
Extensive Genetic Diversity and Epidemiological Patterns of Factor H-Binding Protein Variants among Neisseria meningitidis in China


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Collecting of N. meningitidis Isolates and Multilocus Sequence Typing
2.2. Serogrouping, DNA Preparation and Whole-Genome Sequencing
2.3. Genome Validation and Quality Control
2.4. Detection and Genotyping of fHbp Allelic Variants
2.5. Association Analysis of fHbp Variants, Serogroups, and Clonal Complexes
2.6. Phylogenetic Analysis and Identifying Epidemiological Patterns of fHbp Allelic Variants
3. Results
3.1. fHbp Allelic Variants Existed in Most Chinese N. meningitidis Strains with Extensive Genetic Diversity
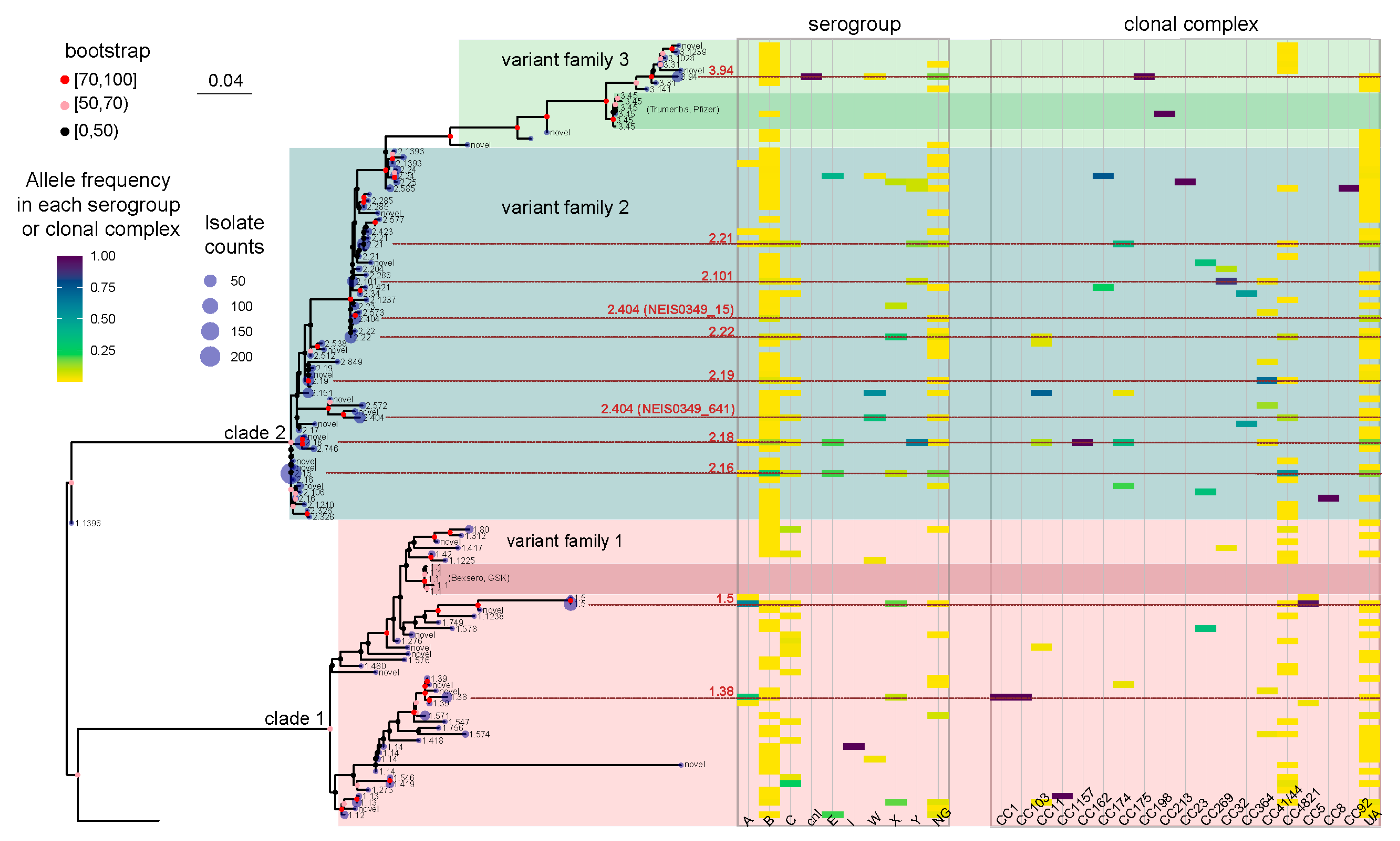
3.2. The Phylogenetic Tree Displayed the Phenotypic and Genetic Characteristics of fHbp Allelic Variants in Chinese N. meningitidis Strains
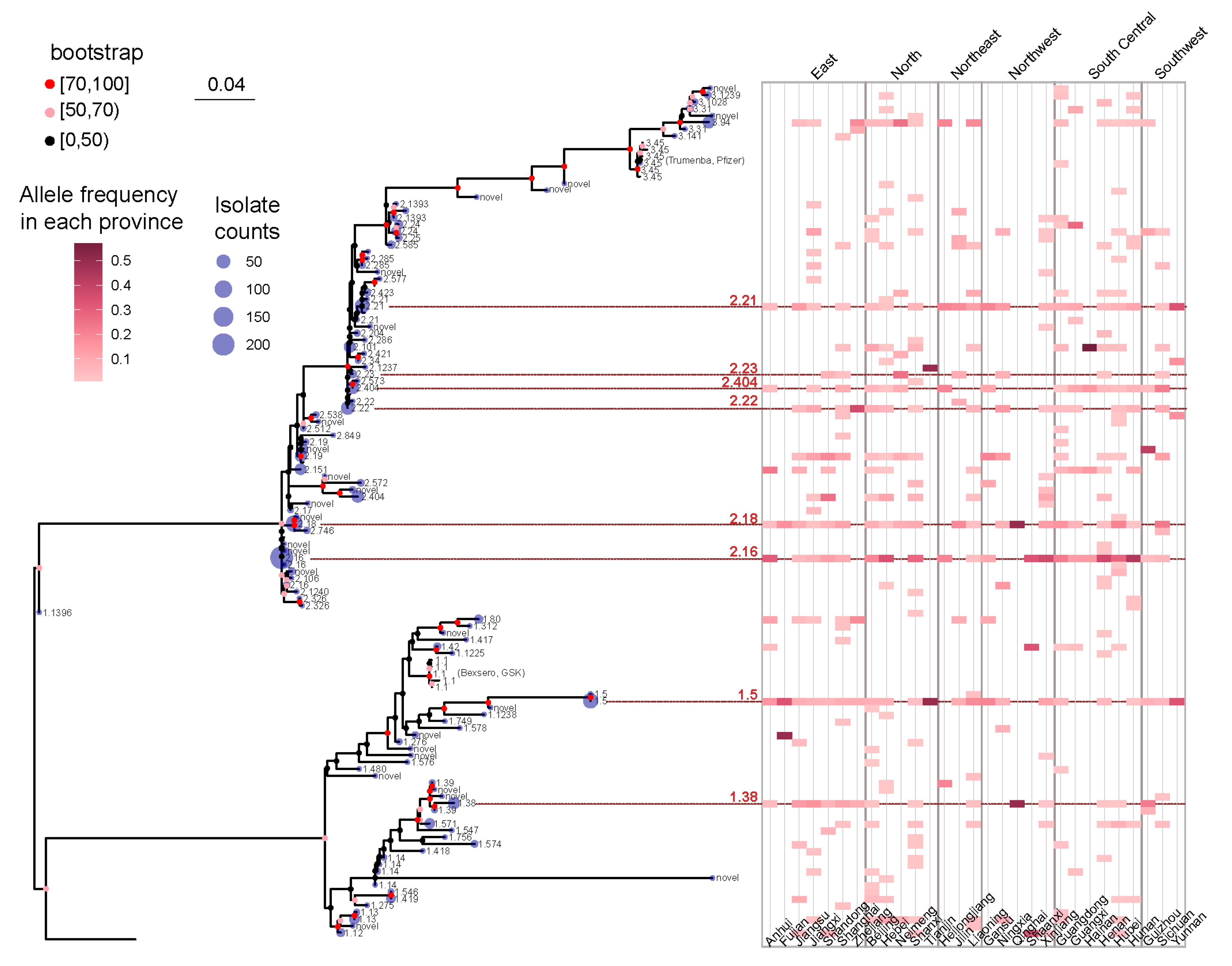
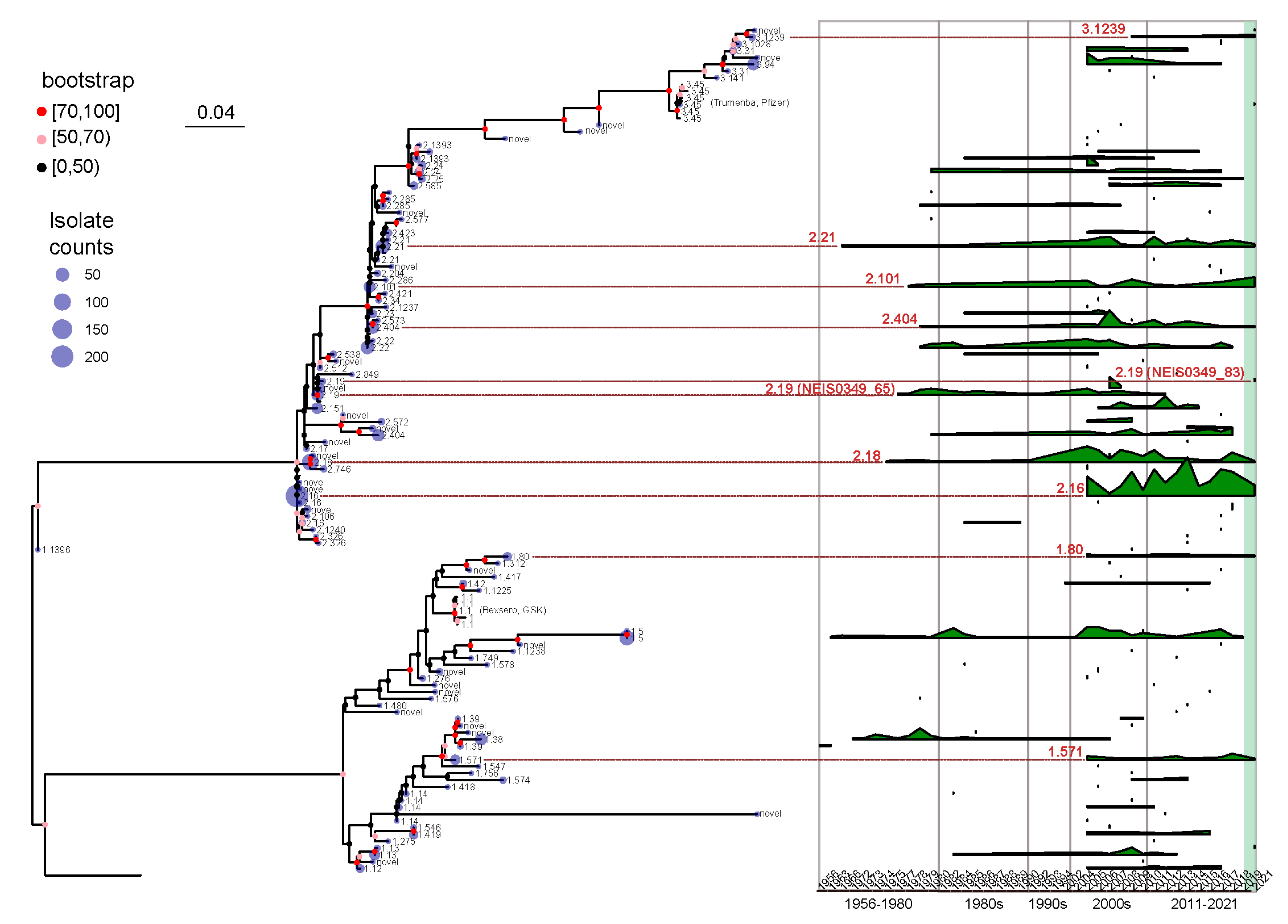
3.3. The Geographical and Temporal Distribution Patterns of fHbp Allelic Variants in Chinese N. meningitidis Strains Uncovered Potential fHbp Types for Vaccine Development
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Van de Beek, D.; Brouwer, M.C.; Koedel, U.; Wall, E.C. Community-acquired bacterial meningitis. Lancet 2021, 398, 1171–1183. [Google Scholar] [CrossRef]
- Caugant, D.A.; Brynildsrud, O.B. Neisseria meningitidis: Using genomics to understand diversity, evolution and pathogenesis. Nat. Rev. Microbiol. 2020, 18, 84–96. [Google Scholar] [CrossRef]
- Dretler, A.W.; Rouphael, N.G.; Stephens, D.S. Progress toward the global control of Neisseria meningitidis: 21st century vaccines, current guidelines, and challenges for future vaccine development. Hum. Vaccines Immunother. 2018, 14, 1146–1160. [Google Scholar] [CrossRef]
- Azmi, F.H.; Lucas, A.H.; Spiegelberg, H.L.; Granoff, D.M. Human immunoglobulin M paraproteins cross-reactive with Neisseria meningitidis group B polysaccharide and fetal brain. Infect. Immun. 1995, 63, 1906–1913. [Google Scholar] [CrossRef]
- Schneider, M.C.; Exley, R.M.; Chan, H.; Feavers, I.; Kang, Y.H.; Sim, R.B.; Tang, C.M. Functional significance of factor H binding to Neisseria meningitidis. J. Immunol. 2006, 176, 7566–7575. [Google Scholar] [CrossRef]
- Sands, N.A.; Beernink, P.T. Two human antibodies to a meningococcal serogroup B vaccine antigen enhance binding of complement Factor H by stabilizing the Factor H binding site. PLoS Pathog. 2021, 17, e1009655. [Google Scholar] [CrossRef]
- Madico, G.; Welsch, J.A.; Lewis, L.A.; McNaughton, A.; Perlman, D.H.; Costello, C.E.; Ngampasutadol, J.; Vogel, U.; Granoff, D.M.; Ram, S. The meningococcal vaccine candidate GNA1870 binds the complement regulatory protein factor H and enhances serum resistance. J. Immunol. 2006, 177, 501–510. [Google Scholar] [CrossRef]
- Fletcher, L.D.; Bernfield, L.; Barniak, V.; Farley, J.E.; Howell, A.; Knauf, M.; Ooi, P.; Smith, R.P.; Weise, P.; Wetherell, M.; et al. Vaccine potential of the Neisseria meningitidis 2086 lipoprotein. Infect. Immun. 2004, 72, 2088–2100. [Google Scholar] [CrossRef]
- Masignani, V.; Comanducci, M.; Giuliani, M.M.; Bambini, S.; Adu-Bobie, J.; Arico, B.; Brunelli, B.; Pieri, A.; Santini, L.; Savino, S.; et al. Vaccination against Neisseria meningitidis using three variants of the lipoprotein GNA1870. J. Exp. Med. 2003, 197, 789–799. [Google Scholar] [CrossRef]
- McNeil, L.K.; Zagursky, R.J.; Lin, S.L.; Murphy, E.; Zlotnick, G.W.; Hoiseth, S.K.; Jansen, K.U.; Anderson, A.S. Role of factor H binding protein in Neisseria meningitidis virulence and its potential as a vaccine candidate to broadly protect against meningococcal disease. Microbiol. Mol. Biol. Rev. MMBR 2013, 77, 234–252. [Google Scholar] [CrossRef]
- Gorringe, A.R.; Pajón, R. Bexsero: A multicomponent vaccine for prevention of meningococcal disease. Hum. Vaccines Immunother. 2012, 8, 174–183. [Google Scholar] [CrossRef]
- Shirley, M.; Dhillon, S. Bivalent rLP2086 Vaccine (Trumenba(®)): A Review in Active Immunization Against Invasive Meningococcal Group B Disease in Individuals Aged 10-25 Years. BioDrugs Clin. Immunother. Biopharm. Gene Ther. 2015, 29, 353–361. [Google Scholar] [CrossRef]
- Marshall, H.S.; Vesikari, T.; Richmond, P.C.; Wysocki, J.; Szenborn, L.; Beeslaar, J.; Maguire, J.D.; Balmer, P.; O’Neill, R.; Anderson, A.S.; et al. Safety and immunogenicity of a primary series and booster dose of the meningococcal serogroup B-factor H binding protein vaccine (MenB-FHbp) in healthy children aged 1–9 years: Two phase 2 randomised, controlled, observer-blinded studies. Lancet Infect. Dis. 2023, 23, 103–116. [Google Scholar] [CrossRef]
- Marshall, H.S.; Richmond, P.C.; Nissen, M.D.; Wouters, A.; Baber, J.; Jiang, Q.; Anderson, A.S.; Jones, T.R.; Harris, S.L.; Jansen, K.U.; et al. A phase 2 open-label safety and immunogenicity study of a meningococcal B bivalent rLP2086 vaccine in healthy adults. Vaccine 2013, 31, 1569–1575. [Google Scholar] [CrossRef]
- Flacco, M.E.; Manzoli, L.; Rosso, A.; Marzuillo, C.; Bergamini, M.; Stefanati, A.; Cultrera, R.; Villari, P.; Ricciardi, W.; Ioannidis, J.P.A.; et al. Immunogenicity and safety of the multicomponent meningococcal B vaccine (4CMenB) in children and adolescents: A systematic review and meta-analysis. Lancet Infect. Dis. 2018, 18, 461–472. [Google Scholar] [CrossRef]
- Valente Pinto, M.; O’Connor, D.; Galal, U.; Clutterbuck, E.A.; Robinson, H.; Plested, E.; Bibi, S.; Camara Pellisso, S.; Hughes, H.; Kerridge, S.; et al. Immunogenicity and reactogenicity of a reduced schedule of a 4-component capsular group B meningococcal vaccine: A randomized controlled trial in infants. Open Forum Infect. Dis. 2020, 7, ofaa143. [Google Scholar] [CrossRef]
- Pajon, R.; Beernink, P.T.; Harrison, L.H.; Granoff, D.M. Frequency of factor H-binding protein modular groups and susceptibility to cross-reactive bactericidal activity in invasive meningococcal isolates. Vaccine 2010, 28, 2122–2129. [Google Scholar] [CrossRef]
- Brehony, C.; Hill, D.M.; Lucidarme, J.; Borrow, R.; Maiden, M.C. Meningococcal vaccine antigen diversity in global databases. Euro Surveill. Bull. Eur. Sur Les Mal. Transm. Eur. Commun. Dis. Bull. 2015, 20, 30084. [Google Scholar] [CrossRef]
- Säll, O.; Olofsson, E.; Jacobsson, S. High genomic-based predicted strain coverage among invasive meningococcal isolates when combining Bexsero and Trumenba vaccines. Vaccine 2020, 38, 4374–4378. [Google Scholar] [CrossRef]
- Shi, F.; Zhang, A.; Zhu, B.; Gao, Y.; Xu, L.; Li, Y.; Yin, Z.; Li, J.; Xie, N.; Shao, Z. Prevalence of factor H binding protein sub-variants among Neisseria meningitidis in China. Vaccine 2017, 35, 2343–2350. [Google Scholar] [CrossRef]
- Watkins, E.R.; Maiden, M.C.J. Persistence of hyperinvasive meningococcal strain types during global spread as recorded in the pubMLST database. PLoS ONE 2012, 7, e45349. [Google Scholar] [CrossRef]
- Bratcher, H.B.; Corton, C.; Jolley, K.A.; Parkhill, J.; Maiden, M.C. A gene-by-gene population genomics platform: De novo assembly, annotation and genealogical analysis of 108 representative Neisseria meningitidis genomes. BMC Genom. 2014, 15, 1138. [Google Scholar] [CrossRef]
- Gandhi, A.; Balmer, P.; York, L.J. Characteristics of a new meningococcal serogroup B vaccine, bivalent rLP2086 (MenB-FHbp; Trumenba®). Postgrad. Med. 2016, 128, 548–556. [Google Scholar] [CrossRef]
- Parks, D.H.; Chuvochina, M.; Rinke, C.; Mussig, A.J.; Chaumeil, P.-A.; Hugenholtz, P. GTDB: An ongoing census of bacterial and archaeal diversity through a phylogenetically consistent, rank normalized and complete genome-based taxonomy. Nucleic Acids Res. 2021, 50, D785–D794. [Google Scholar] [CrossRef]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef]
- Silva, M.; Machado, M.P.; Silva, D.N.; Rossi, M.; Moran-Gilad, J.; Santos, S.; Ramirez, M.; Carriço, J.A. chewBBACA: A complete suite for gene-by-gene schema creation and strain identification. Microb. Genom. 2018, 4, e000166. [Google Scholar] [CrossRef]
- Meyer, D.; Zeileis, A.; Hornik, K. The strucplot framework: Visualizing multi-way contingency tables with vcd. J. Stat. Softw. 2006, 17, 1–48. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef]
- Yu, G. Using ggtree to visualize data on tree-like structures. Curr. Protoc. Bioinform. 2020, 69, e96. [Google Scholar] [CrossRef]
- Xu, S.; Dai, Z.; Guo, P.; Fu, X.; Liu, S.; Zhou, L.; Tang, W.; Feng, T.; Chen, M.; Zhan, L.; et al. ggtreeExtra: Compact visualization of richly annotated phylogenetic data. Mol. Biol. Evol. 2021, 38, 4039–4042. [Google Scholar] [CrossRef]
- Yue, M.; Xu, J.; Yu, J.; Shao, Z. Carriage prevalence of Neisseria meningitidis in China, 2005–2022: A systematic review and meta-analysis. BMC Infect. Dis. 2022, 22, 594. [Google Scholar] [CrossRef]
- Li, J.H.; Wu, D.; Yin, Z.D.; Li, Y.X. Analysis of epidemic characteristics for meningococcal meningitis in China during 2015–2017. Zhonghua Yu Fang Yi Xue Za Zhi [Chin. J. Prev. Med.] 2019, 53, 159–163. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Chen, Y.; Yue, M.; Yu, J.; Han, F.; Xu, L.; Shao, Z. Prevalence of Neisseria meningitidis serogroups in invasive meningococcal disease in China, 2010–2020: A systematic review and meta-analysis. Hum. Vaccines Immunother. 2022, 18, 2071077. [Google Scholar] [CrossRef]
- Xu, Y.; Li, Y.; Wang, S.; Li, M.; Xu, M.; Ye, Q. Meningococcal vaccines in China. Hum. Vaccines Immunother. 2021, 17, 2197–2204. [Google Scholar] [CrossRef]
- Pizza, M.; Bekkat-Berkani, R.; Rappuoli, R. Vaccines against Meningococcal Diseases. Microorganisms 2020, 8, 1521. [Google Scholar] [CrossRef]
- Masignani, V.; Pizza, M.; Moxon, E.R. The development of a vaccine against meningococcus B using reverse vaccinology. Front. Immunol. 2019, 10, 00751. [Google Scholar] [CrossRef]
- Muzzi, A.; Brozzi, A.; Serino, L.; Bodini, M.; Abad, R.; Caugant, D.; Comanducci, M.; Lemos, A.P.; Gorla, M.C.; Křížová, P.; et al. Genetic Meningococcal Antigen Typing System (gMATS): A genotyping tool that predicts 4CMenB strain coverage worldwide. Vaccine 2019, 37, 991–1000. [Google Scholar] [CrossRef]
- Li, J.; Shao, Z.; Liu, G.; Bai, X.; Borrow, R.; Chen, M.; Guo, Q.; Han, Y.; Li, Y.; Taha, M.K.; et al. Meningococcal disease and control in China: Findings and updates from the Global Meningococcal Initiative (GMI). J. Infect. 2018, 76, 429–437. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tan, Z.; Xu, J.; Che, J.; Xu, L.; Yan, D.; Zhang, M.; Shao, Z. Extensive Genetic Diversity and Epidemiological Patterns of Factor H-Binding Protein Variants among Neisseria meningitidis in China. Microorganisms 2024, 12, 481. https://doi.org/10.3390/microorganisms12030481
Tan Z, Xu J, Che J, Xu L, Yan D, Zhang M, Shao Z. Extensive Genetic Diversity and Epidemiological Patterns of Factor H-Binding Protein Variants among Neisseria meningitidis in China. Microorganisms. 2024; 12(3):481. https://doi.org/10.3390/microorganisms12030481
Chicago/Turabian StyleTan, Zhizhou, Juan Xu, Jie Che, Li Xu, Dongshan Yan, Maojun Zhang, and Zhujun Shao. 2024. "Extensive Genetic Diversity and Epidemiological Patterns of Factor H-Binding Protein Variants among Neisseria meningitidis in China" Microorganisms 12, no. 3: 481. https://doi.org/10.3390/microorganisms12030481
APA StyleTan, Z., Xu, J., Che, J., Xu, L., Yan, D., Zhang, M., & Shao, Z. (2024). Extensive Genetic Diversity and Epidemiological Patterns of Factor H-Binding Protein Variants among Neisseria meningitidis in China. Microorganisms, 12(3), 481. https://doi.org/10.3390/microorganisms12030481