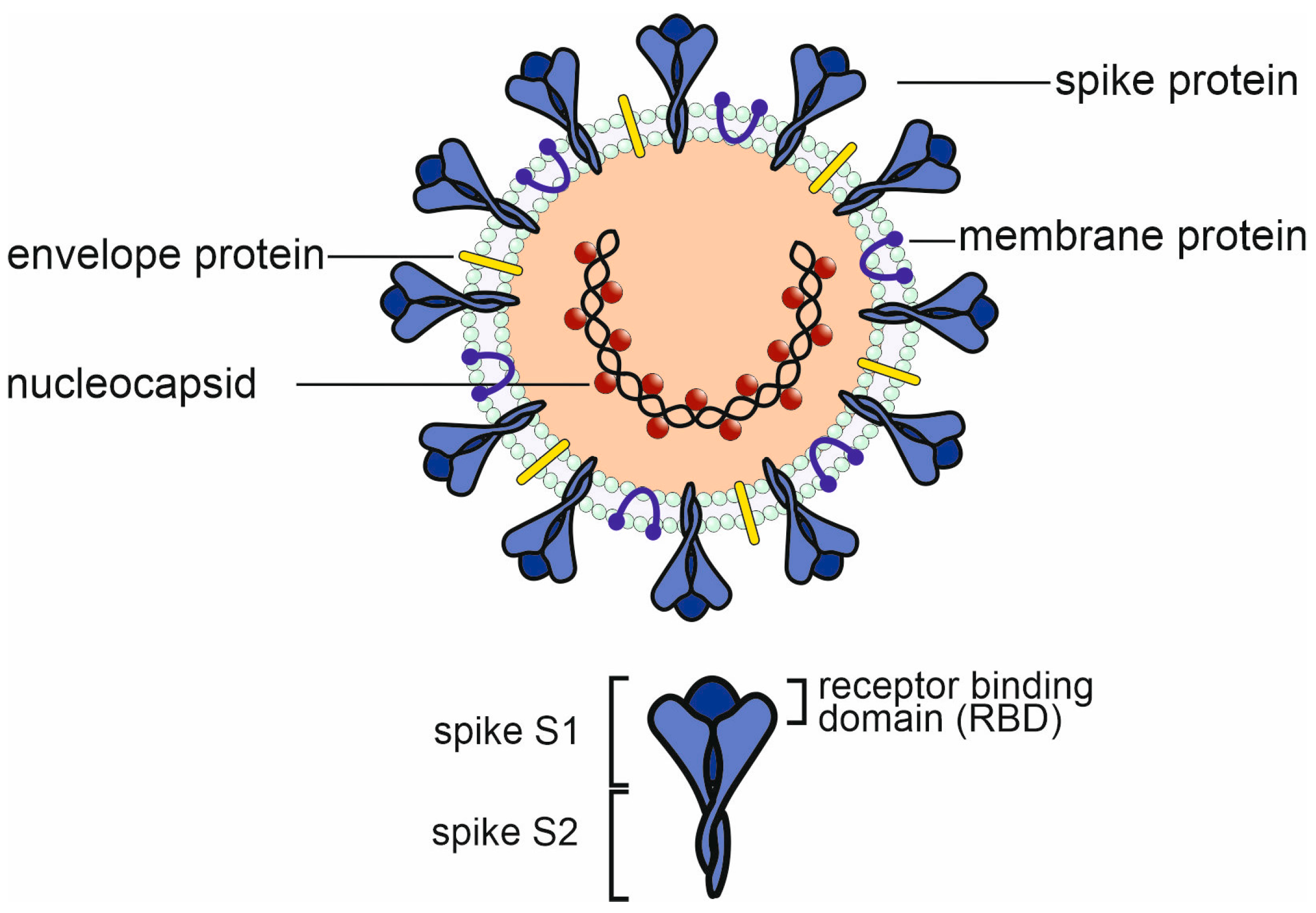
1. Introduction
2. The Immune Response to SARS-CoV-2 Infection and Vaccination
2.1. The Humoral Immune Response to SARS-CoV-2 Infection
COVID-19 and Response to Vaccination in Patients with Humoral Immunodeficiency or B-Cell-Depletion Therapy
2.2. T-Cell-Mediated Immunity against Coronaviruses
Importance of T-Cell Immunity
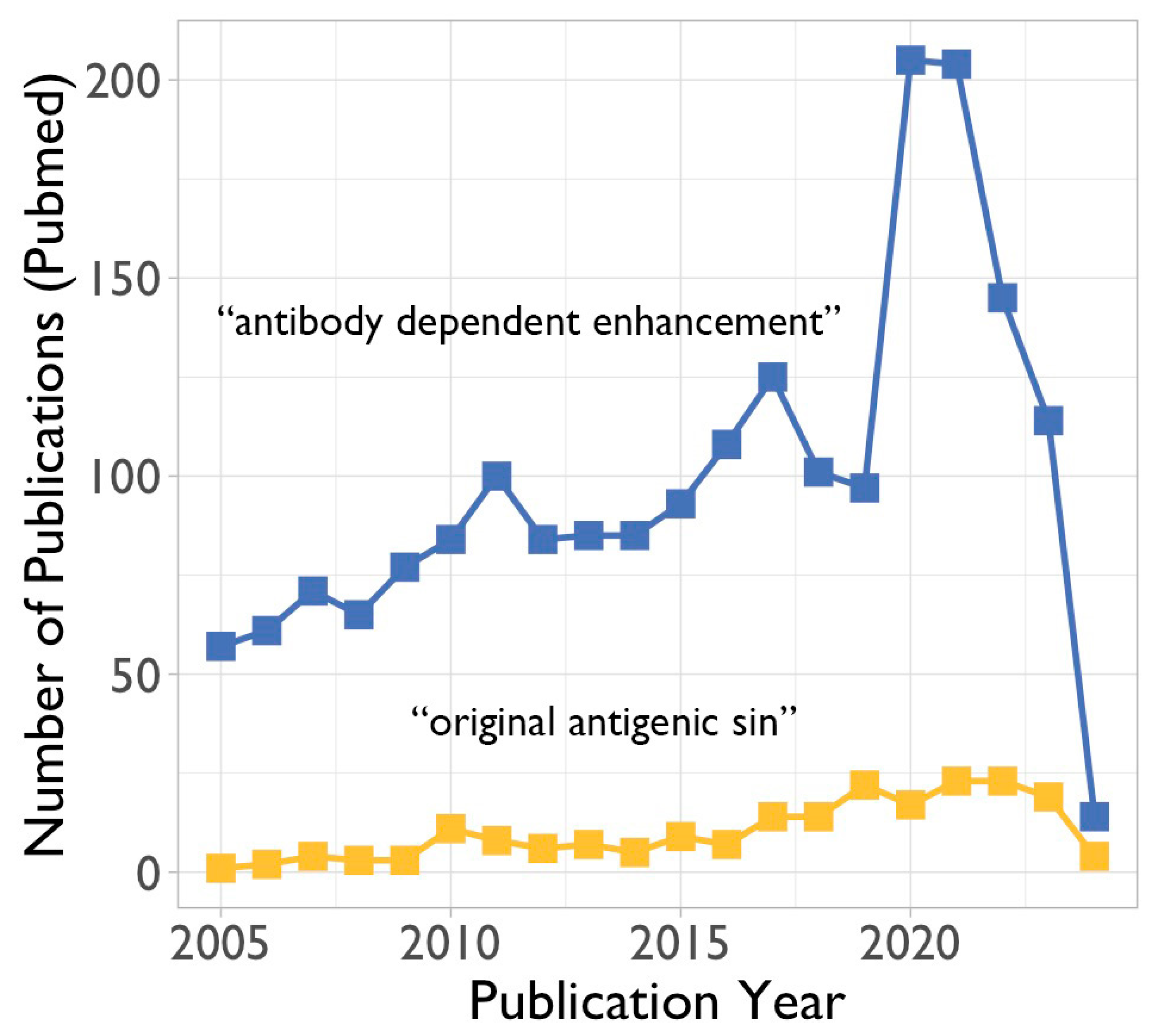
3. Potential Negative Impact of Existing Antibodies and Memory B Cells on Future Viruses
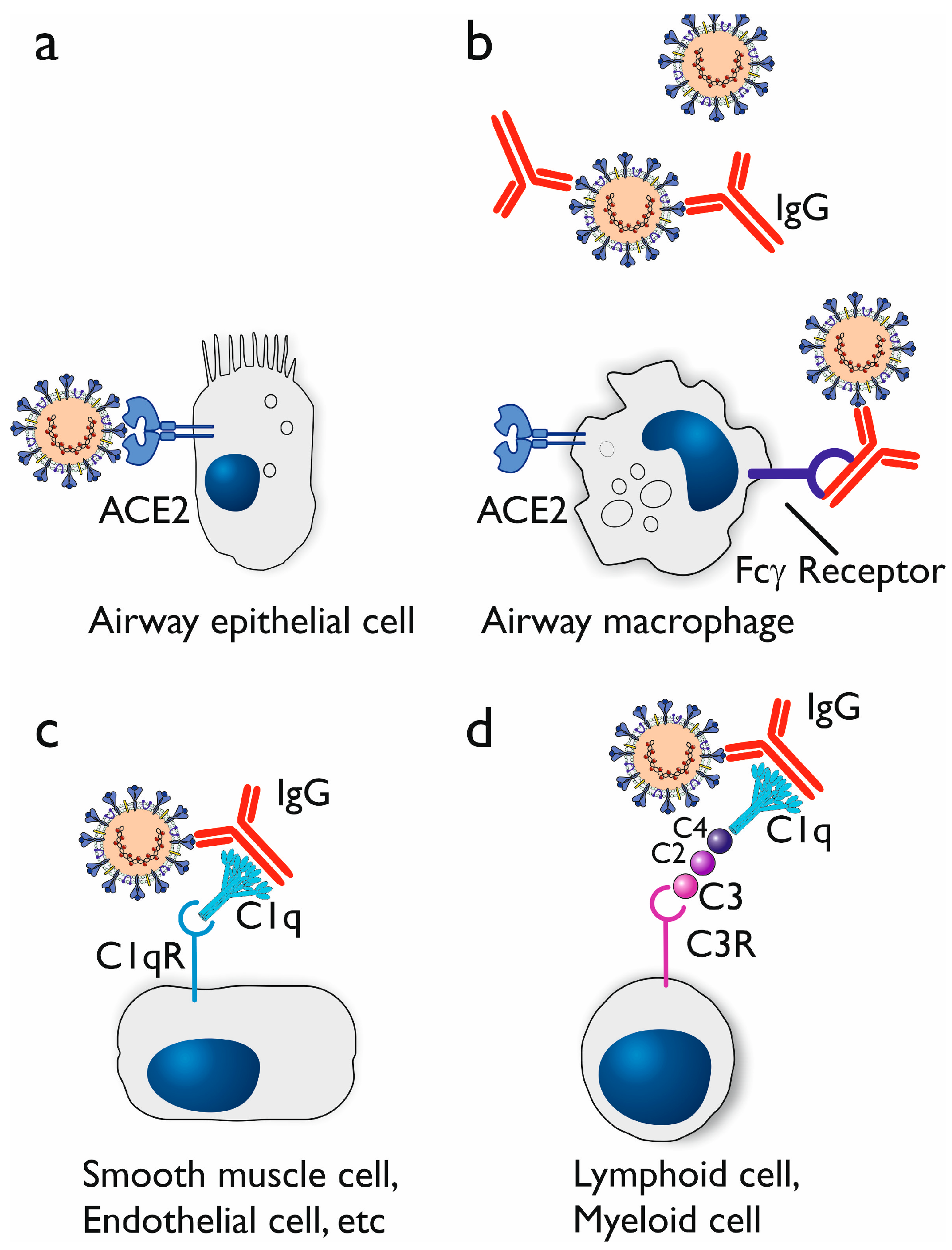
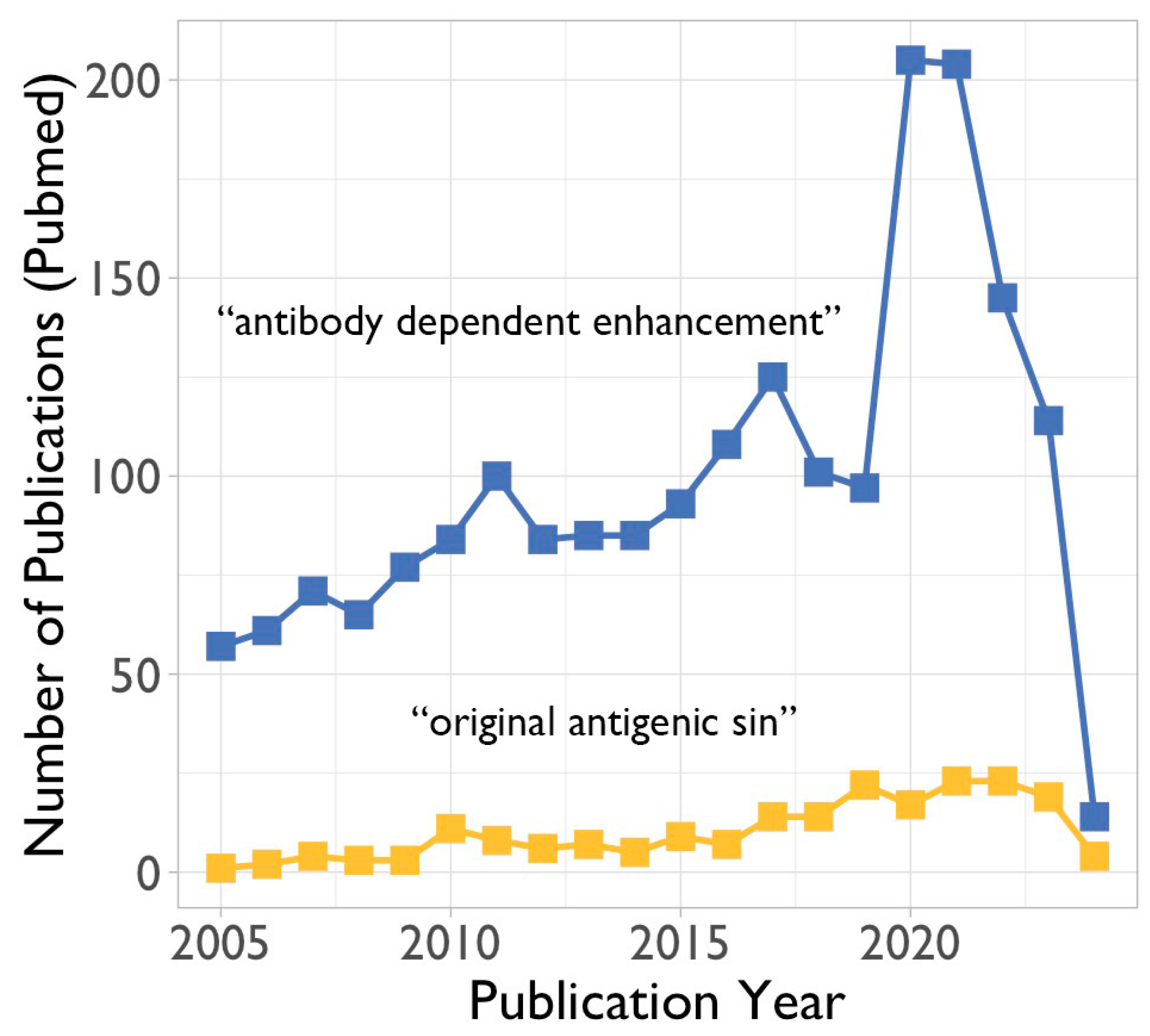
3.1. Antibody-Dependent Enhancement (ADE)
3.1.1. In Vitro Studies on Antibody-Dependent Enhancement
3.1.2. In Vivo Indications for Antibody-Dependent Enhancement
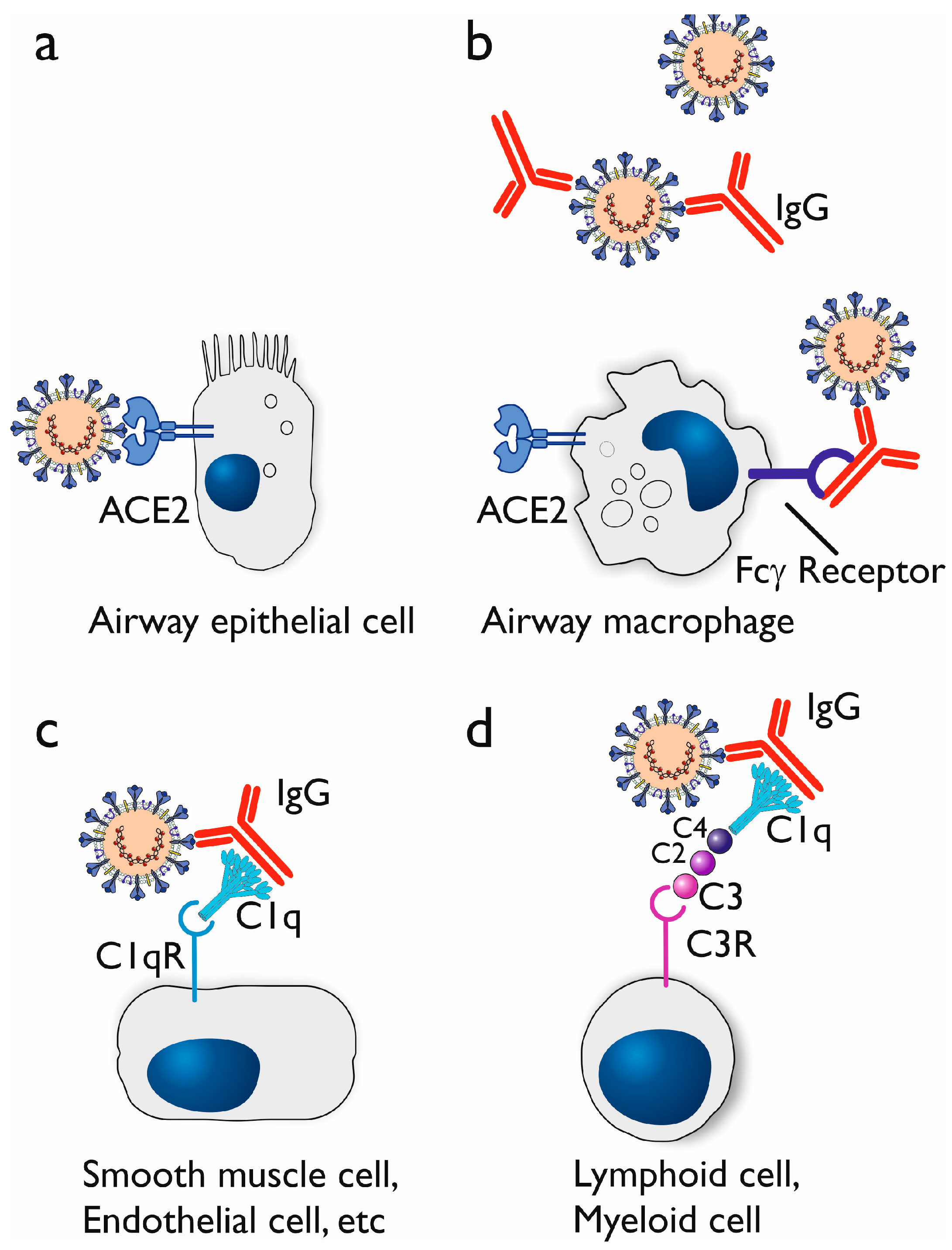
3.1.3. Proposed Mechanisms of Antibody-Dependent Enhancement
3.2. The Original Antigenic Sin
4. Concluding Remarks and Outlook for the Future
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. China Novel Coronavirus Investigating Research Team ANovel Coronavirus from Patients with Pneumonia in China 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef]
- World Health Organization. Virtual Press Conference on COVID-19 & Other Global Health Emergencies. Available online: https://www.who.int/publications/m/item/virtual-press-conference-on-covid-19---other-global-health-emergencies (accessed on 14 January 2024).
- Harris, E. WHO Declares End of COVID-19 Global Health Emergency. JAMA 2023, 329, 1817. [Google Scholar] [CrossRef]
- Cohen, J. COVID’s cold cousins. Four largely ignored coronaviruses circulate in humans without causing great harm and may portend the future for SARS-CoV-2. Science 2024, 383, 141–145. [Google Scholar] [CrossRef]
- Coronaviridae Study Group of the International Committee on Taxonomy of Viruses. The species Severe acute respiratory syndrome-related coronavirus: Classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 2020, 5, 536–544. [Google Scholar] [CrossRef]
- Al-Qahtani, W.S.; Alneghery, L.M.; Alqahtani, A.Q.S.; Al-Kahtani, M.D.; Alkahtani, S. A review of comparison study between corona viruses (SARS-CoV, mers-cov) and novel corona virus (COVID-19). Rev. Mex. Ing. Química 2020, 19 (Suppl. 1), 201–212. [Google Scholar] [CrossRef]
- Yang, H.; Rao, Z. Structural biology of SARS-CoV-2 and implications for therapeutic development. Nat. Rev. Microbiol. 2021, 19, 685–700. [Google Scholar] [CrossRef]
- Tang, G.; Liu, Z.; Chen, D. Human coronaviruses: Origin, host and receptor. J. Clin. Virol. 2022, 155, 105246. [Google Scholar] [CrossRef]
- Rajapakse, N.; Dixit, D. Human and novel coronavirus infections in children: A review. Paediatr. Int. Child Health 2021, 41, 36–55. [Google Scholar] [CrossRef] [PubMed]
- van der Hoek, L.; Pyrc, K.; Jebbink, M.F.; Vermeulen-Oost, W.; Berkhout, R.J.; Wolthers, K.C.; Wertheim-van Dillen, P.M.; Kaandorp, J.; Spaargaren, J.; Berkhout, B. Identification of a new human coronavirus. Nat. Med. 2004, 10, 368–373. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Huang, S.; Yin, L. The cytokine storm and COVID-19. J. Med. Virol. 2021, 93, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Fajgenbaum, D.C.; June, C.H. Cytokine Storm. N. Engl. J. Med. 2020, 383, 2255–2273. [Google Scholar] [CrossRef]
- Lei, X.; Dong, X.; Ma, R.; Wang, W.; Xiao, X.; Tian, Z.; Wang, C.; Wang, Y.; Li, L.; Ren, L.; et al. Activation and evasion of type I interferon responses by SARS-CoV-2. Nat. Commun. 2020, 11, 3810. [Google Scholar] [CrossRef]
- Moss, P. The T cell immune response against SARS-CoV-2. Nat Immunol. 2022, 23, 186–193. [Google Scholar] [CrossRef]
- Woodruff, M.C.; Ramonell, R.P.; Nguyen, D.C.; Cashman, K.S.; Saini, A.S.; Haddad, N.S.; Ley, A.M.; Kyu, S.; Howell, J.C.; Ozturk, T.; et al. Extrafollicular B cell responses correlate with neutralizing antibodies and morbidity in COVID-19. Nat Immunol. 2020, 21, 1506–1516. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Wang, Y.; Qi, C.; Shen, L.; Li, J. Clinical trial analysis of 2019-nCoV therapy registered in China. J. Med. Virol. 2020, 92, 540–545. [Google Scholar] [CrossRef]
- Hashem, A.M.; Algaissi, A.; Almahboub, S.A.; Alfaleh, M.A.; Abujamel, T.S.; Alamri, S.S.; Alluhaybi, K.A.; Hobani, H.I.; AlHarbi, R.H.; Alsulaiman, R.M.; et al. Early Humoral Response Correlates with Disease Severity and Outcomes in COVID-19 Patients. Viruses 2020, 12, 1390. [Google Scholar] [CrossRef] [PubMed]
- Ng, K.W.; Faulkner, N.; Cornish, G.H.; Rosa, A.; Harvey, R.; Hussain, S.; Ulferts, R.; Earl, C.; Wrobel, A.G.; Benton, D.J.; et al. Preexisting and de novo humoral immunity to SARS-CoV-2 in humans. Science 2020, 370, 1339–1343. [Google Scholar] [CrossRef]
- Liu, X.; Wang, J.; Xu, X.; Liao, G.; Chen, Y.; Hu, C.H. Patterns of IgG and IgM antibody response in COVID-19 patients. Emerg. Microbes Infect. 2020, 9, 1269–1274. [Google Scholar] [CrossRef] [PubMed]
- Rijkers, G.; Murk, J.L.; Wintermans, B.; van Looy, B.; van den Berge, M.; Veenemans, J.; Stohr, J.; Reusken, C.; van der Pol, P.; Reimerink, J. Differences in Antibody Kinetics and Functionality Between Severe and Mild Severe Acute Respiratory Syndrome Coronavirus 2 Infections. J. Infect. Dis. 2020, 222, 1265–1269. [Google Scholar] [CrossRef]
- Chen, Y.; Zhao, X.; Zhou, H.; Zhu, H.; Jiang, S.; Wang, P. Broadly neutralizing antibodies to SARS-CoV-2 and other human coronaviruses. Nat. Rev. Immunol. 2023, 23, 189–199. [Google Scholar] [CrossRef]
- Swadling, L.; Maini, M.K. Can T cells abort SARS-CoV-2 and other viral infections? Int. J. Mol. Sci. 2023, 24, 4371. [Google Scholar] [CrossRef] [PubMed]
- Suardana, I.B.K.; Mahardika, B.K.; Pharmawati, M.; Sudipa, P.H.; Sari, T.K.; Mahendra, N.B.; Mahardika, G.N. Whole-Genome Comparison of Representatives of All Variants of SARS-CoV-2, Including Subvariant BA.2 and the GKA Clade. Adv. Virol. 2023, 2023, 6476626. [Google Scholar] [CrossRef] [PubMed]
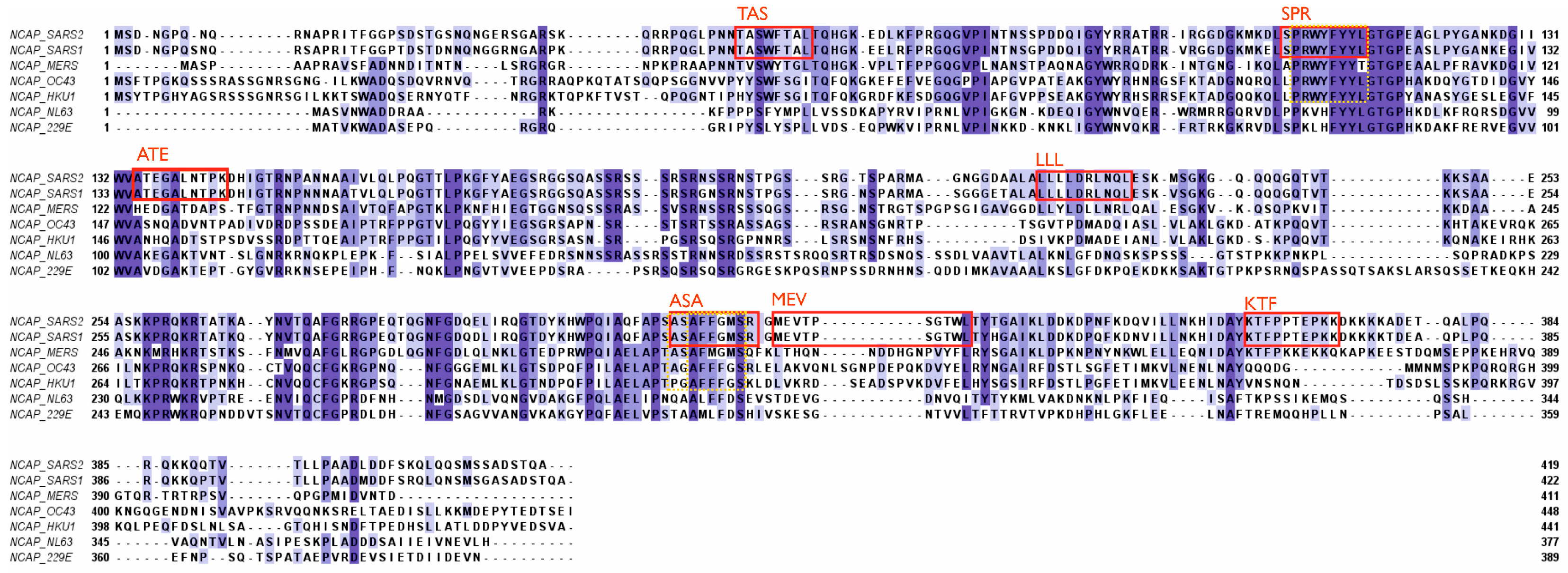
- Bai, Z.; Cao, Y.; Liu, W.; Li, J. The SARS-CoV-2 Nucleocapsid Protein and Its Role in Viral Structure, Biological Functions, and a Potential Target for Drug or Vaccine Mitigation. Viruses 2021, 13, 1115. [Google Scholar] [CrossRef] [PubMed]
- Soresina, A.; Moratto, D.; Chiarini, M.; Paolillo, C.; Baresi, G.; Focà, E.; Bezzi, M.; Baronio, B.; Giacomelli, M.; Badolato, R. Two X-linked agammaglobulinemia patients develop pneumonia as COVID-19 manifestation but recover. Pediatr. Allergy Immunol. 2020, 31, 565–569. [Google Scholar] [CrossRef] [PubMed]
- Devassikutty, F.M.; Jain, A.; Edavazhippurath, A.; Joseph, M.C.; Peedikayil, M.M.T.; Scaria, V.; Sandhya, P.; Govindaraj, G.M. X-Linked Agammaglobulinemia and COVID-19: Two Case Reports and Review of Literature. Pediatr. Allergy Immunol. Pulmonol. 2021, 34, 115–118. [Google Scholar] [CrossRef] [PubMed]
- Quinti, I.; Locatelli, F.; Carsetti, R. The Immune Response to SARS-CoV-2 Vaccination: Insights Learned from Adult Patients with Common Variable Immune Deficiency. Front. Immunol. 2022, 12, 815404. [Google Scholar] [CrossRef] [PubMed]
- Vanni, A.; Salvati, L.; Mazzoni, A.; Lamacchia, G.; Capone, M.; Francalanci, S.; Kiros, S.T.; Cosmi, L.; Puccini, B.; Ciceri, M.; et al. Bendamustine impairs humoral but not cellular immunity to SARS-CoV-2 vaccination in rituximab-treated B-cell lymphoma-affected patients. Front. Immunol. 2023, 14, 1322594. [Google Scholar] [CrossRef]
- Candon, S.; Lemee, V.; Leveque, E.; Etancelin, P.; Paquin, C.; Carette, M.; Contentin, N.; Bobee, V.; Alani, M.; Cardinael, N.; et al. Dissociated humoral and cellular immune responses after a three-dose schema of BNT162b2 vaccine in patients receiving anti-CD20 monoclonal antibody maintenance treatment for B-cell lymphomas. Haematologica 2022, 107, 755–758. [Google Scholar] [CrossRef]
- Ishio, T.; Tsukamoto, S.; Yokoyama, E.; Izumiyama, K.; Saito, M.; Muraki, H.; Kobayashi, M.; Mori, A.; Morioka, M.; Kondo, T. Anti-CD20 antibodies and bendamustine attenuate humoral immunity to COVID-19 vaccination in patients with B-cell non-Hodgkin lymphoma. Ann. Hematol. 2023, 12, 1421–1431. [Google Scholar] [CrossRef]
- Perry, C.; Luttwak, E.; Balaban, R.; Shefer, G.; Morales, M.M.; Aharon, A.; Tabib, Y.; Cohen, Y.C.; Benyamini, N.; Beyar-Katz, O.; et al. Efficacy of the BNT162b2 mRNA COVID-19 vaccine in patients with B-cell non-Hodgkin lymphoma. Blood Adv. 2021, 5, 3053–3061. [Google Scholar] [CrossRef]
- Lu, L.; Chan, C.Y.; Lim, Y.Y.; Than, M.; Teo, S.; Lau, P.Y.W.; Ng, K.H.; Yap, H.K. SARS-CoV-2 Humoral Immunity Persists Following Rituximab Therapy. Vaccines 2023, 11, 1864. [Google Scholar] [CrossRef]
- Bsteh, G.; Assar, H.; Hegen, H.; Heschl, B.; Leutmezer, F.; Di Pauli, F.; Gradl, C.; Traxler, G.; Zulehner, G. AUT-MuSC investigators. COVID-19 severity and mortality in multiple sclerosis are not associated with immunotherapy: Insights from a nation-wide Austrian registry. PLoS ONE 2021, 16, e0255316. [Google Scholar] [CrossRef]
- Avouac, J.; Drumez, E.; Hachulla, E.; Seror, R.; Georgin-Lavialle, S.; El Mahou, S.; Pertuiset, E.; Pham, T.; Marotte, H. FAIR/SFR/SNFMI/SOFREMIP/CRI/IMIDIATE consortium and contributors. COVID-19 outcomes in patients with inflammatory rheumatic and musculoskeletal diseases treated with rituximab: A cohort study. Lancet Rheumatol. 2021, 3, e419–e426. [Google Scholar] [CrossRef]
- Anand, P.; Puranik, A.; Aravamudan, M.; Venkatakrishnan, A.J. Soundararajan VSARS-CoV-2 strategically mimics proteolytic activation of human ENaC. Elife 2020, 9, e58603. [Google Scholar] [CrossRef]
- Kotsias, F.; Cebrian, I.; Alloatti, A. Antigen processing and presentation. Int. Rev. Cell Mol. Biol. 2019, 348, 69–121. [Google Scholar] [CrossRef]
- Nagler, A.; Kalaora, S.; Barbolin, C.; Gangaev, A.; Ketelaars, S.L.C.; Alon, M.; Pai, J.; Benedek, G.; Yahalom-Ronen, Y.; Erez, N.; et al. Identification of presented SARS-CoV-2 HLA class I and HLA class II peptides using HLA peptidomics. Cell Rep. 2021, 35, 109305. [Google Scholar] [CrossRef]
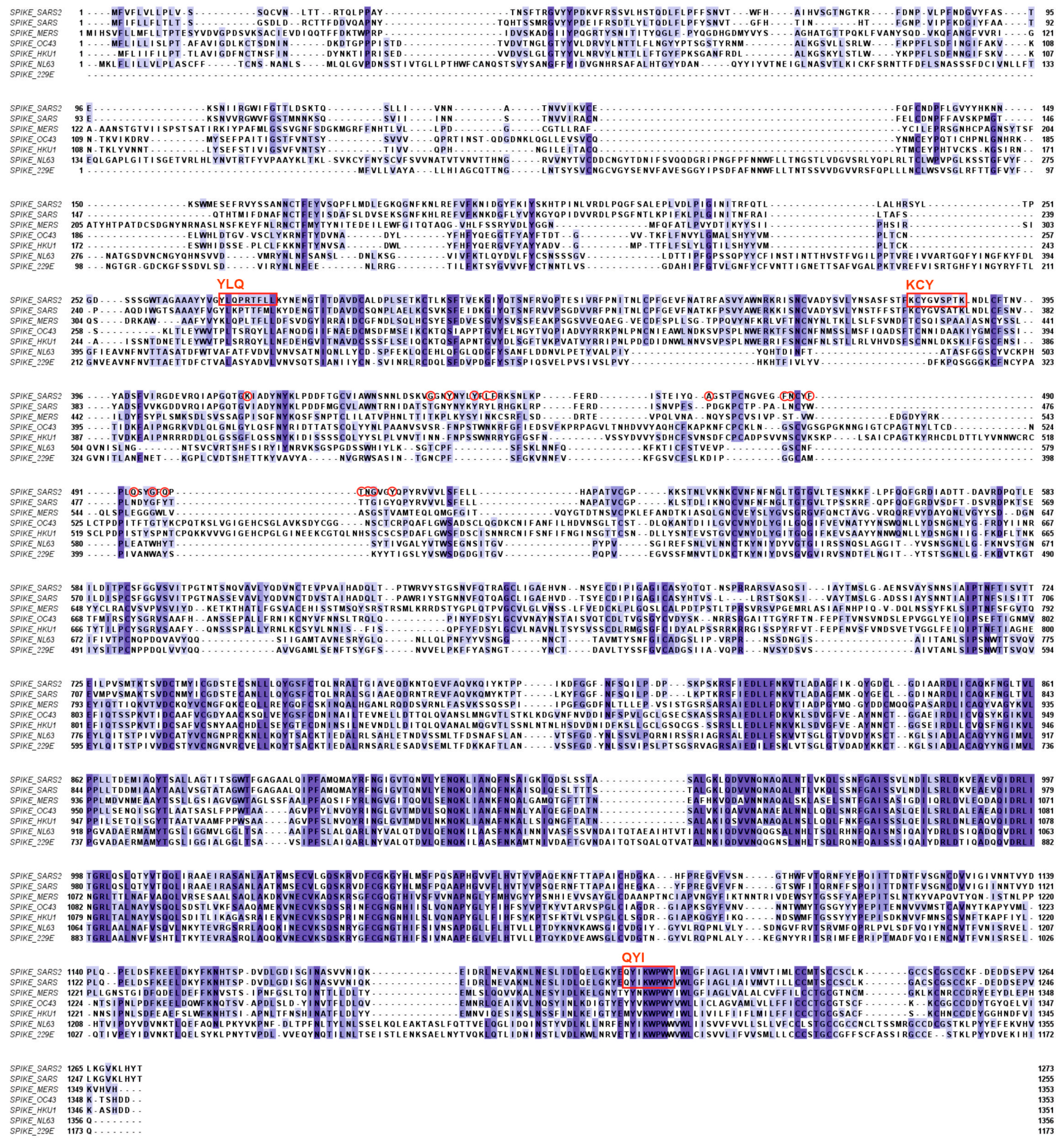
- Tarke, A.; Sidney, J.; Kidd, C.K.; Dan, J.M.; Ramirez, S.I.; Yu, E.D.; Mateus, J.; da Silva Antunes, R.; Moore, E.; Rubiro, P.; et al. Comprehensive analysis of T cell immunodominance and immunoprevalence of SARS-CoV-2 epitopes in COVID-19 cases. Cell Rep. Med. 2021, 2, 100204. [Google Scholar] [CrossRef] [PubMed]
- Bertoletti, A.; Tan, A.T.; Le Bert, N. The T-cell response to SARS-CoV-2: Kinetic and quantitative aspects and the case for their protective role. Oxf. Open Immunol. 2021, 2, iqab006. [Google Scholar] [CrossRef]
- Ferretti, A.P.; Kula, T.; Wang, Y.; Nguyen, D.M.V.; Weinheimer, A.; Dunlap, G.S.; Xu, Q.; Nabilsi, N.; Perullo, C.R.; Cristofaro, A.W.; et al. Unbiased Screens Show CD8+ T Cells of COVID-19 Patients Recognize Shared Epitopes in SARS-CoV-2 that Largely Reside outside the Spike Protein. Immunity 2020, 53, 1095–1107. [Google Scholar] [CrossRef] [PubMed]
- Verma, J.; Kaushal, N.; Manish, M.; Subbarao, N.; Shakirova, V.; Martynova, E.; Liu, R.; Hamza, S.; Rizvanov, A.A.; Khaiboullina, S.F.; et al. Identification of conserved immunogenic peptides of SARS-CoV-2 nucleocapsid protein. J. Biomol. Struct. Dyn. 2023, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Le Bert, N.; Tan, A.T.; Kunasegaran, K.; Tham, C.Y.L.; Hafezi, M.; Chia, A.; Chng, M.H.Y.; Lin, M.; Tan, N.; Linster, M.; et al. SARS-CoV-2-specific T cell immunity in cases of COVID-19 and SARS, and uninfected controls. Nature 2020, 584, 457–462. [Google Scholar] [CrossRef] [PubMed]
- AlKhalifah, J.M.; Seddiq, W.; Alshehri, M.A.; Alhetheel, A.; Albarrag, A.; Meo, S.A.; Al-Tawfiq, J.A.; Barry, M. Impact of MERS-CoV and SARS-CoV-2 Viral Infection on Immunoglobulin-IgG Cross-Reactivity. Vaccines 2023, 11, 552. [Google Scholar] [CrossRef] [PubMed]
- Kesheh, M.M.; Hosseini, P.; Soltani, S.; Zandi, M. An overview on the seven pathogenic human coronaviruses. Rev. Med. Virol. 2022, 32, e2282. [Google Scholar] [CrossRef]
- Kundu, R.; Narean, J.S.; Wang, L.; Fenn, J.; Pillay, T.; Fernandez, N.D.; Conibear, E.; Koycheva, A.; Davies, M.; Tolosa-Wright, M.; et al. Cross-reactive memory T cells associate with protection against SARS-CoV-2 infection in COVID-19 contacts. Nat. Commun. 2022, 13, 80. [Google Scholar] [CrossRef]
- Loyal, L.; Braun, J.; Henze, L.; Kruse, B.; Dingeldey, M.; Reimer, U.; Kern, F.; Schwarz, T.; Mangold, M.; Unger, C.; et al. Cross-reactive CD4+ T cells enhance SARS-CoV-2 immune responses upon infection and vaccination. Science 2021, 374, eabh1823. [Google Scholar] [CrossRef] [PubMed]
- Hewitt, E.W. The MHC class I antigen presentation pathway: Strategies for viral immune evasion. Immunology 2003, 110, 163–169. [Google Scholar] [CrossRef]
- Cassioli, C.; Baldari, C.T. The Expanding Arsenal of Cytotoxic T Cells. Front. Immunol. 2022, 13, 883010. [Google Scholar] [CrossRef]
- Niessl, J.; Sekine, T.; Buggert, M. T cell immunity to SARS-CoV-2. Semin. Immunol. 2021, 55, 101505. [Google Scholar] [CrossRef]
- Tay, C.; Kanellakis, P.; Hosseini, H.; Cao, A.; Toh, B.H.; Bobik, A.; Kyaw, T. B Cell and CD4 T Cell Interactions Promote Development of Atherosclerosis. Front. Immunol. 2020, 10, 3046. [Google Scholar] [CrossRef]
- Kared, H.; Redd, A.D.; Bloch, E.M.; Bonny, T.S.; Sumatoh, H.; Kairi, F.; Carbajo, D.; Abel, B.; Newell, E.W.; Bettinotti, M.P.; et al. SARS-CoV-2-specific CD8+ T cell responses in convalescent COVID-19 individuals. J. Clin. Investig. 2021, 131, e145476. [Google Scholar] [CrossRef]
- Song, G.; He, W.T.; Callaghan, S.; Anzanello, F.; Huang, D.; Ricketts, J.; Torres, J.L.; Beutler, N.; Peng, L.; Vargas, S.; et al. Cross-reactive serum and memory B-cell responses to spike protein in SARS-CoV-2 and endemic coronavirus infection. Nat. Commun. 2021, 12, 2938. [Google Scholar] [CrossRef]
- Vennema, H.; de Groot, R.J.; Harbour, D.A.; Dalderup, M.; Gruffydd-Jones, T.; Horzinek, M.C.; Spaan, W.J. Early death after feline infectious peritonitis virus challenge due to recombinant vaccinia virus immunization. J. Virol. 1990, 64, 1407–1409. [Google Scholar] [CrossRef]
- Olsen, C.W.; Corapi, W.V.; Ngichabe, C.K.; Baines, J.D.; Scott, F.W. Monoclonal antibodies to the spike protein of feline infectious peritonitis virus mediate antibody-dependent enhancement of infection of feline macrophages. J. Virol. 1992, 66, 956–965. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Shang, J.; Sun, S.; Tai, W.; Chen, J.; Geng, Q.; He, L.; Chen, Y.; Wu, J.; Shi, Z.; et al. Molecular Mechanism for Antibody-Dependent Enhancement of Coronavirus Entry. J. Virol. 2020, 94, e02015-19. [Google Scholar] [CrossRef]
- Takada, A.; Kawaoka, Y. Antibody-dependent enhancement of viral infection: Molecular mechanisms and in vivo implications. Rev. Med. Virol. 2003, 13, 387–398. [Google Scholar] [CrossRef]
- Arvin, A.M.; Fink, K.; Schmid, M.A.; Cathcart, A.; Spreafico, R.; Havenar-Daughton, C.; Lanzavecchia, A.; Corti, D.; Virgin, H.W. A perspective on potential antibody-dependent enhancement of SARS-CoV-2. Nature 2020, 584, 353–363. [Google Scholar] [CrossRef]
- Pongracz, T.; Vidarsson, G.; Wuhrer, M. Antibody glycosylation in COVID-19. Glycoconj. J. 2022, 39, 335–344. [Google Scholar] [CrossRef] [PubMed]
- van Osch, T.L.J.; Nouta, J.; Derksen, N.I.L.; van Mierlo, G.; van der Schoot, C.E.; Wuhrer, M.; Rispens, T.; Vidarsson, G. Fc galactosylation promotes hexamerization of human IgG1, leading to enhanced classical complement activation. J. Immunol. 2021, 207, 1545–1554. [Google Scholar] [CrossRef] [PubMed]
- Bergwerk, M.; Gonen, T.; Lustig, Y.; Amit, S.; Lipsitch, M.; Cohen, C.; Mandelboim, M.; Levin, E.G.; Rubin, C.; Indenbaum, V.; et al. COVID-19 Breakthrough Infections in Vaccinated Health Care Workers. N. Engl. J. Med. 2021, 385, 1474–1484. [Google Scholar] [CrossRef]
- Chandan, S.; Khan, S.R.; Deliwala, S.; Mohan, B.P.; Ramai, D.; Chandan, O.C.; Facciorusso, A. Postvaccination SARS-CoV-2 infection among healthcare workers: A systematic review and meta-analysis. J. Med. Virol. 2022, 94, 1428–1441. [Google Scholar] [CrossRef]
- Tenforde, M.W.; Self, W.H.; Adams, K.; Gaglani, M.; Ginde, A.A.; McNeal, T.; Ghamande, S.; Douin, D.J.; Talbot, H.K.; Casey, J.D.; et al. Association Between mRNA Vaccination and COVID-19 Hospitalization and Disease Severity. JAMA 2021, 326, 2043–2054. [Google Scholar] [CrossRef]
- Nakayama, E.E.; Shioda, T. SARS-CoV-2 Related Antibody-Dependent Enhancement Phenomena In Vitro and In Vivo. Microorganisms 2023, 11, 1015. [Google Scholar] [CrossRef] [PubMed]
- Weiskopf, K.; Weissman, I.L. Macrophages are critical effectors of antibody therapies for cancer. MAbs 2015, 7, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Junker, F.; Gordon, J.; Qureshi, O. Fc Gamma Receptors and Their Role in Antigen Uptake, Presentation, and T Cell Activation. Front. Immunol. 2020, 11, 1393. [Google Scholar] [CrossRef] [PubMed]
- von Kietzell, K.; Pozzuto, T.; Heilbronn, R.; Grössl, T.; Fechner, H.; Weger, S. Antibody-mediated enhancement of parvovirus B19 uptake into endothelial cells mediated by a receptor for complement factor C1q. J. Virol. 2014, 88, 8102–8115. [Google Scholar] [CrossRef] [PubMed]
- Eggleton, P.; Tenner, A.J.; Reid, K.B. C1q receptors. Clin. Exp. Immunol. 2000, 120, 406–412. [Google Scholar] [CrossRef] [PubMed]
- Wen, J.; Cheng, Y.; Ling, R.; Dai, Y.; Huang, B.; Huang, W.; Zhang, S.; Jiang, Y. Antibody-dependent enhancement of coronavirus. Int. J. Infect. Dis. 2020, 100, 483–489. [Google Scholar] [CrossRef]
- Thomas, S.; Smatti, M.K.; Ouhtit, A.; Cyprian, F.S.; Almaslamani, M.A.; Thani, A.A.; Yassine, H.M. Antibody-Dependent Enhancement (ADE) and the role of complement system in disease pathogenesis. Mol. Immunol. 2022, 152, 172–182. [Google Scholar] [CrossRef] [PubMed]
- Monto, A.S.; Malosh, R.E.; Petrie, J.G.; Martin, E.T. The Doctrine of Original Antigenic Sin: Separating Good from Evil. J. Infect. Dis. 2017, 215, 1782–1788. [Google Scholar] [CrossRef]
- Francis, T. On the doctrine of original antigenic sin. Proc. Am. Philos. Soc. 1960, 104, 572–578. [Google Scholar]
- Petráš, M.; Králová Lesná, I. SARS-CoV-2 vaccination in the context of original antigenic sin. Hum. Vaccines Immunother. 2022, 18, 1949953. [Google Scholar] [CrossRef]
- Pillai, S. SARS-CoV-2 vaccination washes away original antigenic sin. Trends Immunol. 2022, 43, 271–273. [Google Scholar] [CrossRef]
- Rijkers, G.T.; van Overveld, F.J. The “original antigenic sin” and its relevance for SARS-CoV-2 (COVID-19) vaccination. Clin. Immunol. Commun. 2021, 1, 13–16. [Google Scholar] [CrossRef]
- Xia, C.S.; Zhan, M.; Liu, Y.; Yue, Z.H.; Song, Y.; Zhang, F.; Wang, H. SARS-CoV-2 antibody response in SARS survivors with and without the COVID-19 vaccine. Int. J. Antimicrob. Agent 2023, 62, 106947. [Google Scholar] [CrossRef] [PubMed]
- El-Saed, A.; Othman, F.; Baffoe-Bonnie, H.; Almulhem, R.; Matalqah, M.; Alshammari, L.; Alshamrani, M.M. Symptomatic MERS-CoV infection reduces the risk of future COVID-19 disease; A retrospective cohort study. BMC Infect. Dis. 2023, 23, 757. [Google Scholar] [CrossRef]
- Sette, A.; Sidney, J.; Crotty, S. T Cell Responses to SARS-CoV-2. Annu. Rev. Immunol. 2023, 41, 343–373. [Google Scholar] [CrossRef] [PubMed]
- Appelberg, S.; Ahlén, G.; Yan, J.; Nikouyan, N.; Weber, S.; Larsson, O.; Höglund, U.; Aleman, S.; Weber, F.; Perlhamre, E.; et al. A universal SARS-CoV DNA vaccine inducing highly cross-reactive neutralizing antibodies and T cells. EMBO Mol. Med. 2022, 14, e15821. [Google Scholar] [CrossRef]
- Altmann, D.M.; Boyton, R.J. COVID-19 vaccination: The road ahead. Science 2022, 375, 1127–1132. [Google Scholar] [CrossRef]
- Dolgin, E. Pan-coronavirus vaccine pipeline takes form. Nat. Rev. Drug Discov. 2022, 21, 324–326. [Google Scholar] [CrossRef]
- Temmam, S.; Vongphayloth, K.; Baquero, E.; Munier, S.; Bonomi, M.; Regnault, B.; Douangboubpha, B.; Karami, Y.; Chrétien, D.; Sanamxay, D.; et al. Bat coronaviruses related to SARS-CoV-2 and infectious for human cells. Nature 2022, 604, 330–336. [Google Scholar] [CrossRef]
- Shannon, C.P.; Blimkie, T.M.; Ben-Othman, R.; Gladish, N.; Amenyogbe, N.; Drissler, S.; Edgar, R.D.; Chan, Q.; Krajden, M.; Foster, L.J.; et al. Multi-Omic Data Integration Allows Baseline Immune Signatures to Predict Hepatitis B Vaccine Response in a Small Cohort. Front. Immunol. 2020, 11, 578801. [Google Scholar] [CrossRef] [PubMed]
- Leslie, M. Giant project will chart human immune diversity to improve drugs and vaccines. Science 2024, 383y, 13–14. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bartels, M.; Sala Solé, E.; Sauerschnig, L.M.; Rijkers, G.T. Back to the Future: Immune Protection or Enhancement of Future Coronaviruses. Microorganisms 2024, 12, 617. https://doi.org/10.3390/microorganisms12030617
Bartels M, Sala Solé E, Sauerschnig LM, Rijkers GT. Back to the Future: Immune Protection or Enhancement of Future Coronaviruses. Microorganisms. 2024; 12(3):617. https://doi.org/10.3390/microorganisms12030617
Chicago/Turabian StyleBartels, Merit, Eric Sala Solé, Lotte M. Sauerschnig, and Ger T. Rijkers. 2024. "Back to the Future: Immune Protection or Enhancement of Future Coronaviruses" Microorganisms 12, no. 3: 617. https://doi.org/10.3390/microorganisms12030617
APA StyleBartels, M., Sala Solé, E., Sauerschnig, L. M., & Rijkers, G. T. (2024). Back to the Future: Immune Protection or Enhancement of Future Coronaviruses. Microorganisms, 12(3), 617. https://doi.org/10.3390/microorganisms12030617