

Genomic Surveillance and Mutation Analysis of SARS-CoV-2 Variants among Patients in Saudi Arabia

 , , , , , ,
, , , , , ,
Abstract
1. Introduction
2. Methods
2.1. Ethical Approval
2.2. Clinical Specimen and Data Collection
2.3. Whole Genome Sequencing
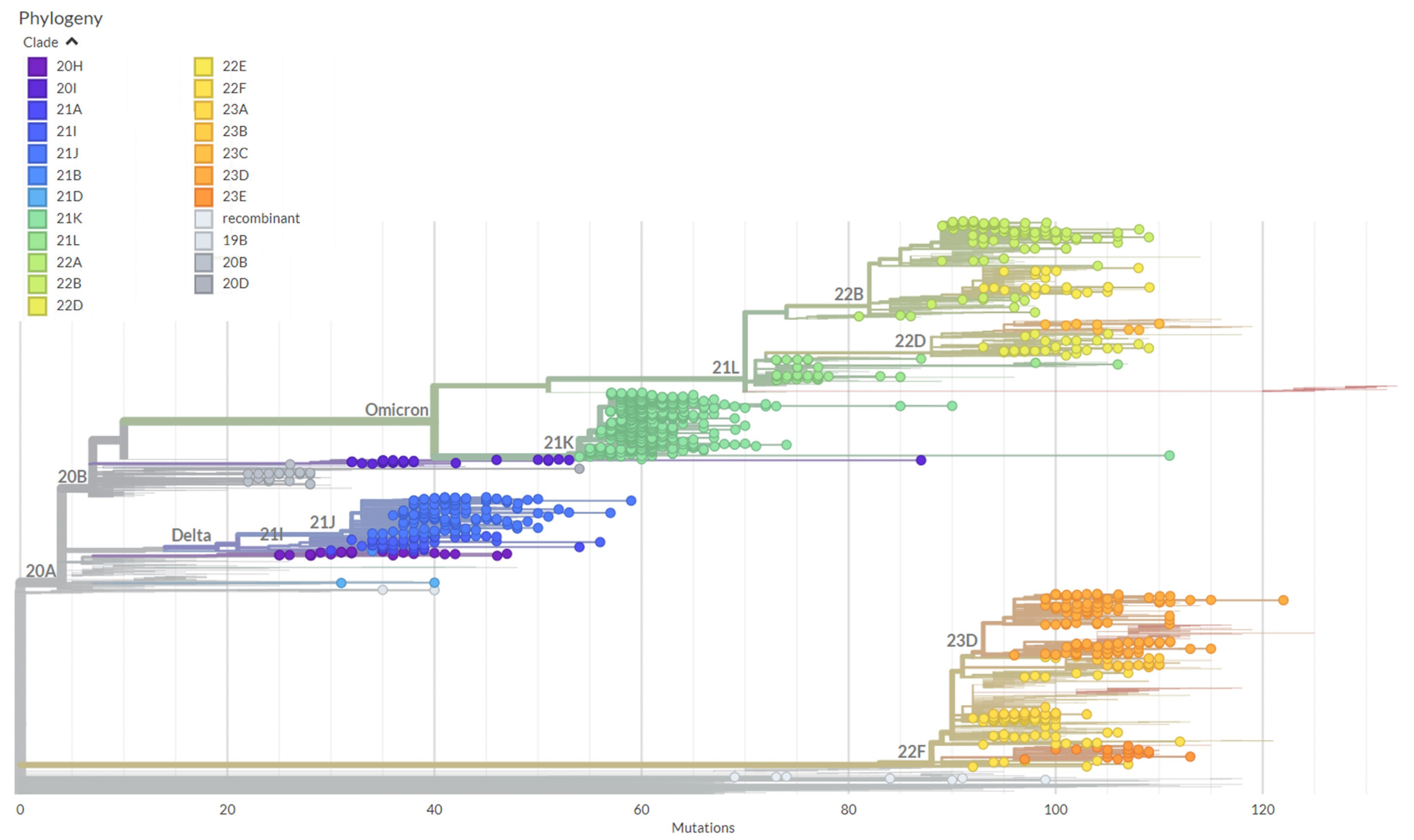
2.4. Data Analysis and Phylogenetic Analysis
2.5. Data Statistical Analysis
3. Results
3.1. Demographic and Clinical Characteristics of this Study’s Patients
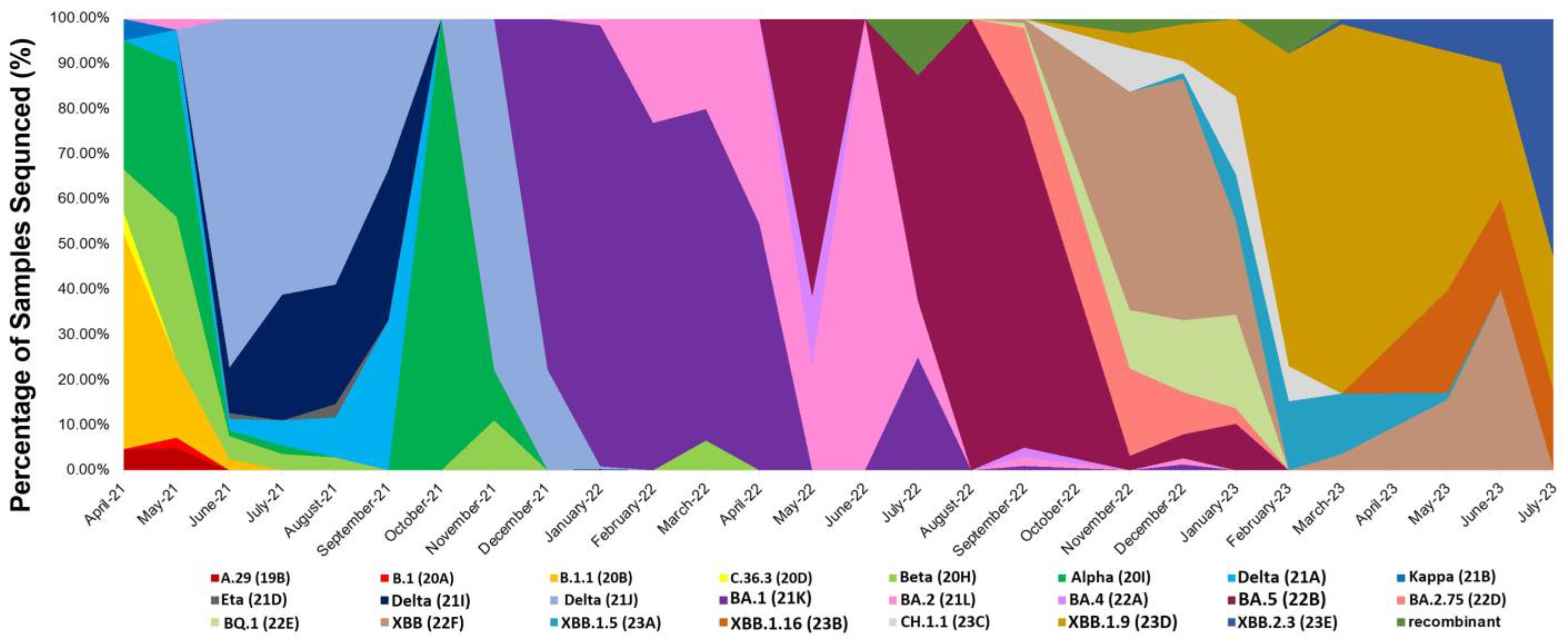
3.2. Distribution of SARS-CoV-2 Variants among the Genomic Sequences of the Patients
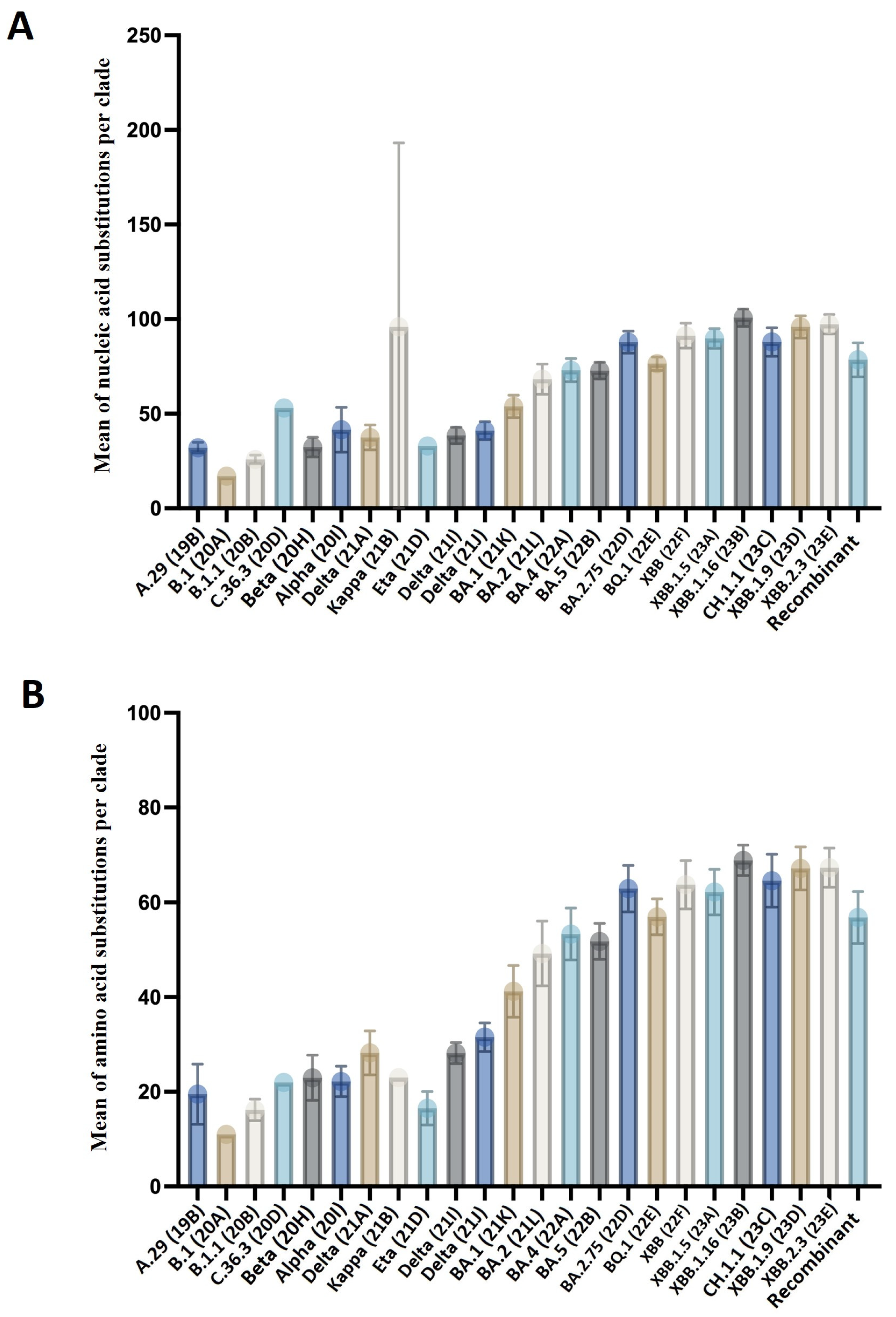
3.3. Genetic Diversity among the SARS-CoV-2 Variants
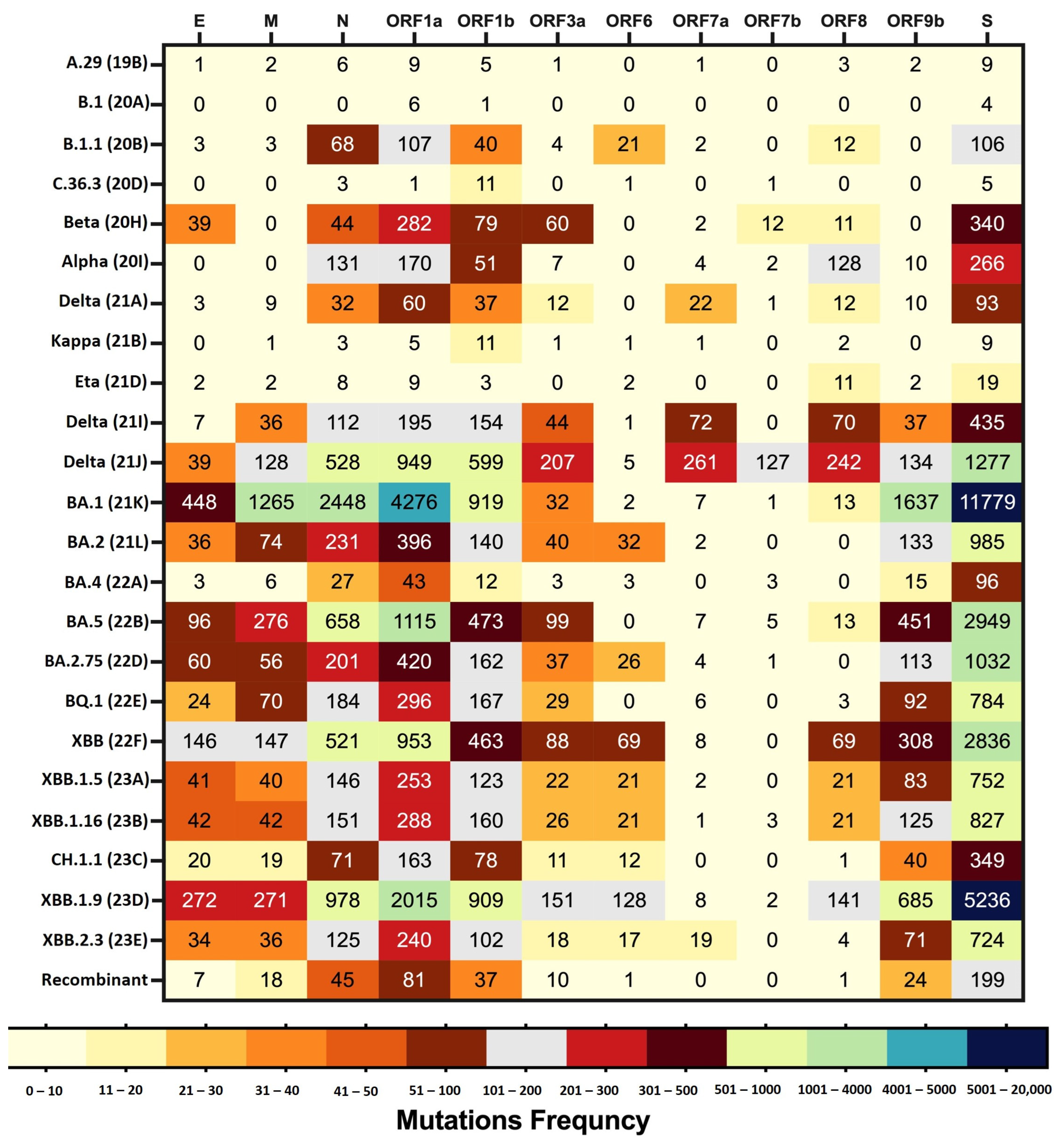
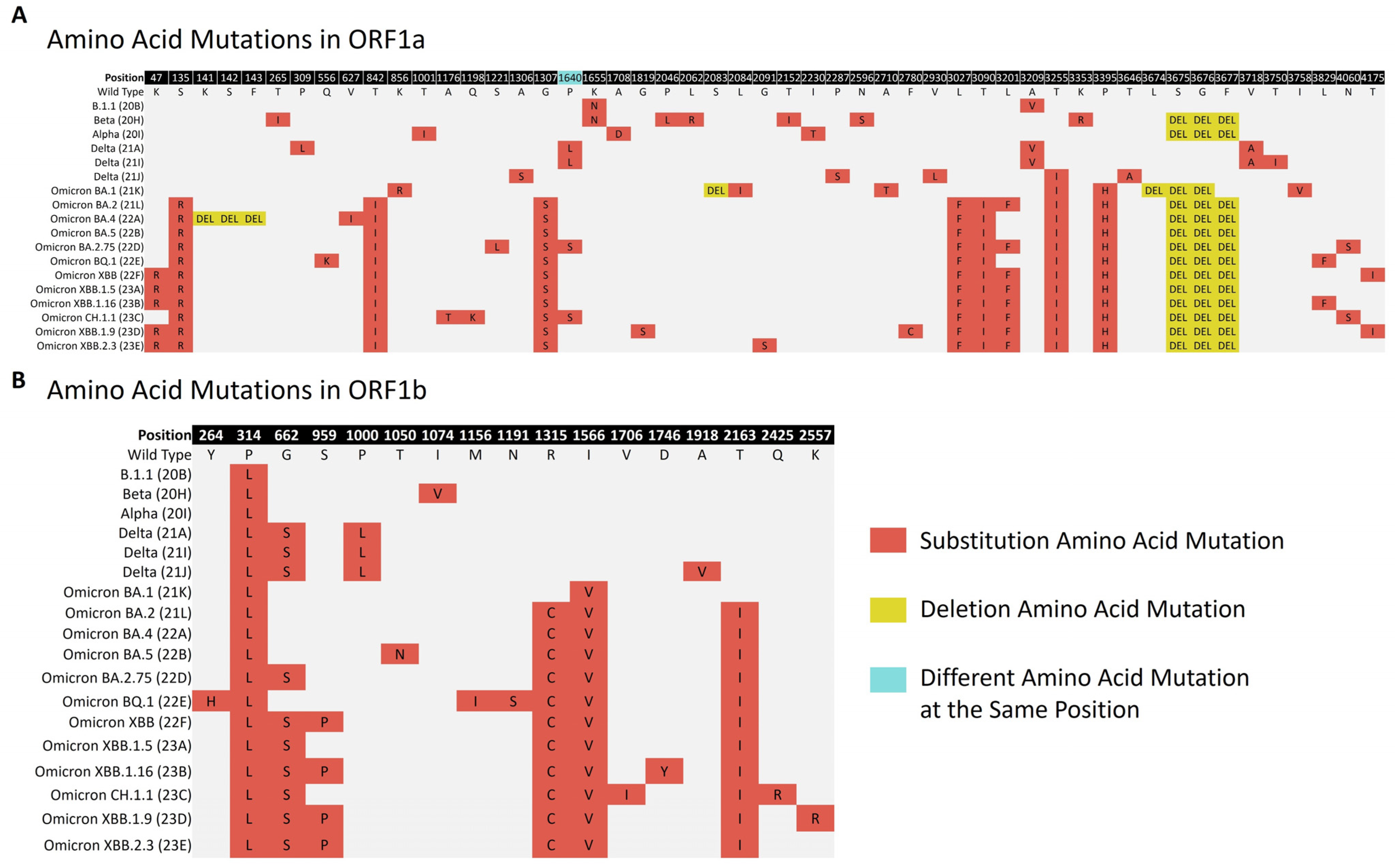
3.4. Amino Acid Mutation Analysis across the Variants of SARS-CoV-2
3.4.1. Amino Acid Mutations in Non-Structural Polyproteins
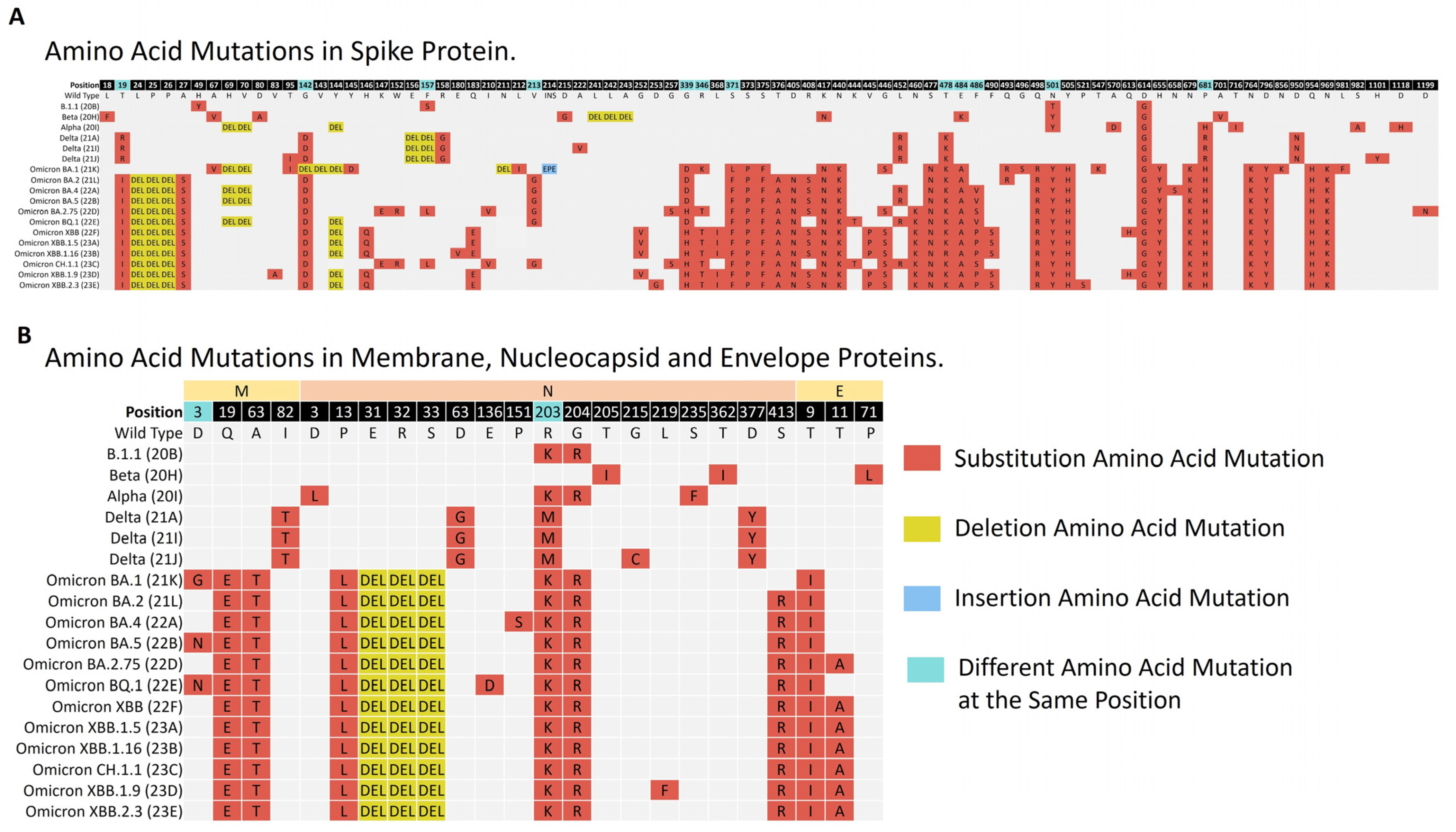
3.4.2. Amino Acid Mutations Analysis of Structural Proteins (S, E, M, and N)
Amino Acid Mutations Analysis of the Regions of the Structural Proteins
3.4.3. Amino Acid Mutations Analysis of Accessory Proteins
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ramanathan, K.; Antognini, D.; Combes, A.; Paden, M.; Zakhary, B.; Ogino, M.; Maclaren, G.; Brodie, D. A Familial Cluster of Pneumonia Associated with the 2019 Novel Coronavirus Indicating Person to Person Transmission: A Study of a Family Cluster. Lancet 2020, 395, 514–523. [Google Scholar]
- Markov, P.V.; Ghafari, M.; Beer, M.; Lythgoe, K.; Simmonds, P.; Stilianakis, N.I.; Katzourakis, A. The Evolution of SARS-CoV-2. Nat. Rev. Microbiol. 2023, 21, 361–379. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Mossman, K.; Grandvaux, N. Molecular Determinants of SARS-CoV-2 Variants. Trends Microbiol. 2021, 29, 871–873. [Google Scholar] [CrossRef] [PubMed]
- Di Giorgio, S.; Martignano, F.; Torcia, M.G.; Mattiuz, G.; Conticello, S.G. Evidence for Host-Dependent RNA Editing in the Transcriptome of SARS-CoV-2. Sci. Adv. 2020, 6, 423–428. [Google Scholar] [CrossRef]
- Morang’a, C.M.; Ngoi, J.M.; Gyamfi, J.; Amuzu, D.S.Y.; Nuertey, B.D.; Soglo, P.M.; Appiah, V.; Asante, I.A.; Owusu-Oduro, P.; Armoo, S.; et al. Genetic Diversity of SARS-CoV-2 Infections in Ghana from 2020–2021. Nat. Commun. 2022, 13, 2494. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Lee, J.Y.; Yang, J.S.; Kim, J.W.; Kim, V.N.; Chang, H. The Architecture of SARS-CoV-2 Transcriptome. Cell 2020, 181, 914–921.e10. [Google Scholar] [CrossRef] [PubMed]
- Cao, C.; Cai, Z.; Xiao, X.; Rao, J.; Chen, J.; Hu, N.; Yang, M.; Xing, X.; Wang, Y.; Li, M.; et al. The Architecture of the SARS-CoV-2 RNA Genome inside Virion. Nat. Commun. 2021, 12, 3917. [Google Scholar] [CrossRef] [PubMed]
- Kandeel, M.; Ibrahim, A.; Fayez, M.; Al-Nazawi, M. From SARS and MERS CoVs to SARS-CoV-2: Moving toward More Biased Codon Usage in Viral Structural and Nonstructural Genes. J. Med. Virol. 2020, 92, 660–666. [Google Scholar] [CrossRef]
- Ellis, P.; Somogyvári, F.; Virok, D.P.; Noseda, M.; Mclean, G.R. Decoding COVID-19 with the SARS-CoV-2 Genome. Curr. Genet. Med. Rep. 2021, 9, 1–12. [Google Scholar] [CrossRef]
- Chatterjee, S.; Kim, C.M.; Lee, Y.M.; Seo, J.W.; Kim, D.Y.; Yun, N.R.; Kim, D.M. Whole-Genome Analysis and Mutation Pattern of SARS-CoV-2 during First and Second Wave Outbreak in Gwangju, Republic of Korea. Sci. Rep. 2022, 12, 11354. [Google Scholar] [CrossRef]
- Yoshimoto, F.K. The Proteins of Severe Acute Respiratory Syndrome Coronavirus-2 (SARS-CoV-2 or n-COV19), the Cause of COVID-19. Protein J. 2020, 39, 198–216. [Google Scholar] [CrossRef]
- Shin, D.; Mukherjee, R.; Grewe, D.; Bojkova, D.; Baek, K.; Bhattacharya, A.; Schulz, L.; Widera, M.; Mehdipour, A.R.; Tascher, G.; et al. Papain-like Protease Regulates SARS-CoV-2 Viral Spread and Innate Immunity. Nature 2020, 587, 657–662. [Google Scholar] [CrossRef]
- Jungreis, I.; Sealfon, R. SARS-CoV-2 Gene Content and COVID-19 Mutation Impact by Comparing 44 Sarbecovirus Genomes. Nat. Commun. 2021, 12, 2642. [Google Scholar] [CrossRef]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 Spike Receptor-Binding Domain Bound to the ACE2 Receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef]
- Cubuk, J.; Alston, J.J.; Incicco, J.J.; Singh, S.; Stuchell-Brereton, M.D.; Ward, M.D.; Zimmerman, M.I.; Vithani, N.; Griffith, D.; Wagoner, J.A.; et al. The SARS-CoV-2 Nucleocapsid Protein Is Dynamic, Disordered, and Phase Separates with RNA. Nat. Commun. 2021, 12, 1936. [Google Scholar] [CrossRef]
- Mohideen, A.K.S. Molecular Docking Analysis of Phytochemical Thymoquinone as a Therapeutic Agent on SARS-Cov-2 Envelope Protein. Biointerface Res. Appl. Chem. 2021, 11, 8389–8401. [Google Scholar] [CrossRef]
- Lubin, J.H.; Zardecki, C.; Dolan, E.M.; Lu, C.; Shen, Z.; Dutta, S.; Westbrook, J.D.; Hudson, B.P.; Goodsell, D.S.; Williams, J.K.; et al. Evolution of the SARS-CoV-2 Proteome in Three Dimensions (3D) during the First 6 Months of the COVID-19 Pandemic. Proteins Struct. Funct. Bioinform. 2022, 90, 1054–1080. [Google Scholar] [CrossRef] [PubMed]
- Liang, F. Quantitative Mutation Analysis of Genes and Proteins of Major SARS-CoV-2 Variants of Concern and Interest. Viruses 2023, 15, 1193. [Google Scholar] [CrossRef] [PubMed]
- Olukitibi, T.A.; Ao, Z.; Warner, B.; Unat, R.; Kobasa, D.; Yao, X. Significance of Conserved Regions in Coronavirus Spike Protein for Developing a Novel Vaccine against SARS-CoV-2 Infection. Vaccines 2023, 11, 545. [Google Scholar] [CrossRef] [PubMed]
- Sanjuán, R.; Domingo-Calap, P. Genetic Diversity and Evolution of Viral Populations. Encycl. Virol. 2021, 1, 53–61. [Google Scholar] [CrossRef]
- Alisoltani, A.; Jaroszewski, L.; Iyer, M.; Iranzadeh, A.; Godzik, A. Increased Frequency of Indels in Hypervariable Regions of SARS-CoV-2 Proteins—A Possible Signature of Adaptive Selection. Front. Genet. 2022, 13, 875406. [Google Scholar] [CrossRef]
- Abbasian, M.H.; Mahmanzar, M.; Rahimian, K.; Mahdavi, B.; Tokhanbigli, S.; Moradi, B.; Sisakht, M.M.; Deng, Y. Global Landscape of SARS-CoV-2 Mutations and Conserved Regions. J. Transl. Med. 2023, 21, 152. [Google Scholar] [CrossRef]
- World Health Organization. Available online: https://www.who.int/ (accessed on 16 November 2023).
- Rambaut, A.; Holmes, E.C.; O’Toole, Á.; Hill, V.; McCrone, J.T.; Ruis, C.; du Plessis, L.; Pybus, O.G. A Dynamic Nomenclature Proposal for SARS-CoV-2 Lineages to Assist Genomic Epidemiology. Nat. Microbiol. 2020, 5, 1403–1407. [Google Scholar] [CrossRef]
- Aksamentov, I.; Roemer, C.; Hodcroft, E.; Neher, R. Nextclade: Clade Assignment, Mutation Calling and Quality Control for Viral Genomes. J. Open Source Softw. 2021, 6, 3773. [Google Scholar] [CrossRef]
- Thakur, B.; Dubey, P.; Benitez, J.; Torres, J.P.; Reddy, S.; Shokar, N.; Aung, K.; Mukherjee, D.; Dwivedi, A.K. A Systematic Review and Meta-Analysis of Geographic Differences in Comorbidities and Associated Severity and Mortality among Individuals with COVID-19. Sci. Rep. 2021, 11, 8562. [Google Scholar] [CrossRef]
- Liu, K.; Chen, Y.; Lin, R.; Han, K. Clinical Features of COVID-19 in Elderly Patients: A Comparison with Young and Middle-Aged Patients. J. Infect. 2020, 80, e14–e18. [Google Scholar] [CrossRef]
- Akinosoglou, K.; Schinas, G.; Panagiota, M. The Impact of Age on Intensive Care. Ageing Res. Rev. J. 2023, 84, 101832. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.A.; Patel, K.; Patton, M.E.; Reingold, A.; Kawasaki, B.; Meek, J.; Openo, K.; Ryan, P.A.; Falkowski, A.; Bye, E.; et al. COVID-19–Associated Hospitalizations Among U.S. Adults Aged ≥65 Years—COVID-NET, 13 States, January–August 2023. MMWR Recomm. Rep. 2023, 72, 1089–1094. [Google Scholar] [CrossRef] [PubMed]
- Houhamdi, L.; Gautret, P.; Hoang, V.T.; Fournier, P.E.; Colson, P.; Raoult, D. Characteristics of the First 1119 SARS-CoV-2 Omicron Variant Cases, in Marseille, France, November−December 2021. J. Med. Virol. 2022, 94, 2290–2295. [Google Scholar] [CrossRef] [PubMed]
- Lauring, A.S.; Tenforde, M.W.; Chappell, J.D.; Gaglani, M.; Ginde, A.A.; Mcneal, T.; Ghamande, S.; Douin, D.J.; Talbot, H.K.; Casey, J.D.; et al. Clinical Severity of, and Effectiveness of MRNA Vaccines against, COVID-19 from Omicron, Delta, and Alpha SARS-CoV-2 Variants in the United States: Prospective Observational Study. BMJ 2022, 376, e069761. [Google Scholar] [CrossRef] [PubMed]
- Modes, M.E.; Directo, M.P.; Melgar, M.; Johnson, L.R.; Yang, H.; Chaudhary, P.; Bartolini, S.; Kho, N.; Noble, P.W.; Isonaka, S.; et al. Clinical Characteristics and Outcomes Among Adults Hospitalized with Laboratory-Confirmed SARS-CoV-2 Infection During Periods of B.1.617.2 (Delta) and B.1.1.529 (Omicron) Variant Predominance—One Hospital, California, July 15–September 23, 2021, and December 21, 2021–January 27, 2022. CDC Morb. Mortal. Wkly. Rep. 2022, 71, 217–223. [Google Scholar] [CrossRef]
- Van Goethem, N.; Chung, P.Y.J.; Meurisse, M.; Vandromme, M.; De Mot, L.; Brondeel, R.; Stouten, V.; Klamer, S.; Cuypers, L.; Braeye, T.; et al. Clinical Severity of SARS-CoV-2 Omicron Variant Compared with Delta among Hospitalized COVID-19 Patients in Belgium during Autumn and Winter Season 2021–2022. Viruses 2022, 14, 1297. [Google Scholar] [CrossRef]
- Mattiuzzia, C.; Lippib, G. Timeline Analysis of Clinical Severity of COVID-19 in the General Population. Eur. J. Intern. Med. 2023, 110, 97–98. [Google Scholar] [CrossRef]
- Bahl, A.; Mielke, N.; Johnson, S.; Desai, A.; Qu, L. Severe COVID-19 Outcomes in Pediatrics: An Observational Cohort Analysis Comparing Alpha, Delta, and Omicron Variants. Lancet Reg. Health Am. 2023, 18, 100405. [Google Scholar] [CrossRef]
- Kant, R.; Nguyen, P.T.; Blomqvist, S.; Erdin, M.; Alburkat, H.; Suvanto, M.; Zakham, F.; Salminen, V.; Olander, V.; Paloniemi, M.; et al. Incidence Trends for SARS-CoV-2 Alpha and Beta Variants, Finland, Spring 2021. Emerg. Infect. Dis. 2021, 27, 3137–3141. [Google Scholar] [CrossRef]
- Cochin, M.; Luciani, L.; Touret, F.; Driouich, J.S.; Petit, P.R.; Moureau, G.; Baronti, C.; Laprie, C.; Thirion, L.; Maes, P.; et al. The SARS-CoV-2 Alpha Variant Exhibits Comparable Fitness to the D614G Strain in a Syrian Hamster Model. Commun. Biol. 2022, 5, 225. [Google Scholar] [CrossRef] [PubMed]
- King, K.L.; Wilson, S.; Napolitano, J.M.; Sell, K.J.; Rennert, L.; Parkinson, C.L.; Dean, D. SARS-CoV-2 Variants of Concern Alpha and Delta Show Increased Viral Load in Saliva. PLoS ONE 2022, 17, e0267750. [Google Scholar] [CrossRef] [PubMed]
- Michaelsen, T.Y.; Bennedbæk, M.; Christiansen, L.E.; Jørgensen, M.S.F.; Møller, C.H.; Sørensen, E.A.; Knutsson, S.; Brandt, J.; Jensen, T.B.N.; Chiche-Lapierre, C.; et al. Introduction and Transmission of SARS-CoV-2 Lineage B.1.1.7, Alpha Variant, in Denmark. Genome Med. 2022, 14, 47. [Google Scholar] [CrossRef] [PubMed]
- Trobajo-Sanmartín, C.; Martínez-Baz, I.; Miqueleiz, A.; Fernández-Huerta, M.; Burgui, C.; Casado, I.; Baigorría, F.; Navascués, A.; Castilla, J.; Ezpeleta, C. Differences in Transmission between SARS-CoV-2 Alpha (B.1.1.7) and Delta (B.1.617.2) Variants. Microbiol. Spectr. 2022, 10, e00008-22. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.T.; Tsai, J.J.; Chang, K.; Chen, C.H.; Lin, P.C.; Tsai, C.Y.; Tsai, Y.Y.; Hsu, M.C.; Chuang, W.L.; Chang, J.M.; et al. Identification and Analysis of SARS-CoV-2 Alpha Variants in the Largest Taiwan COVID-19 Outbreak in 2021. Front. Med. 2022, 9, 869818. [Google Scholar] [CrossRef]
- Elliott, P.; Haw, D.; Wang, H.; Eales, O.; Walters, C.E.; Ainslie, K.E.C.; Atchison, C.; Fronterre, C.; Diggle, P.J.; Page, A.J.; et al. Exponential Growth, High Prevalence of SARS-CoV-2, and Vaccine Effectiveness Associated with the Delta Variant. Science 2021, 374, eabl9551. [Google Scholar] [CrossRef]
- Bolze, A.; Luo, S.; White, S.; Cirulli, E.T.; Wyman, D.; Dei Rossi, A.; Machado, H.; Cassens, T.; Jacobs, S.; Schiabor Barrett, K.M.; et al. SARS-CoV-2 Variant Delta Rapidly Displaced Variant Alpha in the United States and Led to Higher Viral Loads. Cell Rep. Med. 2022, 3, 100564. [Google Scholar] [CrossRef] [PubMed]
- Eggink, D.; Andeweg, S.P.; Vennema, H.; van Maarseveen, N.; Vermaas, K.; Vlaemynck, B.; Schepers, R.; van Gageldonk-Lafeber, A.B.; van den Hof, S.; Reusken, C.B.E.M.; et al. Increased Risk of Infection with SARS-CoV-2 Omicron BA.1 Compared with Delta in Vaccinated and Previously Infected Individuals, the Netherlands, 22 November 2021 to 19 January 2022. Eurosurveillance 2022, 27, 2101196. [Google Scholar] [CrossRef]
- Sayan, M.; Arikan, A.; Isbilen, M. Circulating Dynamics of SARS-CoV-2 Variants between April 2021 and February 2022 in Turkey. Can. J. Infect. Dis. Med. Microbiol. 2022, 2022, 4677720. [Google Scholar] [CrossRef]
- Sievers, C.; Zacher, B.; Ullrich, A.; Huska, M.; Fuchs, S.; Buda, S.; Haas, W.; Diercke, M.; an der Heiden, M.; Kröger, S. SARS-CoV-2 Omicron Variants BA.1 and BA.2 Both Show Similarly Reduced Disease Severity of COVID-19 Compared to Delta, Germany, 2021 to 2022. Eurosurveillance 2022, 27, 2200396. [Google Scholar] [CrossRef] [PubMed]
- Veneti, L.; Bøås, H.; Kristoffersen, A.B.; Stålcrantz, J.; Bragstad, K.; Hungnes, O.; Storm, M.L.; Aasand, N.; Rø, G.; Starrfelt, J.; et al. Reduced Risk of Hospitalisation among Reported COVID-19 Cases Infected with the SARS-CoV-2 Omicron BA.1 Variant Compared with the Delta Variant, Norway, December 2021 to January 2022. Eurosurveillance 2022, 27, 2200077. [Google Scholar] [CrossRef]
- Mohapatra, R.K.; Kandi, V.; Sarangi, A.K.; Verma, S. The Recently Emerged BA.4 and BA.5 Lineages of Omicron and Their Global Health Concerns amid the Ongoing Wave of COVID-19 Pandemic. Int. J. Surg. 2022, 103, 2020–2023. [Google Scholar] [CrossRef]
- Tegally, H.; Moir, M.; Everatt, J.; Giovanetti, M.; Scheepers, C.; Wilkinson, E.; Subramoney, K.; Makatini, Z.; Moyo, S.; Amoako, D.G.; et al. Emergence of SARS-CoV-2 Omicron Lineages BA.4 and BA.5 in South Africa. Nat. Med. 2022, 28, 1785–1790. [Google Scholar] [CrossRef] [PubMed]
- Lewnard, J.A.; Hong, V.; Kim, J.S.; Shaw, S.F.; Lewin, B.; Takhar, H.; Tartof, S.Y. Association of SARS-CoV-2 BA.4/BA.5 Omicron Lineages with Immune Escape and Clinical Outcome. Nat. Commun. 2023, 14, 1407. [Google Scholar] [CrossRef]
- Chen, J.J.; Li, L.B.; Peng, H.H.; Tian, S.; Ji, B.; Shi, C.; Qian, C.; Jiang, W.G.; Liu, M.C.; Li, T.T.; et al. Neutralization against XBB.1 and XBB.1.5 after Omicron Subvariants Breakthrough Infection or Reinfection. Lancet Reg. Health West. Pac. 2023, 33, 100759. [Google Scholar] [CrossRef]
- Tamura, T.; Ito, J.; Uriu, K.; Zahradnik, J.; Kida, I.; Anraku, Y.; Nasser, H.; Shofa, M.; Oda, Y.; Lytras, S.; et al. Virological Characteristics of the SARS-CoV-2 XBB Variant Derived from Recombination of Two Omicron Subvariants. Nat. Commun. 2023, 14, 13–15. [Google Scholar] [CrossRef]
- Nagaraja, M.; Sireesha, K.; Srikar, A.; Sudheer Kumar, K.; Mohan, A.; Vengamma, B.; Tirumala, C.; Verma, A.; Kalawat, U. Mutation Analysis of SARS-CoV-2 Variants Isolated from Symptomatic Cases from Andhra Pradesh, India. Viruses 2023, 15, 1656. [Google Scholar] [CrossRef]
- Tian, D.; Sun, Y.; Xu, H.; Ye, Q. The Emergence and Epidemic Characteristics of the Highly Mutated SARS-CoV-2 Omicron Variant. J. Med. Virol. 2022, 94, 2376–2383. [Google Scholar] [CrossRef]
- Chavda, V.P.; Bezbaruah, R.; Deka, K.; Nongrang, L.; Kalita, T. The Delta and Omicron Variants of SARS-CoV-2: What We Know So Far. Vaccines 2022, 10, 1926. [Google Scholar] [CrossRef]
- Kumar, J.; Sengupta, A.; Pal, P.; Roy, S. Characterizing Genomic Variants and Mutations in SARS-CoV-2 Proteins from Indian Isolates. Gene Rep. 2021, 25, 101044. [Google Scholar] [CrossRef]
- Wabalo, E.K.; Dubiwak, A.D.; Senbetu, M.W.; Gizaw, T.S. Effect of Genomic and Amino Acid Sequence Mutation on Virulence and Therapeutic Target of Severe Acute Respiratory Syndrome Coronavirus-2 (SARS-Cov-2). Infect. Drug Resist. 2021, 14, 2187–2192. [Google Scholar] [CrossRef] [PubMed]
- Menasria, T.; Aguilera, M. Genomic Diversity of SARS-CoV-2 in Algeria and North African Countries: What We Know So Far and What We Expect? Microorganisms 2022, 10, 467. [Google Scholar] [CrossRef] [PubMed]
- Walls, A.C.; Park, Y.-J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Gobeil, S.M.C.; Janowska, K.; McDowell, S.; Mansouri, K.; Parks, R.; Stalls, V.; Kopp, M.F.; Manne, K.; Li, D.; Wiehe, K.; et al. Effect of Natural Mutations of SARS-CoV-2 on Spike Structure, Conformation, and Antigenicity. Science 2021, 373, eabi6226. [Google Scholar] [CrossRef] [PubMed]
- Ozono, S.; Zhang, Y.; Ode, H.; Sano, K.; Tan, T.S.; Imai, K.; Miyoshi, K.; Kishigami, S.; Ueno, T.; Iwatani, Y.; et al. SARS-CoV-2 D614G Spike Mutation Increases Entry Efficiency with Enhanced ACE2-Binding Affinity. Nat. Commun. 2021, 12, 848. [Google Scholar] [CrossRef]
- Bhattacharya, M.; Chatterjee, S.; Sharma, A.R.; Agoramoorthy, G.; Chakraborty, C. D614G Mutation and SARS-CoV-2: Impact on S-Protein Structure, Function, Infectivity, and Immunity. Appl. Microbiol. Biotechnol. 2021, 105, 9035–9045. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.E.; Zhang, X.; Case, J.B.; Winkler, E.S.; Liu, Y.; VanBlargan, L.A.; Liu, J.; Errico, J.M.; Xie, X.; Suryadevara, N.; et al. Resistance of SARS-CoV-2 Variants to Neutralization by Monoclonal and Serum-Derived Polyclonal Antibodies. Nat. Med. 2021, 27, 717–726. [Google Scholar] [CrossRef]
- Lista, M.J.; Winstone, H.; Wilson, H.D.; Dyer, A.; Pickering, S.; Galao, R.P.; De Lorenzo, G.; Cowton, V.M.; Furnon, W.; Suarez, N.; et al. The P681H Mutation in the Spike Glycoprotein of the Alpha Variant of SARS-CoV-2 Escapes IFITM Restriction and Is Necessary for Type I Interferon Resistance. J. Virol. 2022, 96, e01250-22. [Google Scholar] [CrossRef] [PubMed]
- Obeid, D.; Al-Qahtani, A.; Almaghrabi, R.; Alghamdi, S.; Alsanea, M.; Alahideb, B.; Almutairi, S.; Alsuwairi, F.; Al-Abdulkareem, M.; Asiri, M.; et al. Analysis of SARS-CoV-2 Genomic Surveillance Data during the Delta and Omicron Waves at a Saudi Tertiary Referral Hospital. J. Infect. Public Health 2023, 16, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Jawad, B.; Adhikari, P.; Podgornik, R.; Ching, W.Y. Key Interacting Residues between RBD of SARS-CoV-2 and ACE2 Receptor: Combination of Molecular Dynamics Simulation and Density Functional Calculation. J. Chem. Inf. Model. 2021, 61, 4425–4441. [Google Scholar] [CrossRef] [PubMed]
- Verma, J.; Subbarao, N. Insilico Study on the Effect of SARS-CoV-2 RBD Hotspot Mutants’ Interaction with ACE2 to Understand the Binding Affinity and Stability. Virology 2021, 561, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Zhou, L.; Mo, M.; Liu, T.; Wu, C.; Gong, C.; Lu, K.; Gong, L.; Zhu, W.; Xu, Z. SARS-CoV-2 Omicron RBD Shows Weaker Binding Affinity than the Currently Dominant Delta Variant to Human ACE2. Signal Transduct. Target. Ther. 2022, 7, 2021–2023. [Google Scholar] [CrossRef]
- Starr, T.N.; Greaney, A.J.; Hilton, S.K.; Ellis, D.; Crawford, K.H.D.; Dingens, A.S.; Navarro, M.J.; Bowen, J.E.; Tortorici, M.A.; Walls, A.C.; et al. Deep Mutational Scanning of SARS-CoV-2 Receptor Binding Domain Reveals Constraints on Folding and ACE2 Binding. Cell 2020, 182, 1295–1310.e20. [Google Scholar] [CrossRef]
- Kidd, M.; Richter, A.; Best, A.; Cumley, N.; Mirza, J.; Percival, B.; Mayhew, M.; Megram, O.; Ashford, F.; White, T.; et al. S-Variant SARS-CoV-2 Lineage B1.1.7 Is Associated With Significantly Higher Viral Load in Samples Tested by TaqPath Polymerase Chain Reaction. J. Infect. Dis. 2021, 223, 1666–1670. [Google Scholar] [CrossRef]
- Cui, Z.; Liu, P.; Wang, N.; Wang, L.; Fan, K.; Zhu, Q.; Wang, K.; Chen, R.; Feng, R.; Jia, Z.; et al. Structural and Functional Characterizations of Infectivity and Immune Evasion of SARS-CoV-2 Omicron. Cell 2022, 185, 860–871.e13. [Google Scholar] [CrossRef] [PubMed]
- Asif, A.; Ilyas, I.; Abdullah, M.; Sarfraz, S.; Mustafa, M.; Mahmood, A. The Comparison of Mutational Progression in SARS-CoV-2: A Short Updated Overview. J. Mol. Pathol. 2022, 3, 201–218. [Google Scholar] [CrossRef]
- Kang, S.; Yang, M.; Hong, Z.; Zhang, L.; Huang, Z.; Chen, X.; He, S.; Zhou, Z.; Zhou, Z.; Chen, Q.; et al. Crystal Structure of SARS-CoV-2 Nucleocapsid Protein RNA Binding Domain Reveals Potential Unique Drug Targeting Sites. Acta Pharm. Sin. B 2020, 10, 1228–1238. [Google Scholar] [CrossRef]
- Chang, C.; Hou, M.; Chang, C.; Hsiao, C.; Huang, T. The SARS Coronavirus Nucleocapsid Protein—Forms and Functions. Antivir. Res. 2014, 103, 39–50. [Google Scholar] [CrossRef]
- Wu, W.; Cheng, Y.; Zhou, H.; Sun, C.; Zhang, S. The SARS-CoV-2 Nucleocapsid Protein: Its Role in the Viral Life Cycle, Structure and Functions, and Use as a Potential Target in the Development of Vaccines and Diagnostics. Virol. J. 2023, 20, 6. [Google Scholar] [CrossRef]
- Alsuwairi, F.A.; Alsaleh, A.N.; Alsanea, M.S.; Al-Qahtani, A.A.; Obeid, D.; Almaghrabi, R.S.; Alahideb, B.M.; AlAbdulkareem, M.A.; Mutabagani, M.S.; Althawadi, S.I.; et al. Association of SARS-CoV-2 Nucleocapsid Protein Mutations with Patient Demographic and Clinical Characteristics during the Delta and Omicron Waves. Microorganisms 2023, 11, 1288. [Google Scholar] [CrossRef] [PubMed]
- Mourier, T.; Shuaib, M.; Hala, S.; Mfarrej, S.; Alofi, F.; Naeem, R.; Alsomali, A.; Jorgensen, D.; Subudhi, A.K.; Ben Rached, F.; et al. SARS-CoV-2 Genomes from Saudi Arabia Implicate Nucleocapsid Mutations in Host Response and Increased Viral Load. Nat. Commun. 2022, 13, 601. [Google Scholar] [CrossRef] [PubMed]
- Shuaib, M.; Adroub, S.; Mourier, T.; Mfarrej, S.; Zhang, H.; Esau, L.; Alsomali, A.; Alofi, F.S.; Ahmad, A.N.; Shamsan, A.; et al. Impact of the SARS-CoV-2 Nucleocapsid 203K/204R Mutations on the Inflammatory Immune Response in COVID-19 Severity. Genome Med. 2023, 15, 54. [Google Scholar] [CrossRef]
- Lopez-Munoz, A.D.; Kosik, I.; Holly, J.; Yewdell, J.W. Cell Surface SARS-CoV-2 Nucleocapsid Protein Modulates Innate and Adaptive Immunity. Sci. Adv. 2022, 8, eabp9770. [Google Scholar] [CrossRef]
- Mou, K.; Abdalla, M.; Qing, D.; Tahir, M. Emerging Mutations in Envelope Protein of SARS-CoV-2 and Their Effect on Thermodynamic Properties. Informatics Med. Unlocked 2021, 25, 100675. [Google Scholar] [CrossRef]
- Rahman, M.S.; Hoque, M.N.; Islam, M.R.; Islam, I. Mutational Insights into the Envelope Protein of SARS-CoV-2. Gene Rep. 2021, 22, 100997. [Google Scholar] [CrossRef] [PubMed]
- Rizwan, T.; Kothidar, A.; Meghwani, H.; Sharma, V.; Shobhawat, R.; Saini, R.; Vaishnav, H.K.; Singh, V.; Pratap, M.; Sihag, H.; et al. Comparative Analysis of SARS-CoV-2 Envelope Viroporin Mutations from COVID-19 Deceased and Surviving Patients Revealed Implications on Its Ion-Channel Activities and Correlation with Patient Mortality. J. Biomol. Struct. Dyn. 2021, 40, 10454–10469. [Google Scholar] [CrossRef]
- Zhou, S.; Lv, P.; Li, M.; Chen, Z.; Xin, H.; Reilly, S.; Zhang, X. SARS-CoV-2 E Protein: Pathogenesis and Potential Therapeutic Development. Biomed. Pharmacother. 2023, 159, 114242. [Google Scholar] [CrossRef]
- Shen, L.; Bard, J.D.; Triche, T.J.; Judkins, A.R.; Biegel, J.A.; Gai, X. Emerging Variants of Concern in SARS-CoV-2 Membrane Protein: A Highly Conserved Target with Potential Pathological and Therapeutic Implications. Emerg. Microbes Infect. 2021, 10, 885–893. [Google Scholar] [CrossRef]
- Abavisani, M.; Rahimian, K.; Mahdavi, B.; Tokhanbigli, S.; Mollapour Siasakht, M.; Farhadi, A.; Kodori, M.; Mahmanzar, M.; Meshkat, Z. Mutations in SARS-CoV-2 Structural Proteins: A Global Analysis. Virol. J. 2022, 19, 220. [Google Scholar] [CrossRef]
- Chen, J.; Deng, Y.; Huang, B.; Han, D.; Wang, W.; Huang, M.; Zhai, C.; Zhao, Z.; Yang, R.; Zhao, Y.; et al. DNA Vaccines Expressing the Envelope and Membrane Proteins Provide Partial Protection Against SARS-CoV-2 in Mice. Front. Immunol. 2022, 13, 827605. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Zhuang, M.W.; Deng, J.; Zheng, Y.; Zhang, J.; Nan, M.L.; Zhang, X.J.; Gao, C.; Wang, P.H. SARS-CoV-2 ORF9b Antagonizes Type I and III Interferons by Targeting Multiple Components of the RIG-I/MDA-5–MAVS, TLR3–TRIF, and CGAS–STING Signaling Pathways. J. Med. Virol. 2021, 93, 5376–5389. [Google Scholar] [CrossRef] [PubMed]
- Redondo, N.; Zaldívar-López, S.; Garrido, J.J.; Montoya, M. SARS-CoV-2 Accessory Proteins in Viral Pathogenesis: Knowns and Unknowns. Front. Immunol. 2021, 12, 708264. [Google Scholar] [CrossRef]
- Wu, J.; Shi, Y.; Pan, X.; Wu, S.; Hou, R.; Zhang, Y.; Zhong, T.; Tang, H.; Du, W.; Wang, L.; et al. SARS-CoV-2 ORF9b Inhibits RIG-I-MAVS Antiviral Signaling by Interrupting K63-Linked Ubiquitination of NEMO. Cell Rep. 2021, 34, 108761. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Zhu, K.; Qin, B.; Olieric, V.; Wang, M.; Cui, S. Crystal Structure of SARS-CoV-2 Orf9b in Complex with Human TOM70 Suggests Unusual Virus-Host Interactions. Nat. Commun. 2021, 12, 2843. [Google Scholar] [CrossRef]
- Thorne, L.G.; Bouhaddou, M.; Reuschl, A.K.; Zuliani-Alvarez, L.; Polacco, B.; Pelin, A.; Batra, J.; Whelan, M.V.X.; Hosmillo, M.; Fossati, A.; et al. Evolution of Enhanced Innate Immune Evasion by SARS-CoV-2. Nature 2022, 602, 487–495. [Google Scholar] [CrossRef] [PubMed]
- Archana, A.; Long, C.; Chandran, K. Analysis of SARS-CoV-2 Amino Acid Mutations in New York City Metropolitan Wastewater (2020–2022) Reveals Multiple Traits with Human Health Implications across the Genome and Environment-Specific Distinctions. medRxiv 2022. [Google Scholar] [CrossRef]
- Hossain, A.; Akter, S.; Rashid, A.A.; Khair, S.; Alam, A.R.U. Unique Mutations in SARS-CoV-2 Omicron Subvariants’ Non-Spike Proteins: Potential Impacts on Viral Pathogenesis and Host Immune Evasion. Microb. Pathog. 2022, 170, 105699. [Google Scholar] [CrossRef]
- Obermeyer, F.; Jankowiak, M.; Barkas, N.; Schaffner, S.F.; Pyle, J.D.; Yurkovetskiy, L.; Bosso, M.; Park, D.J.; Babadi, M. Analysis of 6.4 Million SARS-CoV-2 Genomes Identifies Mutations Associated with Fitness. Science 2022, 376, 1327–1332. [Google Scholar] [CrossRef]
- Bianchi, M.; Borsetti, A.; Ciccozzi, M.; Pascarella, S. SARS-CoV-2 ORF3a: Mutability and Function. Int. J. Biol. Macromol. 2021, 170, 820–826. [Google Scholar] [CrossRef]
- Zhang, J.; Ejikemeuwa, A.; Gerzanich, V.; Nasr, M.; Tang, Q.; Simard, J.M.; Zhao, R.Y. Understanding the Role of SARS-CoV-2 ORF3a in Viral Pathogenesis and COVID-19. Front. Microbiol. 2022, 13, 854567. [Google Scholar] [CrossRef]
- Chaudhari, A.M.; Singh, I.; Joshi, M.; Patel, A.; Joshi, C. Defective ORF8 Dimerization in SARS-CoV-2 Delta Variant Leads to a Better Adaptive Immune Response Due to Abrogation of ORF8-MHC1 Interaction. Mol. Divers. 2023, 27, 45–57. [Google Scholar] [CrossRef]
- Arduini, A.; Laprise, F.; Liang, C. SARS-CoV-2 ORF8: A Rapidly Evolving Immune and Viral Modulator in COVID-19. Viruses 2023, 15, 871. [Google Scholar] [CrossRef] [PubMed]
- Tam, D.; Lorenzo-leal, A.C.; Ricardo, L. Targeting SARS-CoV-2 Non-Structural Proteins. Int. J. Mol. Sci. 2023, 24, 13002. [Google Scholar] [CrossRef]
- Dai, L.; Gao, G.F. Viral Targets for Vaccines against COVID-19. Nat. Rev. Immunol. 2021, 21, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Islam, J.; Nawal, N.; Alom, S.; Kabir, M. A Review on Structural, Non-Structural, and Accessory Proteins of SARS-CoV-2: Highlighting Drug Target Sites. Immunobiology 2023, 228, 152302. [Google Scholar] [CrossRef]
- Chen, K.; Shi, D.; Li, C.; Fang, Z.; Guo, Y.; Jiang, W.; Li, J.; Li, H.; Yao, H. Adenovirus Vaccine Containing Truncated SARS-CoV-2 Spike Protein S1 Subunit Leads to a Specific Immune Response in Mice. Vaccines 2023, 11, 429. [Google Scholar] [CrossRef] [PubMed]
- Arora, P.; Rocha, C.; Kempf, A.; Nehlmeier, I.; Graichen, L.; Winkler, M.S.; Lier, M.; Schulz, S.; Jäck, H.M.; Cossmann, A.; et al. The Spike Protein of SARS-CoV-2 Variant A.30 Is Heavily Mutated and Evades Vaccine-Induced Antibodies with High Efficiency. Cell. Mol. Immunol. 2021, 18, 2673–2675. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clade | N (%) |
|---|---|
| A.29 (19B) | 3 (0.3) |
| B.1 (20A) | 1 (0.1) |
| B.1.1 (20B) | 19 (1.6) |
| C.36.3 (20D) | 1 (0.1) |
| Beta (20H) | 24 (2.1) |
| Alpha (20I) | 24 (2.1) |
| Delta (21A) | 12 (1.0) |
| Kappa (21B) | 1 (0.1) |
| Eta (21D) | 2 (0.2) |
| Delta (21I) | 34 (2.9) |
| Delta (21J) | 139 (12.0) |
| Omicron BA.1 (21K) | 430 (37.0) |
| Omicron BA.2 (21L) | 33 (2.8) |
| Omicron BA.4 (22A) | 4 (0.3) |
| Omicron BA.5 (22B) | 94 (8.1) |
| Omicron BA.2.75 (22D) | 34 (2.9) |
| Omicron BQ.1 (22E) | 23 (2.0) |
| Omicron XBB (22F) | 80 (6.9) |
| Omicron XBB.1.5 (23A) | 18 (1.6) |
| Omicron XBB.1.16 (23B) | 21 (1.8) |
| Omicron CH.1.1 (23C) | 11 (1.0) |
| Omicron XBB.1.9 (23D) | 133 (11.5) |
| Omicron XBB.2.3 (23E) | 16 (1.4) |
| Recombinant | 4 (0.3) |
| Protein | N (%) |
|---|---|
| S | 31,182 (47.6) |
| ORF1a | 12,369 (18.9) |
| N | 6739 (10.3) |
| ORF1b | 4751 (7.2) |
| ORF9b | 3977 (6.1) |
| M | 2503 (3.8) |
| E | 1327 (2.0) |
| ORF3a | 905 (1.4) |
| ORF8 | 784 (1.2) |
| ORF7a | 429 (0.6) |
| ORF6 | 366 (0.5) |
| ORF7b | 158 (0.2) |
| Total | 65,490 |
| Protein | Functional Domain | Range | N (%) |
|---|---|---|---|
| Spike | Signal peptide | Residues 1–13 | 4 (0.01) |
| N-terminal S1 subunit | Residues 14–685 | 26,431 (63.3) | |
| C-terminal S2 subunit | Residues 686–1273 | 4757 (11.4) | |
| Nucleocapsid | N-arm | Residues 1–43 | 3497 (8.4) |
| NTD (RNA binding domain) | Residues 44–174 | 322 (0.8) | |
| SR-rich region | Residues 175–204 | 1966 (4.7) | |
| LKR | Residues 205–254 | 224 (0.5) | |
| CTD (dimerization domain) | Residues 255–364 | 56 (0.1) | |
| C-tail | Residues 365–419 | 679 (1.6) | |
| Membrane | N-terminal ectodomain | Residues 1–25 | 1366 (3.3) |
| Triple-membrane-spanning domain | Residues 26–101 | 1127 (2.7) | |
| C-terminal endodomain | Residues 102–222 | 11 (0.03) | |
| Envelope | N-terminal domain | Residues 1–8 | 0 |
| Transmembrane domain | Residues 9–38 | 1272 (3.1) | |
| C-terminal tail | Residues 39–75 | 55 (0.1) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alsuwairi, F.A.; Alsaleh, A.N.; Obeid, D.A.; Al-Qahtani, A.A.; Almaghrabi, R.S.; Alahideb, B.M.; AlAbdulkareem, M.A.; Alsanea, M.S.; Alharbi, L.A.; Althawadi, S.I.; et al. Genomic Surveillance and Mutation Analysis of SARS-CoV-2 Variants among Patients in Saudi Arabia. Microorganisms 2024, 12, 467. https://doi.org/10.3390/microorganisms12030467
Alsuwairi FA, Alsaleh AN, Obeid DA, Al-Qahtani AA, Almaghrabi RS, Alahideb BM, AlAbdulkareem MA, Alsanea MS, Alharbi LA, Althawadi SI, et al. Genomic Surveillance and Mutation Analysis of SARS-CoV-2 Variants among Patients in Saudi Arabia. Microorganisms. 2024; 12(3):467. https://doi.org/10.3390/microorganisms12030467
Chicago/Turabian StyleAlsuwairi, Feda A., Asma N. Alsaleh, Dalia A. Obeid, Ahmed A. Al-Qahtani, Reem S. Almaghrabi, Basma M. Alahideb, Maha A. AlAbdulkareem, Madain S. Alsanea, Layla A. Alharbi, Sahar I. Althawadi, and et al. 2024. "Genomic Surveillance and Mutation Analysis of SARS-CoV-2 Variants among Patients in Saudi Arabia" Microorganisms 12, no. 3: 467. https://doi.org/10.3390/microorganisms12030467
APA StyleAlsuwairi, F. A., Alsaleh, A. N., Obeid, D. A., Al-Qahtani, A. A., Almaghrabi, R. S., Alahideb, B. M., AlAbdulkareem, M. A., Alsanea, M. S., Alharbi, L. A., Althawadi, S. I., Altamimi, S. A., Alshukairi, A. N., & Alhamlan, F. S. (2024). Genomic Surveillance and Mutation Analysis of SARS-CoV-2 Variants among Patients in Saudi Arabia. Microorganisms, 12(3), 467. https://doi.org/10.3390/microorganisms12030467