

Early Circulating Edema Factor in Inhalational Anthrax Infection: Does It Matter?

 and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Bacterial Strains
2.2. Reagents and Antibodies
2.3. Cloning, Expression, and Purification of Fluorescent Edema Factor
2.4. Effect of Inhibitors on Cell Viability and Endocytosis Pathways
2.5. Confocal Microscopy
2.6. Flow Cytometry Analysis
2.7. cAMP Assay
2.8. Statistical Analysis
3. Results
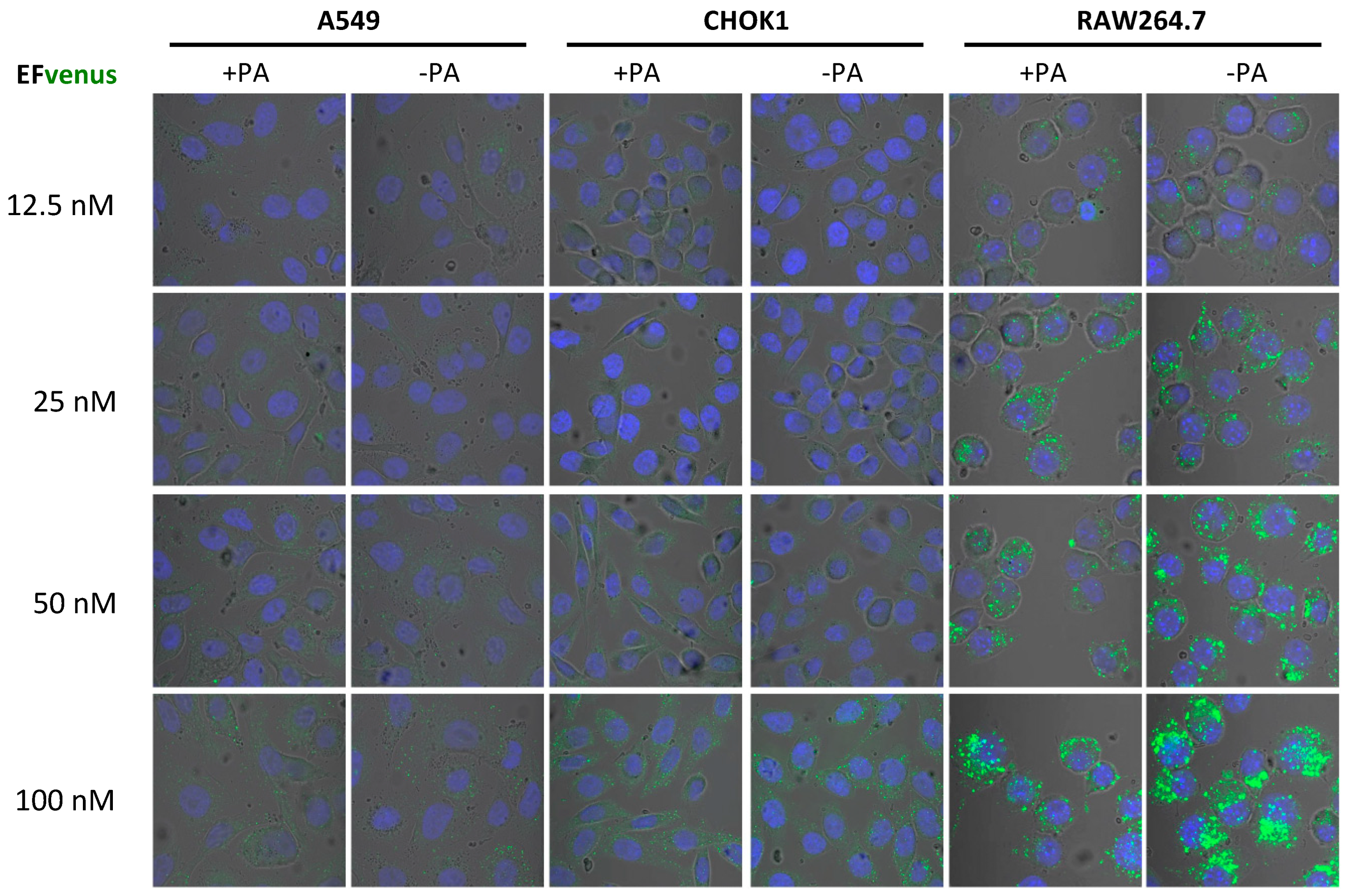
3.1. EFvenus Fluorescent Chimera Alone Could Be Found Inside Cells
3.1.1. The Chimera EFvenus Shows Signal Specificity
3.1.2. EFvenus Alone Enters Cells and Shows Pole-Specificity
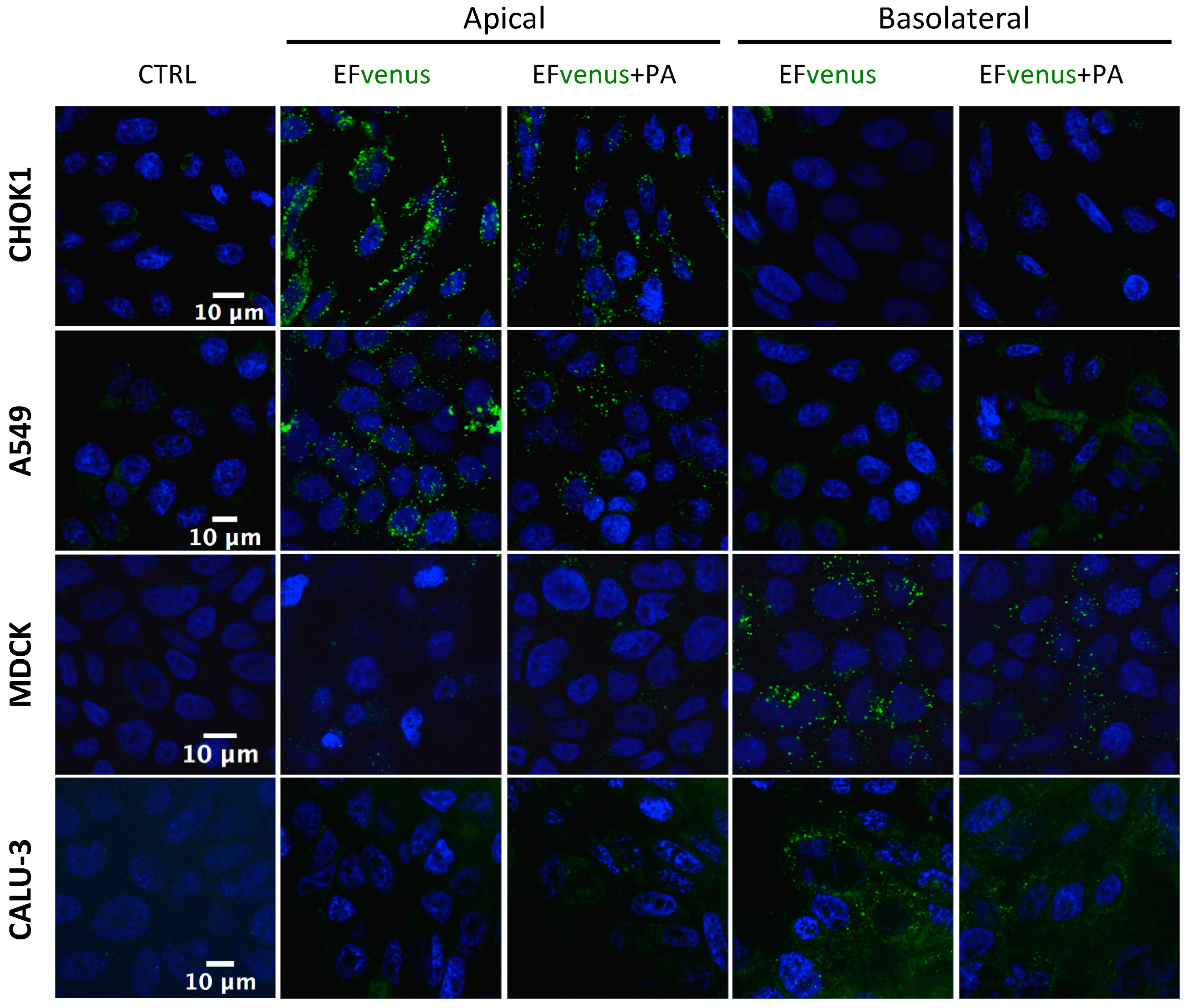
3.1.3. EFvenus Displays a Pole-Specificity in Epithelial Cells, Independently of the Presence of PA
3.2. Entry of the Fluorescent EFvenus Chimera Is Mediated by a Dynamic Process
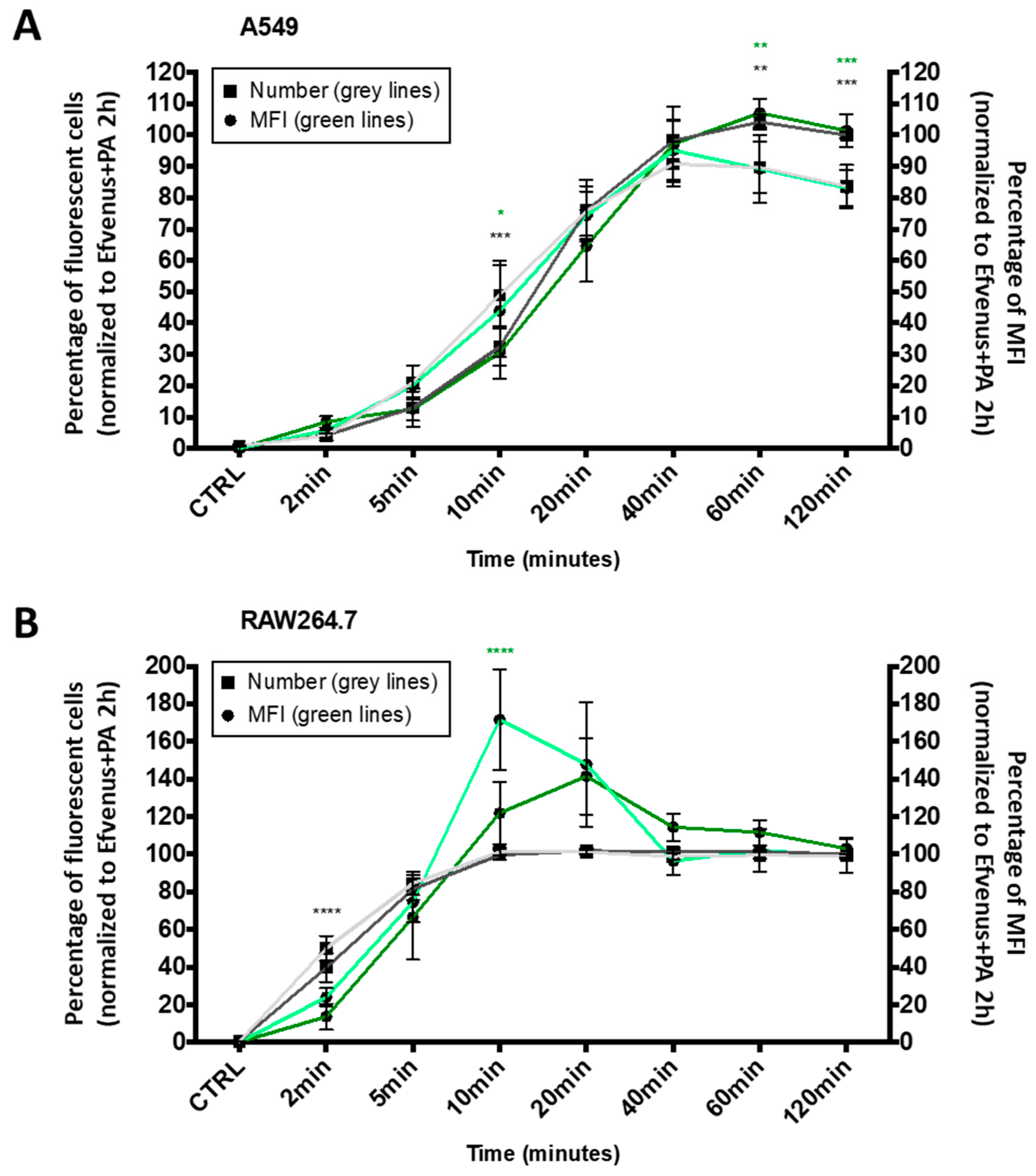
3.2.1. EFvenus Enters Cells Rapidly
3.2.2. EFvenus Is Endocytosed and Follows the Same Endosomal Trafficking as Efvenus + PA
3.3. EF Requirs PA for Intracellular Activity
3.3.1. EFvenus Displays an Enzymatic Activity in the Presence of PA Only
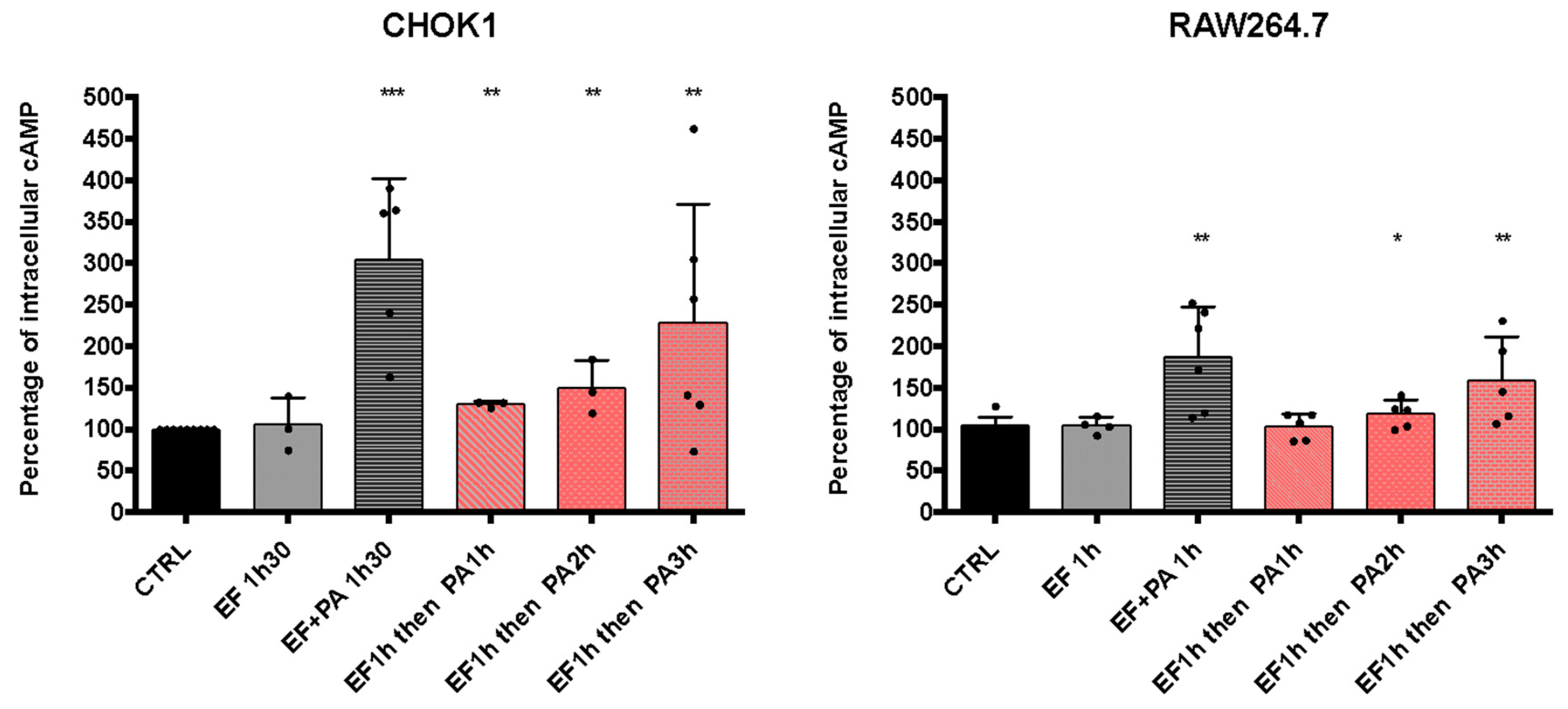
3.3.2. Sequential Entry of EF and PA Allows an Increase in cAMP Levels
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Baldwin, V.M. You Can’t B. cereus—A Review of Bacillus cereus Strains That Cause Anthrax-Like Disease. Front. Microbiol. 2020, 11, 1731. [Google Scholar] [CrossRef]
- Hoffmaster, A.R.; Ravel, J.; Rasko, D.A.; Chapman, G.D.; Chute, M.D.; Marston, C.K.; De, B.K.; Sacchi, C.T.; Fitzgerald, C.; Mayer, L.W.; et al. Identification of anthrax toxin genes in a Bacillus cereus associated with an illness resembling inhalation anthrax. Proc. Natl. Acad. Sci. USA 2004, 101, 8449–8454. [Google Scholar] [CrossRef]
- Klee, S.R.; Ozel, M.; Appel, B.; Boesch, C.; Ellerbrok, H.; Jacob, D.; Holland, G.; Leendertz, F.H.; Pauli, G.; Grunow, R.; et al. Characterization of Bacillus anthracis-like bacteria isolated from wild great apes from Cote d’Ivoire and Cameroon. J. Bacteriol. 2006, 188, 5333–5344. [Google Scholar] [CrossRef]
- Leendertz, F.H.; Yumlu, S.; Pauli, G.; Boesch, C.; Couacy-Hymann, E.; Vigilant, L.; Junglen, S.; Schenk, S.; Ellerbrok, H. A new Bacillus anthracis found in wild chimpanzees and a gorilla from West and Central Africa. PLoS Pathog. 2006, 2, e8. [Google Scholar] [CrossRef]
- Leendertz, F.H.; Ellerbrok, H.; Boesch, C.; Couacy-Hymann, E.; Mätz-Rensing, K.; Hakenbeck, R.; Bergmann, C.; Abaza, P.; Junglen, S.; Moebius, Y.; et al. Anthrax kills wild chimpanzees in a tropical rainforest. Nature 2004, 430, 451–452. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, C.; Zimmermann, F.; Biek, R.; Kuehl, H.; Nowak, K.; Mundry, R.; Agbor, A.; Angedakin, S.; Arandjelovic, M.; Blankenburg, A.; et al. Persistent anthrax as a major driver of wildlife mortality in a tropical rainforest. Nature 2017, 548, 82–86. [Google Scholar] [CrossRef]
- Drum, C.L.; Yan, S.-Z.; Bard, J.; Shen, Y.-Q.; Lu, D.; Soelaiman, S.; Grabarek, Z.; Bohm, A.; Tang, W.-J. Structural basis for the activation of anthrax adenylyl cyclase exotoxin by calmodulin. Nature 2002, 415, 396–402. [Google Scholar] [CrossRef]
- Leppla, S.H. Anthrax toxin edema factor: A bacterial adenylate cyclase that increases cyclic AMP concentrations of eukaryotic cells. Proc. Natl. Acad. Sci. USA 1982, 79, 3162–3166. [Google Scholar] [CrossRef] [PubMed]
- Duesbery, N.S.; Webb, C.P.; Leppla, S.H.; Gordon, V.M.; Klimpel, K.R.; Copeland, T.D.; Ahn, N.G.; Oskarsson, M.K.; Fukasawa, K.; Paull, K.D.; et al. Proteolytic inactivation of MAP-kinase-kinase by anthrax lethal factor. Science 1998, 280, 734–737. [Google Scholar] [CrossRef]
- Vitale, G.; Pellizzari, R.; Recchi, C.; Napolitani, G.; Mock, M.; Montecucco, C. Anthrax lethal factor cleaves the N-terminus of MAPKKs and induces tyrosine/threonine phosphorylation of MAPKs in cultured macrophages. Biochem. Biophys. Res. Commun. 1998, 248, 706–711. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Wilcox-Adelman, S.; Sano, Y.; Tang, W.-J.; Collier, R.J.; Park, J.M. Antiinflammatory cAMP signaling and cell migration genes co-opted by the anthrax bacillus. Proc. Natl. Acad. Sci. USA 2008, 105, 6150–6155. [Google Scholar] [CrossRef]
- Park, J.M.; Greten, F.R.; Wong, A.; Westrick, R.J.; Arthur, J.S.C.; Otsu, K.; Hoffmann, A.; Montminy, M.; Karin, M. Signaling Pathways and Genes that Inhibit Pathogen-Induced Macrophage Apoptosis— CREB and NF-κB as Key Regulators. Immunity 2005, 23, 319–329. [Google Scholar] [CrossRef]
- Puhar, A.; Dal Molin, F.; Horvath, S.; Ladant, D.; Ladants, D.; Montecucco, C. Anthrax edema toxin modulates PKA- and CREB-dependent signaling in two phases. PLoS ONE 2008, 3, e3564. [Google Scholar] [CrossRef]
- Raymond, B.; Leduc, D.; Ravaux, L.; Le Goffic, R.; Candela, T.; Raymondjean, M.; Goossens, P.L.; Touqui, L. Edema toxin impairs anthracidal phospholipase A2 expression by alveolar macrophages. PLoS Pathog. 2007, 3, e187. [Google Scholar] [CrossRef] [PubMed]
- Tonello, F.; Montecucco, C. The anthrax lethal factor and its MAPK kinase-specific metalloprotease activity. Mol. Aspects Med. 2009, 30, 431–438. [Google Scholar] [CrossRef]
- Liu, S.; Moayeri, M.; Leppla, S.H. Anthrax lethal and edema toxins in anthrax pathogenesis. Trends Microbiol. 2014, 22, 317–325. [Google Scholar] [CrossRef]
- Moayeri, M.; Leppla, S.H.; Vrentas, C.; Pomerantsev, A.P.; Liu, S. Anthrax Pathogenesis. Annu. Rev. Microbiol. 2015, 69, 185–208. [Google Scholar] [CrossRef]
- Bradley, K.A.; Mogridge, J.; Mourez, M.; Collier, R.J.; Young, J.A. Identification of the cellular receptor for anthrax toxin. Nature 2001, 414, 225–229. [Google Scholar] [CrossRef] [PubMed]
- Scobie, H.M.; Rainey, G.J.A.; Bradley, K.A.; Young, J.A.T. Human capillary morphogenesis protein 2 functions as an anthrax toxin receptor. Proc. Natl. Acad. Sci. USA 2003, 100, 5170–5174. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Crown, D.; Miller-Randolph, S.; Moayeri, M.; Wang, H.; Hu, H.; Morley, T.; Leppla, S.H. Capillary morphogenesis protein-2 is the major receptor mediating lethality of anthrax toxin in vivo. Proc. Natl. Acad. Sci. USA 2009, 106, 12424–12429. [Google Scholar] [CrossRef]
- Liu, S.; Zhang, Y.; Hoover, B.; Leppla, S.H. The Receptors that Mediate the Direct Lethality of Anthrax Toxin. Toxins 2012, 5, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Scobie, H.M.; Wigelsworth, D.J.; Marlett, J.M.; Thomas, D.; Rainey, G.J.A.; Lacy, D.B.; Manchester, M.; Collier, R.J.; Young, J.A.T. Anthrax toxin receptor 2-dependent lethal toxin killing in vivo. PLoS Pathog. 2006, 2, e111. [Google Scholar] [CrossRef] [PubMed]
- Martchenko, M.; Jeong, S.-Y.; Cohen, S.N. Heterodimeric integrin complexes containing beta1-integrin promote internalization and lethality of anthrax toxin. Proc. Natl. Acad. Sci. USA 2010, 107, 15583–15588. [Google Scholar] [CrossRef] [PubMed]
- Ezzell, J.W.; Abshire, T.G. Serum protease cleavage of Bacillus anthracis protective antigen. J. Gen. Microbiol. 1992, 138, 543–549. [Google Scholar] [CrossRef]
- Panchal, R.G.; Halverson, K.M.; Ribot, W.; Lane, D.; Kenny, T.; Abshire, T.G.; Ezzell, J.W.; Hoover, T.A.; Powell, B.; Little, S.; et al. Purified Bacillus anthracis lethal toxin complex formed in vitro and during infection exhibits functional and biological activity. J. Biol. Chem. 2005, 280, 10834–10839. [Google Scholar] [CrossRef]
- Moayeri, M.; Wiggins, J.F.; Leppla, S.H. Anthrax Protective Antigen Cleavage and Clearance from the Blood of Mice and Rats. Infect. Immun. 2007, 75, 5175–5184. [Google Scholar] [CrossRef]
- Klimpel, K.R.; Molloy, S.S.; Thomas, G.; Leppla, S.H. Anthrax toxin protective antigen is activated by a cell surface protease with the sequence specificity and catalytic properties of furin. Proc. Natl. Acad. Sci. USA 1992, 89, 10277–10281. [Google Scholar] [CrossRef]
- Novak, J.M.; Stein, M.P.; Little, S.F.; Leppla, S.H.; Friedlander, A.M. Functional characterization of protease-treated Bacillus anthracis protective antigen. J. Biol. Chem. 1992, 267, 17186–17193. [Google Scholar] [CrossRef]
- Milne, J.C.; Furlong, D.; Hanna, P.C.; Wall, J.S.; Collier, R.J. Anthrax protective antigen forms oligomers during intoxication of mammalian cells. J. Biol. Chem. 1994, 269, 20607–20612. [Google Scholar] [CrossRef]
- Petosa, C.; Collier, R.J.; Klimpel, K.R.; Leppla, S.H.; Liddington, R.C. Crystal structure of the anthrax toxin protective antigen. Nature 1997, 385, 833–838. [Google Scholar] [CrossRef]
- Kintzer, A.F.; Thoren, K.L.; Sterling, H.J.; Dong, K.C.; Feld, G.K.; Tang, I.I.; Zhang, T.T.; Williams, E.R.; Berger, J.M.; Krantz, B.A. The protective antigen component of anthrax toxin forms functional octameric complexes. J. Mol. Biol. 2009, 392, 614–629. [Google Scholar] [CrossRef]
- Abrami, L.; Bischofberger, M.; Kunz, B.; Groux, R.; van der Goot, F.G. Endocytosis of the anthrax toxin is mediated by clathrin, actin and unconventional adaptors. PLoS Pathog. 2010, 6, e1000792. [Google Scholar] [CrossRef] [PubMed]
- Abrami, L.; Liu, S.; Cosson, P.; Leppla, S.H.; van der Goot, F.G. Anthrax toxin triggers endocytosis of its receptor via a lipid raft-mediated clathrin-dependent process. J. Cell Biol. 2003, 160, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Young, J.A.T.; Collier, R.J. Anthrax toxin: Receptor binding, internalization, pore formation, and translocation. Annu. Rev. Biochem. 2007, 76, 243–265. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.J.; Elliott, J.L.; Collier, R.J. Anthrax protective antigen: Prepore-to-pore conversion. Biochemistry 1999, 38, 10432–10441. [Google Scholar] [CrossRef] [PubMed]
- Wesche, J.; Elliott, J.L.; Falnes, P.Ø.; Olsnes, S.; Collier, R.J. Characterization of Membrane Translocation by Anthrax Protective Antigen. Biochemistry 1998, 37, 15737–15746. [Google Scholar] [CrossRef]
- Milne, J.C.; Collier, R.J. pH-dependent permeabilization of the plasma membrane of mammalian cells by anthrax protective antigen. Mol. Microbiol. 1993, 10, 647–653. [Google Scholar] [CrossRef] [PubMed]
- Zornetta, I.; Brandi, L.; Janowiak, B.; Dal Molin, F.; Tonello, F.; Collier, R.J.; Montecucco, C. Imaging the cell entry of the anthrax oedema and lethal toxins with fluorescent protein chimeras. Cell. Microbiol. 2010, 12, 1435–1445. [Google Scholar] [CrossRef] [PubMed]
- Guidi-Rontani, C.; Weber-Levy, M.; Mock, M.; Cabiaux, V. Translocation of Bacillus anthracis lethal and oedema factors across endosome membranes. Cell. Microbiol. 2000, 2, 259–264. [Google Scholar] [CrossRef] [PubMed]
- Dal Molin, F.; Tonello, F.; Ladant, D.; Zornetta, I.; Zamparo, I.; Di Benedetto, G.; Zaccolo, M.; Montecucco, C. Cell entry and cAMP imaging of anthrax edema toxin. EMBO J. 2006, 25, 5405–5413. [Google Scholar] [CrossRef] [PubMed]
- Abrami, L.; Brandi, L.; Moayeri, M.; Brown, M.J.; Krantz, B.A.; Leppla, S.H.; van der Goot, F.G. Hijacking multivesicular bodies enables long-term and exosome-mediated long-distance action of anthrax toxin. Cell Rep. 2013, 5, 986–996. [Google Scholar] [CrossRef]
- Rivera, J.; Cordero, R.J.B.; Nakouzi, A.S.; Frases, S.; Nicola, A.; Casadevall, A. Bacillus anthracis produces membrane-derived vesicles containing biologically active toxins. Proc. Natl. Acad. Sci. USA 2010, 107, 19002–19007. [Google Scholar] [CrossRef] [PubMed]
- Tournier, J.-N.; Rossi Paccani, S.; Quesnel-Hellmann, A.; Baldari, C.T. Anthrax toxins: A weapon to systematically dismantle the host immune defenses. Mol. Aspects Med. 2009, 30, 456–466. [Google Scholar] [CrossRef] [PubMed]
- Moayeri, M.; Leppla, S.H. Cellular and systemic effects of anthrax lethal toxin and edema toxin. Mol. Aspects Med. 2009, 30, 439–455. [Google Scholar] [CrossRef]
- Lowe, D.E.; Glomski, I.J. Cellular and Physiological Effects of Anthrax Exotoxin and Its Relevance to Disease. Front. Cell. Infect. Microbiol. 2012, 2, 76. [Google Scholar] [CrossRef] [PubMed]
- Maddugoda, M.P.; Stefani, C.; Gonzalez-Rodriguez, D.; Saarikangas, J.; Torrino, S.; Janel, S.; Munro, P.; Doye, A.; Prodon, F.; Aurrand-Lions, M.; et al. cAMP signaling by anthrax edema toxin induces transendothelial cell tunnels, which are resealed by MIM via Arp2/3-driven actin polymerization. Cell Host Microbe 2011, 10, 464–474. [Google Scholar] [CrossRef]
- Liu, S.; Zhang, Y.; Moayeri, M.; Liu, J.; Crown, D.; Fattah, R.J.; Wein, A.N.; Yu, Z.-X.; Finkel, T.; Leppla, S.H. Key tissue targets responsible for anthrax-toxin-induced lethality. Nature 2013, 501, 63–68. [Google Scholar] [CrossRef]
- Lovchik, J.A.; Drysdale, M.; Koehler, T.M.; Hutt, J.A.; Lyons, C.R. Expression of either Lethal Toxin or Edema Toxin by Bacillus anthracis Is Sufficient for Virulence in a Rabbit Model of Inhalational Anthrax. Infect. Immun. 2012, 80, 2414–2425. [Google Scholar] [CrossRef] [PubMed]
- Hutt, J.A.; Lovchik, J.A.; Drysdale, M.; Sherwood, R.L.; Brasel, T.; Lipscomb, M.F.; Lyons, C.R. Lethal factor, but not edema factor, is required to cause fatal anthrax in cynomolgus macaques after pulmonary spore challenge. Am. J. Pathol. 2014, 184, 3205–3216. [Google Scholar] [CrossRef]
- Lins, R.C.; Boyer, A.E.; Kuklenyik, Z.; Woolfitt, A.R.; Goldstein, J.; Hoffmaster, A.R.; Gallegos-Candela, M.; Leysath, C.E.; Chen, Z.; Brumlow, J.O.; et al. Zeptomole per milliliter detection and quantification of edema factor in plasma by LC-MS/MS yields insights into toxemia and the progression of inhalation anthrax. Anal. Bioanal. Chem. 2019, 411, 2493–2509. [Google Scholar] [CrossRef]
- Rougeaux, C.; Becher, F.; Goossens, P.L.; Tournier, J.-N. Very Early Blood Diffusion of the Active Lethal and Edema Factors of Bacillus anthracis After Intranasal Infection. J. Infect. Dis. 2020, 221, 660–667. [Google Scholar] [CrossRef]
- Rougeaux, C.; Becher, F.; Ezan, E.; Tournier, J.-N.; Goossens, P.L. In vivo dynamics of active edema and lethal factors during anthrax. Sci. Rep. 2016, 6, 23346. [Google Scholar] [CrossRef] [PubMed]
- Solano, M.I.; Woolfitt, A.R.; Boyer, A.E.; Lins, R.C.; Isbell, K.; Gallegos-Candela, M.; Moura, H.; Pierce, C.L.; Barr, J.R. Accurate and selective quantification of anthrax protective antigen in plasma by immunocapture and isotope dilution mass spectrometry. Analyst 2019, 144, 2264–2274. [Google Scholar] [CrossRef]
- Mabry, R.; Brasky, K.; Geiger, R.; Carrion, R.; Hubbard, G.B.; Leppla, S.; Patterson, J.L.; Georgiou, G.; Iverson, B.L. Detection of anthrax toxin in the serum of animals infected with Bacillus anthracis by using engineered immunoassays. Clin. Vaccine Immunol. CVI 2006, 13, 671–677. [Google Scholar] [CrossRef] [PubMed]
- Boyer, A.E.; Quinn, C.P.; Hoffmaster, A.R.; Kozel, T.R.; Saile, E.; Marston, C.K.; Percival, A.; Plikaytis, B.D.; Woolfitt, A.R.; Gallegos, M.; et al. Kinetics of Lethal Factor and Poly-d-Glutamic Acid Antigenemia during Inhalation Anthrax in Rhesus Macaques. Infect. Immun. 2009, 77, 3432–3441. [Google Scholar] [CrossRef] [PubMed]
- Kobiler, D.; Weiss, S.; Levy, H.; Fisher, M.; Mechaly, A.; Pass, A.; Altboum, Z. Protective antigen as a correlative marker for anthrax in animal models. Infect. Immun. 2006, 74, 5871–5876. [Google Scholar] [CrossRef] [PubMed]
- Molin, F.D.; Fasanella, A.; Simonato, M.; Garofolo, G.; Montecucco, C.; Tonello, F. Ratio of lethal and edema factors in rabbit systemic anthrax. Toxicon Off. J. Int. Soc. Toxinology 2008, 52, 824–828. [Google Scholar] [CrossRef] [PubMed]
- Tournier, J.-N.; Rougeaux, C. Anthrax Toxin Detection: From In Vivo Studies to Diagnostic Applications. Microorganisms 2020, 8, 1103. [Google Scholar] [CrossRef] [PubMed]
- Stanley, J.L.; Smith, H. Purification of factor I and recognition of a third factor of the anthrax toxin. J. Gen. Microbiol. 1961, 26, 49–63. [Google Scholar] [CrossRef]
- Friebe, S.; van der Goot, F.G.; Bürgi, J. The Ins and Outs of Anthrax Toxin. Toxins 2016, 8, E69. [Google Scholar] [CrossRef]
- Kumar, P.; Ahuja, N.; Bhatnagar, R. Purification of anthrax edema factor from Escherichia coli and identification of residues required for binding to anthrax protective antigen. Infect. Immun. 2001, 69, 6532–6536. [Google Scholar] [CrossRef]
- Little, S.F.; Leppla, S.H.; Cora, E. Production and characterization of monoclonal antibodies to the protective antigen component of Bacillus anthracis toxin. Infect. Immun. 1988, 56, 1807–1813. [Google Scholar] [CrossRef]
- Rennick, J.J.; Johnston, A.P.R.; Parton, R.G. Key principles and methods for studying the endocytosis of biological and nanoparticle therapeutics. Nat. Nanotechnol. 2021, 16, 266–276. [Google Scholar] [CrossRef]
- Beitzinger, C.; Stefani, C.; Kronhardt, A.; Rolando, M.; Flatau, G.; Lemichez, E.; Benz, R. Role of N-terminal His6-Tags in binding and efficient translocation of polypeptides into cells using anthrax protective antigen (PA). PLoS ONE 2012, 7, e46964. [Google Scholar] [CrossRef]
- Neumeyer, T.; Tonello, F.; Dal Molin, F.; Schiffler, B.; Orlik, F.; Benz, R. Anthrax lethal factor (LF) mediated block of the anthrax protective antigen (PA) ion channel: Effect of ionic strength and voltage. Biochemistry 2006, 45, 3060–3068. [Google Scholar] [CrossRef]
- Cranfill, P.J.; Sell, B.R.; Baird, M.A.; Allen, J.R.; Lavagnino, Z.; de Gruiter, H.M.; Kremers, G.-J.; Davidson, M.W.; Ustione, A.; Piston, D.W. Quantitative Assessment of Fluorescent Proteins. Nat. Methods 2016, 13, 557–562. [Google Scholar] [CrossRef]
- Day, R.N.; Davidson, M.W. The fluorescent protein palette: Tools for cellular imaging. Chem. Soc. Rev. 2009, 38, 2887–2921. [Google Scholar] [CrossRef] [PubMed]
- Fujikura, D.; Toyomane, K.; Kamiya, K.; Mutoh, M.; Mifune, E.; Ohnuma, M.; Higashi, H. ANTXR-1 and -2 independent modulation of a cytotoxicity mediated by anthrax toxin in human cells. J. Vet. Med. Sci. 2016, 78, 1311–1317. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Guichard, A.; Jain, P.; Moayeri, M.; Schwartz, R.; Chin, S.; Zhu, L.; Cruz-Moreno, B.; Liu, J.Z.; Aguilar, B.; Hollands, A.; et al. Anthrax edema toxin disrupts distinct steps in Rab11-dependent junctional transport. PLoS Pathog. 2017, 13, e1006603. [Google Scholar] [CrossRef] [PubMed]
- Powell, J.D.; Hutchison, J.R.; Hess, B.M.; Straub, T.M. Bacillus anthracis spores germinate extracellularly at air-liquid interface in an in vitro lung model under serum-free conditions. J. Appl. Microbiol. 2015, 119, 711–723. [Google Scholar] [CrossRef]
- Beauregard, K.E.; Wimer-Mackin, S.; Collier, R.J.; Lencer, W.I. Anthrax toxin entry into polarized epithelial cells. Infect. Immun. 1999, 67, 3026–3030. [Google Scholar] [CrossRef]
- Gordon, V.M.; Young, W.W.; Lechler, S.M.; Gray, M.C.; Leppla, S.H.; Hewlett, E.L. Adenylate cyclase toxins from Bacillus anthracis and Bordetella pertussis. Different processes for interaction with and entry into target cells. J. Biol. Chem. 1989, 264, 14792–14796. [Google Scholar] [CrossRef]
- Gordon, V.M.; Leppla, S.H.; Hewlett, E.L. Inhibitors of receptor-mediated endocytosis block the entry of Bacillus anthracis adenylate cyclase toxin but not that of Bordetella pertussis adenylate cyclase toxin. Infect. Immun. 1988, 56, 1066–1069. [Google Scholar] [CrossRef] [PubMed]
- Ménard, A.; Altendorf, K.; Breves, D.; Mock, M.; Montecucco, C. The vacuolar ATPase proton pump is required for the cytotoxicity of Bacillus anthracis lethal toxin. FEBS Lett. 1996, 386, 161–164. [Google Scholar] [CrossRef]
- Mock, M.; Labruyère, E.; Glaser, P.; Danchin, A.; Ullmann, A. Cloning and expression of the calmodulin-sensitive Bacillus anthracis adenylate cyclase in Escherichia coli. Gene 1988, 64, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Ahuja, N.; Bhatnagar, R. Anthrax Edema Toxin Requires Influx of Calcium for Inducing Cyclic AMP Toxicity in Target Cells. Infect. Immun. 2002, 70, 4997–5007. [Google Scholar] [CrossRef] [PubMed]
- Rainey, G.J.A.; Wigelsworth, D.J.; Ryan, P.L.; Scobie, H.M.; Collier, R.J.; Young, J.A.T. Receptor-specific requirements for anthrax toxin delivery into cells. Proc. Natl. Acad. Sci. USA 2005, 102, 13278–13283. [Google Scholar] [CrossRef]
- Khandia, R.; Pattnaik, B.; Rajukumar, K.; Pateriya, A.K.; Puranik, N.; Bhatia, S.; Murugkar, H. Identification of other Cellular Receptors for Edema Factor of Bacillus anthracis by Independent Inhibition of Protective Antigen Evidenced by Inhibition of Embryo Growth and Angiogenesis. Arch. Razi Inst. 2021, 76, 847–855. [Google Scholar] [CrossRef]
- Ménard, L.; Parker, P.J.; Kermorgant, S. Receptor tyrosine kinase c-Met controls the cytoskeleton from different endosomes via different pathways. Nat. Commun. 2014, 5, 3907. [Google Scholar] [CrossRef]
- Wang, X.M.; Wattiez, R.; Mock, M.; Falmagne, P.; Ruysschaert, J.M.; Cabiaux, V. Structure and interaction of PA63 and EF (edema toxin) of Bacillus anthracis with lipid membrane. Biochemistry 1997, 36, 14906–14913. [Google Scholar] [CrossRef]
- Kochi, S.K.; Martin, I.; Schiavo, G.; Mock, M.; Cabiaux, V. The effects of pH on the interaction of anthrax toxin lethal and edema factors with phospholipid vesicles. Biochemistry 1994, 33, 2604–2609. [Google Scholar] [CrossRef] [PubMed]
- Hasan, S.; Kulkarni, N.N.; Asbjarnarson, A.; Linhartova, I.; Osicka, R.; Sebo, P.; Gudmundsson, G.H. Bordetella pertussis Adenylate Cyclase Toxin Disrupts Functional Integrity of Bronchial Epithelial Layers. Infect. Immun. 2018, 86, e00445-17. [Google Scholar] [CrossRef]
- Donato, G.M.; Goldsmith, C.S.; Paddock, C.D.; Eby, J.C.; Gray, M.C.; Hewlett, E.L. Delivery of Bordetella pertussis adenylate cyclase toxin to target cells via outer membrane vesicles. FEBS Lett. 2012, 586, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.P.; Mintern, J.D.; Gleeson, P.A. Macropinocytosis in Different Cell Types: Similarities and Differences. Membranes 2020, 10, E177. [Google Scholar] [CrossRef] [PubMed]
- Steinman, R.M.; Brodie, S.E.; Cohn, Z.A. Membrane flow during pinocytosis. A stereologic analysis. J. Cell Biol. 1976, 68, 665–687. [Google Scholar] [CrossRef]
- Thottacherry, J.J.; Sathe, M.; Prabhakara, C.; Mayor, S. Spoiled for Choice: Diverse Endocytic Pathways Function at the Cell Surface. Annu. Rev. Cell Dev. Biol. 2019, 35, 55–84. [Google Scholar] [CrossRef]
- Lajoie, P.; Nabi, I.R. Lipid rafts, caveolae, and their endocytosis. Int. Rev. Cell Mol. Biol. 2010, 282, 135–163. [Google Scholar] [CrossRef]
- Johannes, L.; Mayor, S. Induced domain formation in endocytic invagination, lipid sorting, and scission. Cell 2010, 142, 507–510. [Google Scholar] [CrossRef]
- Lencer, W.I.; Moe, S.; Rufo, P.A.; Madara, J.L. Transcytosis of cholera toxin subunits across model human intestinal epithelia. Proc. Natl. Acad. Sci. USA 1995, 92, 10094–10098. [Google Scholar] [CrossRef] [PubMed]
- Malyukova, I.; Murray, K.F.; Zhu, C.; Boedeker, E.; Kane, A.; Patterson, K.; Peterson, J.R.; Donowitz, M.; Kovbasnjuk, O. Macropinocytosis in Shiga toxin 1 uptake by human intestinal epithelial cells and transcellular transcytosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 296, G78–G92. [Google Scholar] [CrossRef]
- Saslowsky, D.E.; Te Welscher, Y.M.; Chinnapen, D.J.-F.; Wagner, J.S.; Wan, J.; Kern, E.; Lencer, W.I. Ganglioside GM1-mediated transcytosis of cholera toxin bypasses the retrograde pathway and depends on the structure of the ceramide domain. J. Biol. Chem. 2013, 288, 25804–25809. [Google Scholar] [CrossRef]
- Couesnon, A.; Pereira, Y.; Popoff, M.R. Receptor-mediated transcytosis of botulinum neurotoxin A through intestinal cell monolayers. Cell. Microbiol. 2008, 10, 375–387. [Google Scholar] [CrossRef]
- Lencer, W.I.; Delp, C.; Neutra, M.R.; Madara, J.L. Mechanism of cholera toxin action on a polarized human intestinal epithelial cell line: Role of vesicular traffic. J. Cell Biol. 1992, 117, 1197–1209. [Google Scholar] [CrossRef] [PubMed]
- Richard, J.F.; Mainguy, G.; Gibert, M.; Marvaud, J.C.; Stiles, B.G.; Popoff, M.R. Transcytosis of iota-toxin across polarized CaCo-2 cells. Mol. Microbiol. 2002, 43, 907–917. [Google Scholar] [CrossRef] [PubMed]
- Eby, J.C.; Ciesla, W.P.; Hamman, W.; Donato, G.M.; Pickles, R.J.; Hewlett, E.L.; Lencer, W.I. Selective Translocation of the Bordetella pertussis Adenylate Cyclase Toxin across the Basolateral Membranes of Polarized Epithelial Cells. J. Biol. Chem. 2010, 285, 10662–10670. [Google Scholar] [CrossRef] [PubMed]
- Kirby, J.E. Anthrax Lethal Toxin Induces Human Endothelial Cell Apoptosis. Infect. Immun. 2004, 72, 430–439. [Google Scholar] [CrossRef] [PubMed]
- Xie, T.; Auth, R.D.; Frucht, D.M. The Effects of Anthrax Lethal Toxin on Host Barrier Function. Toxins 2011, 3, 591–607. [Google Scholar] [CrossRef] [PubMed]
- Weiner, Z.P.; Glomski, I.J. Updating Perspectives on the Initiation of Bacillus anthracis Growth and Dissemination through Its Host. Infect. Immun. 2012, 80, 1626–1633. [Google Scholar] [CrossRef]
- Chitlaru, T.; Gat, O.; Gozlan, Y.; Ariel, N.; Shafferman, A. Differential Proteomic Analysis of the Bacillus anthracis Secretome: Distinct Plasmid and Chromosome CO2-Dependent Cross Talk Mechanisms Modulate Extracellular Proteolytic Activities. J. Bacteriol. 2006, 188, 3551–3571. [Google Scholar] [CrossRef]
- Chitlaru, T.; Gat, O.; Grosfeld, H.; Inbar, I.; Gozlan, Y.; Shafferman, A. Identification of in vivo-expressed immunogenic proteins by serological proteome analysis of the Bacillus anthracis secretome. Infect. Immun. 2007, 75, 2841–2852. [Google Scholar] [CrossRef] [PubMed]
- Bishop, B.L.; Lodolce, J.P.; Kolodziej, L.E.; Boone, D.L.; Tang, W.J. The role of anthrolysin O in gut epithelial barrier disruption during Bacillus anthracis infection. Biochem. Biophys. Res. Commun. 2010, 394, 254–259. [Google Scholar] [CrossRef]
- Bourdeau, R.W.; Malito, E.; Chenal, A.; Bishop, B.L.; Musch, M.W.; Villereal, M.L.; Chang, E.B.; Mosser, E.M.; Rest, R.F.; Tang, W.-J. Cellular functions and X-ray structure of anthrolysin O, a cholesterol-dependent cytolysin secreted by Bacillus anthracis. J. Biol. Chem. 2009, 284, 14645–14656. [Google Scholar] [CrossRef]
- Titball, R.W. Bacterial phospholipases. Symp. Ser. Soc. Appl. Microbiol. 1998, 27, 127S–137S. [Google Scholar] [PubMed]
- Heffernan, B.J.; Thomason, B.; Herring-Palmer, A.; Shaughnessy, L.; McDonald, R.; Fisher, N.; Huffnagle, G.B.; Hanna, P. Bacillus anthracis Phospholipases C Facilitate Macrophage-Associated Growth and Contribute to Virulence in a Murine Model of Inhalation Anthrax. Infect. Immun. 2006, 74, 3756–3764. [Google Scholar] [CrossRef]
- Kiss, A.L. Caveolae and the regulation of endocytosis. Adv. Exp. Med. Biol. 2012, 729, 14–28. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.; Zhang, G.; Li, J.; Chen, P.R. Monitoring endocytic trafficking of anthrax lethal factor by precise and quantitative protein labeling. Angew. Chem. Int. Ed. Engl. 2014, 53, 6449–6453. [Google Scholar] [CrossRef]
- Leysath, C.E.; Phillips, D.D.; Crown, D.; Fattah, R.J.; Moayeri, M.; Leppla, S.H. Anthrax edema factor toxicity is strongly mediated by the N-end rule. PLoS ONE 2013, 8, e74474. [Google Scholar] [CrossRef]
- Moffat, J.M.; Mintern, J.D.; Villadangos, J.A. Control of MHC II antigen presentation by ubiquitination. Curr. Opin. Immunol. 2013, 25, 109–114. [Google Scholar] [CrossRef]
- Huotari, J.; Helenius, A. Endosome maturation. EMBO J. 2011, 30, 3481–3500. [Google Scholar] [CrossRef] [PubMed]
- Abrami, L.; Lindsay, M.; Parton, R.G.; Leppla, S.H.; van der Goot, F.G. Membrane insertion of anthrax protective antigen and cytoplasmic delivery of lethal factor occur at different stages of the endocytic pathway. J. Cell Biol. 2004, 166, 645–651. [Google Scholar] [CrossRef]
- Wosen, J.E.; Mukhopadhyay, D.; Macaubas, C.; Mellins, E.D. Epithelial MHC Class II Expression and Its Role in Antigen Presentation in the Gastrointestinal and Respiratory Tracts. Front. Immunol. 2018, 9, 2144. [Google Scholar] [CrossRef] [PubMed]
- Kurosawa, S.; Myers, A.C.; Chen, L.; Wang, S.; Ni, J.; Plitt, J.R.; Heller, N.M.; Bochner, B.S.; Schleimer, R.P. Expression of the costimulatory molecule B7-H2 (inducible costimulator ligand) by human airway epithelial cells. Am. J. Respir. Cell Mol. Biol. 2003, 28, 563–573. [Google Scholar] [CrossRef] [PubMed]
- Lo, B.; Hansen, S.; Evans, K.; Heath, J.K.; Wright, J.R. Alveolar epithelial type II cells induce T cell tolerance to specific antigen. J. Immunol. Baltim. 2008, 180, 881–888. [Google Scholar] [CrossRef] [PubMed]
- Corbière, V.; Dirix, V.; Norrenberg, S.; Cappello, M.; Remmelink, M.; Mascart, F. Phenotypic characteristics of human type II alveolar epithelial cells suitable for antigen presentation to T lymphocytes. Respir. Res. 2011, 12, 15. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Sequence | Matrix | Target | Resulting Plasmid |
|---|---|---|---|---|
| Fwd-pQE60 | GGATCTCATCACCATCACC | pQE60 | pQE60 open with His-tag Cter | pQE60-cya-mVenus-His-tag |
| Rev-pQE60 | CATGGTTAATTTCTCCTCTTTAATGAATTCTGTG | |||
| Fwd-(pQE60)-Cya | AAGAGGAGAAATTAACCATGAATGAACATTACACTGAGAG | B. cereus G9241 genomic DNA | Cya gene | |
| Rev-Cya-(Fluo+) | TGCTCACCATGAGCTCTTTTTCATCAATAATTTTTTGGAAG | |||
| Fwd-(Cya)-Fluo+ | TGATGAAAAAGAGCTCATGGTGAGCAAGGGCGAG | pET28-mVenus | mVenus | |
| Rev-Fluo+-(pQE60) | ATGGTGATGGTGATGAGATCCCTTGTACAGCTCGTCCATGC | |||
| Fwd-pQE60Fluo | GCTGTACAAGTAAGCTTAATTAGCTGAGC | pQE60-EFmVenusHis | pQE60 open with His-Tag Nter | pQE60-His-tag-cya-mVenus |
| Rev-pQE60Cya NterHis | AATGTTCATTGTGATGGTGATGGTGATGCATGGTTAATTTCTCCTCTTTAATG | |||
| Fwd-EFvenus- NterHis | ATTAACCATGCATCACCATCACCATCACAATGAACATTACACTGAGAGTG | pQE60-EFmVenusHis | Cya-mVenus with His-Tag Nter, without His-Tag Cter | |
| Rev-EFvenus- Nter | ATTAAGCTTACTTGTACAGCTCGTCCATG |
| Inhibitors | Targets and Actions | RAW264.7 Macrophages | A549 Epithelial Cells | ||
|---|---|---|---|---|---|
| Concentration | Viability | Concentration | Viability | ||
| Bafilomycin A | Endosomal acidification (vacuolar H+-ATPases) | 0.4 µM | 92.9% | 0.4 µM | 98.5% |
| Chloroquine | Endosomal acidification (maturation) | 400 µM | 104.3% | 500 µM | 83.1% |
| Cytochalasin D | Actin polymerization (most endocytic pathways) | 10 µM | 93.5% | 5 µM | 75.7% |
| Nocodazole | Microtubules depolymerization (trafficking from early to late endosomes) | 5 µM | 51.2% | 10 µM | 95.9% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tessier, E.; Cheutin, L.; Garnier, A.; Vigne, C.; Tournier, J.-N.; Rougeaux, C. Early Circulating Edema Factor in Inhalational Anthrax Infection: Does It Matter? Microorganisms 2024, 12, 308. https://doi.org/10.3390/microorganisms12020308
Tessier E, Cheutin L, Garnier A, Vigne C, Tournier J-N, Rougeaux C. Early Circulating Edema Factor in Inhalational Anthrax Infection: Does It Matter? Microorganisms. 2024; 12(2):308. https://doi.org/10.3390/microorganisms12020308
Chicago/Turabian StyleTessier, Emilie, Laurence Cheutin, Annabelle Garnier, Clarisse Vigne, Jean-Nicolas Tournier, and Clémence Rougeaux. 2024. "Early Circulating Edema Factor in Inhalational Anthrax Infection: Does It Matter?" Microorganisms 12, no. 2: 308. https://doi.org/10.3390/microorganisms12020308
APA StyleTessier, E., Cheutin, L., Garnier, A., Vigne, C., Tournier, J.-N., & Rougeaux, C. (2024). Early Circulating Edema Factor in Inhalational Anthrax Infection: Does It Matter? Microorganisms, 12(2), 308. https://doi.org/10.3390/microorganisms12020308