
Ecological Interaction between Bacteriophages and Bacteria in Sub-Arctic Kongsfjorden Bay, Svalbard, Norway

 , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Metaviromic Analysis of DNA Viruses
2.2. Metabarcoding Analyses of Bacteria
2.3. Statistical Analysis
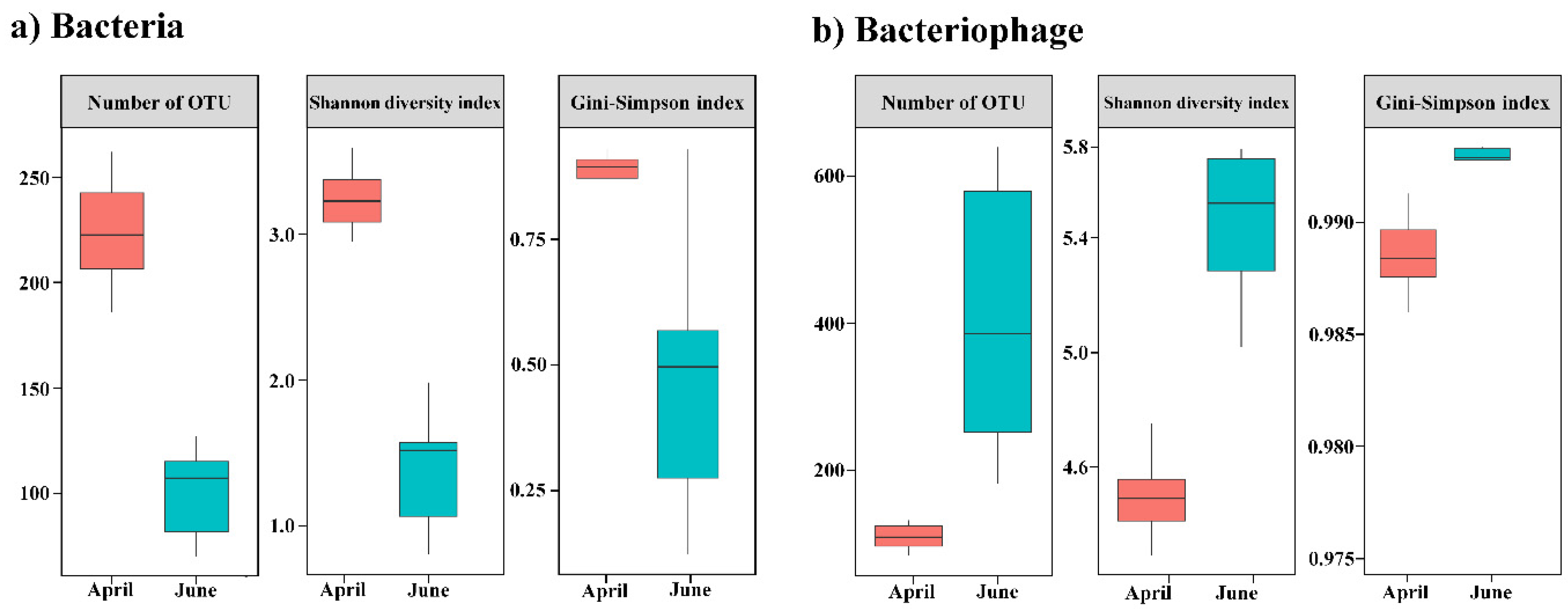
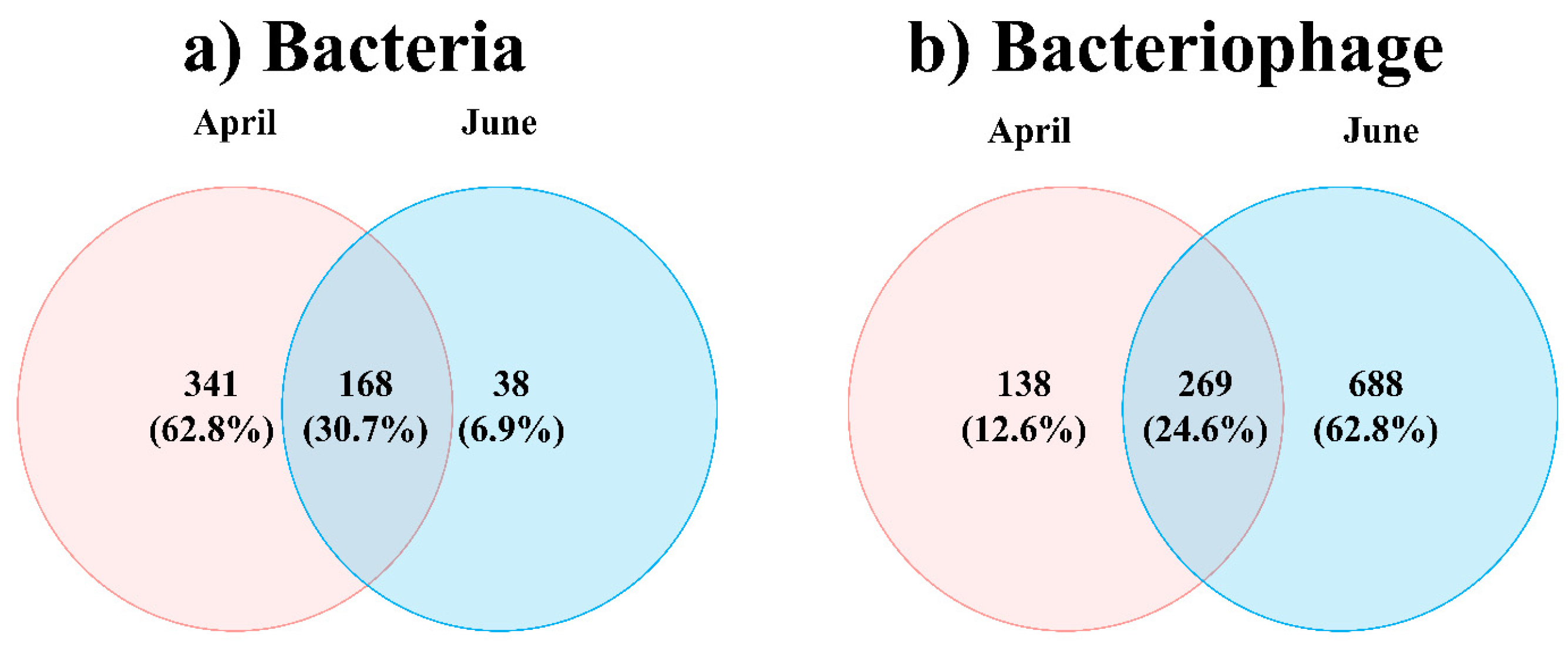
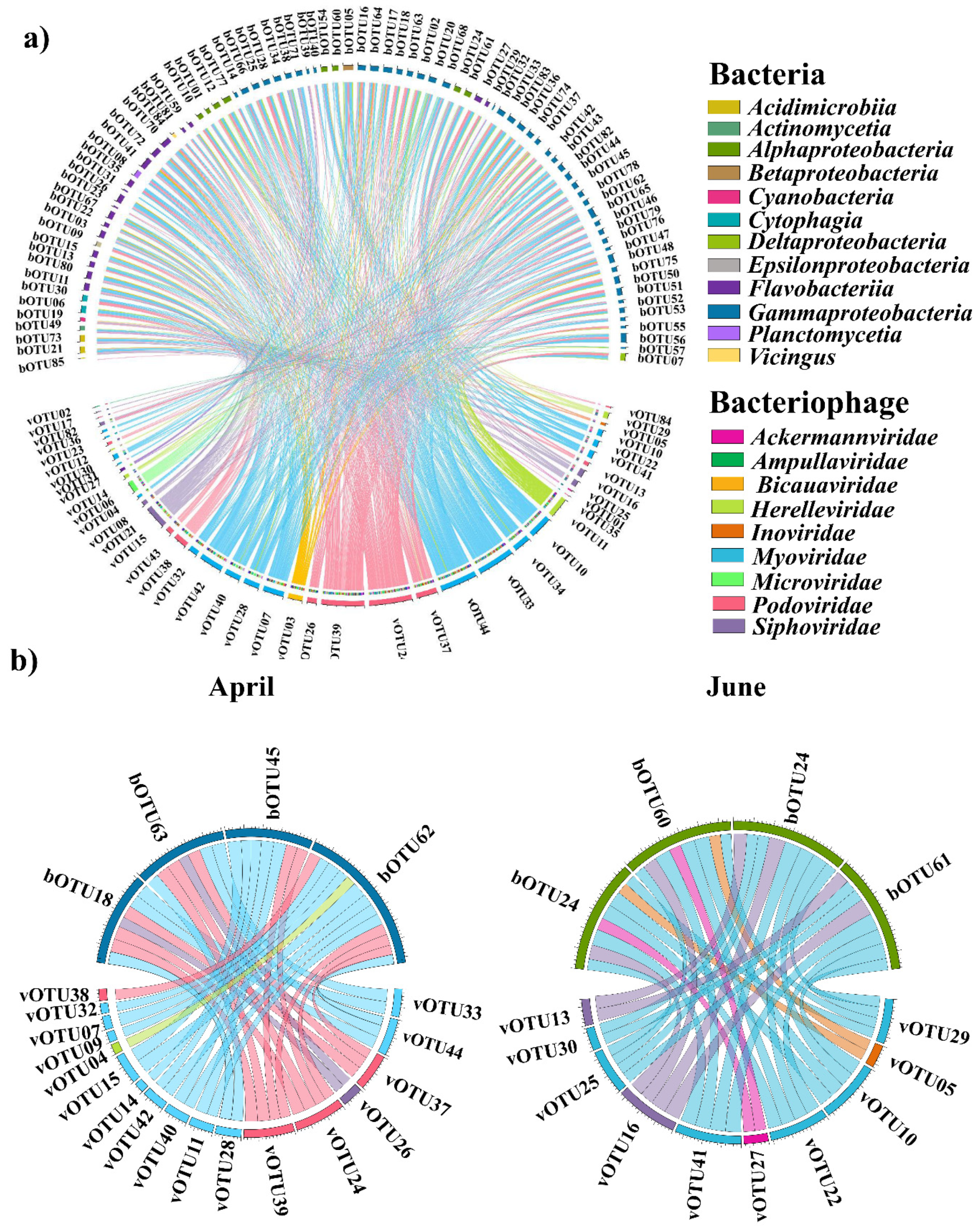
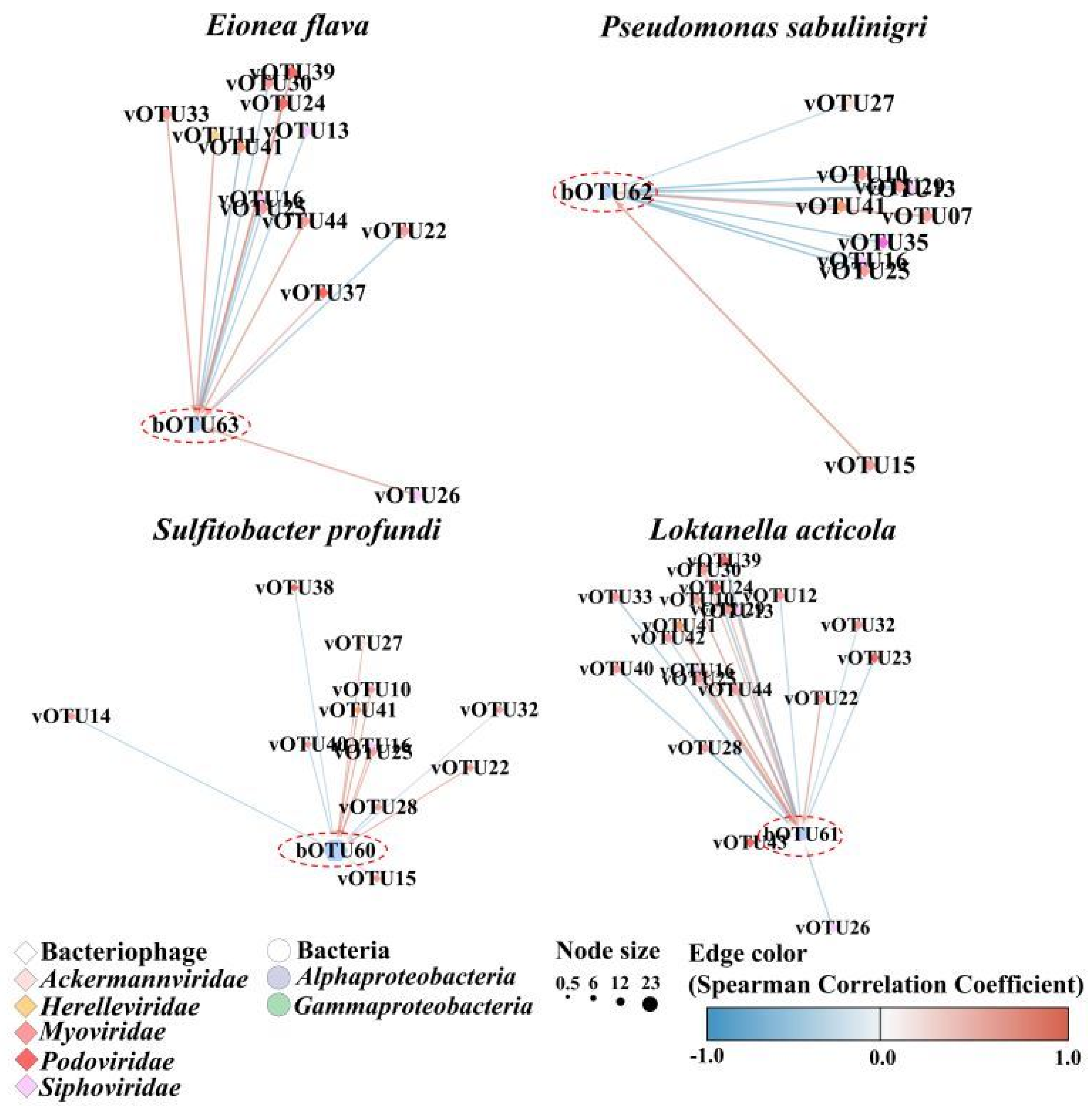
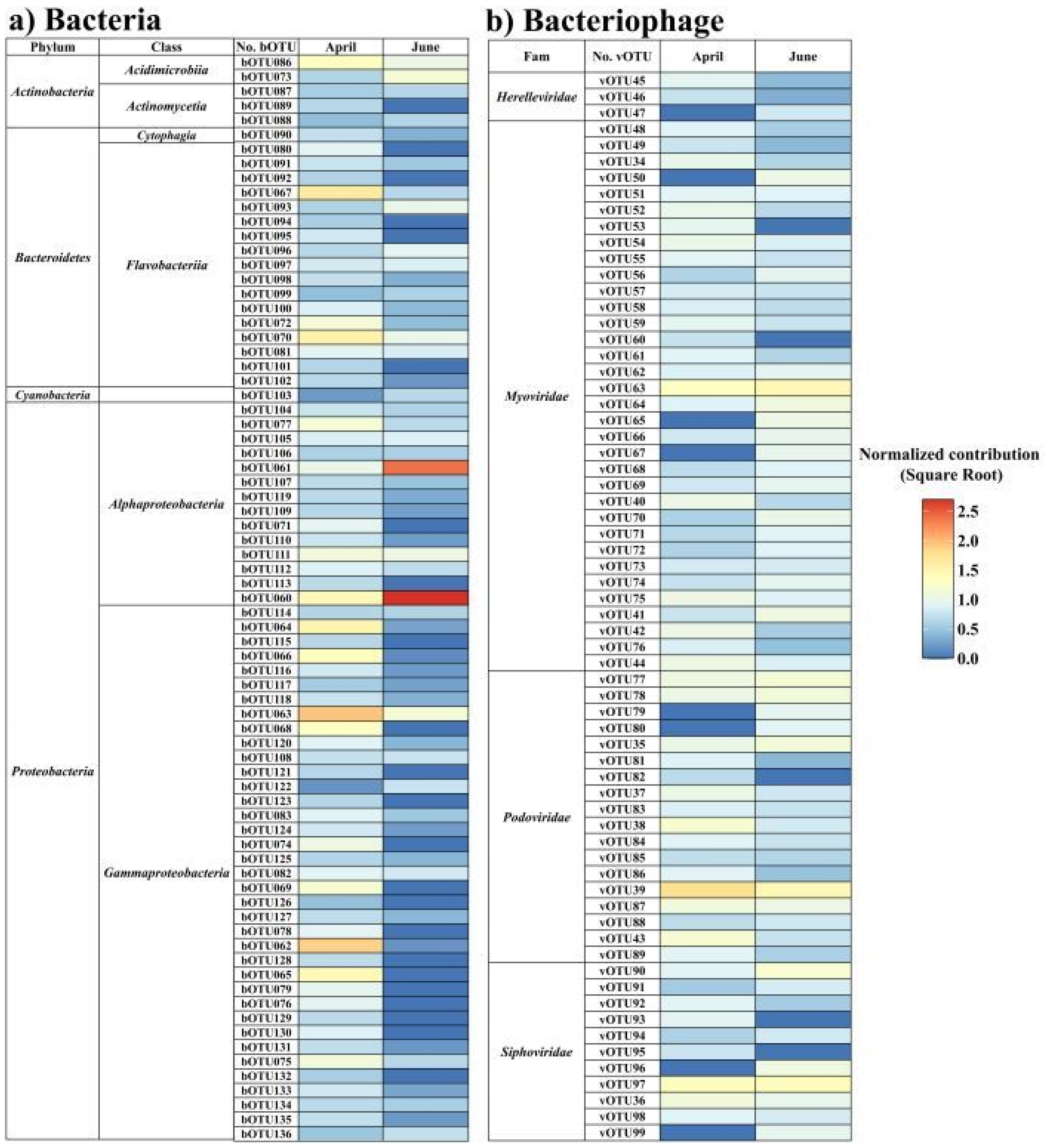
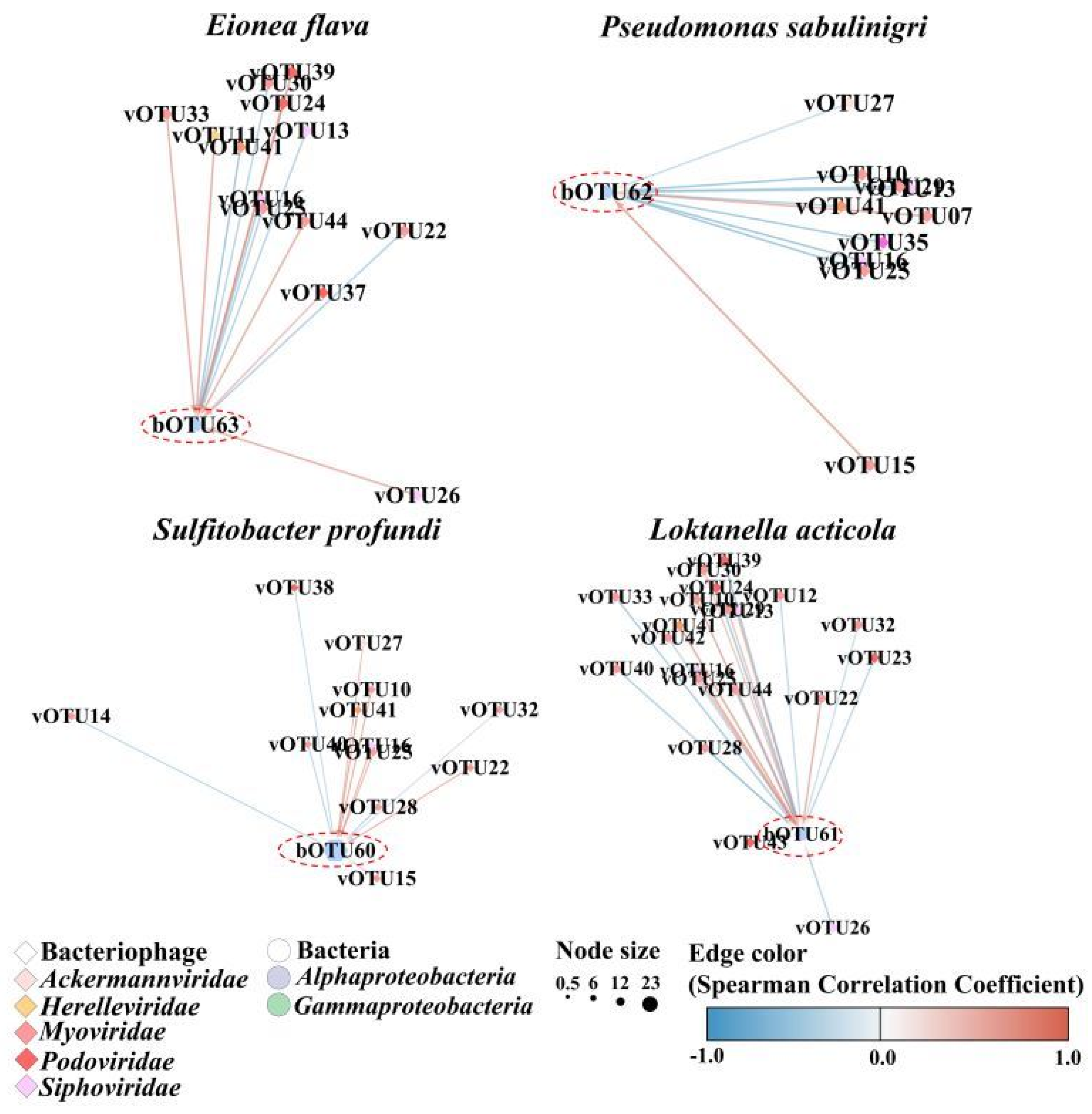
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Suttle, C.A. Marine Viruses—Major Players in the Global Ecosystem. Nat. Rev. Microbiol. 2007, 5, 801–812. [Google Scholar] [CrossRef]
- Edwards, R.A.; Rohwer, F. Viral Metagenomics. Nat. Rev. Microbiol. 2005, 3, 504–510. [Google Scholar] [CrossRef]
- Wommack, K.E.; Colwell, R.R. Virioplankton: Viruses in Aquatic Ecosystems. Microbiol. Mol. Biol. Rev. 2000, 64, 69–114. [Google Scholar] [CrossRef]
- Suttle, C.A. Viruses in the Sea. Nature 2005, 437, 356–361. [Google Scholar] [CrossRef]
- Zhu, Y.; Shang, J.; Peng, C.; Sun, Y. Phage Family Classification under Caudoviricetes: A Review of Current Tools Using the Latest ICTV Classification Framework. Front. Microbiol. 2022, 13, 1032186. [Google Scholar] [CrossRef]
- Jung, S.W.; Kim, K.E.; Kim, H.J.; Lee, T.K. Metavirome Profiling and Dynamics of the DNA Viral Community in Seawater in Chuuk State, Federated States Of Micronesia. Viruses 2023, 15, 1293. [Google Scholar] [CrossRef]
- Endo, H.; Blanc-Mathieu, R.; Li, Y.; Salazar, G.; Henry, N.; Labadie, K.; de Vargas, C.; Sullivan, M.B.; Bowler, C.; Wincker, P.; et al. Biogeography of Marine Giant Viruses Reveals Their Interplay with Eukaryotes and Ecological Functions. Nat. Ecol. Evol. 2020, 4, 1639–1649. [Google Scholar] [CrossRef]
- Weinbauer, M.G.; Rassoulzadegan, F. Are Viruses Driving Microbial Diversification and Diversity? Environ. Microbiol. 2004, 6, 1–11. [Google Scholar] [CrossRef]
- Weinbauer, M.G. Ecology of Prokaryotic Viruses. FEMS Microbiol. Rev. 2004, 28, 127–181. [Google Scholar] [CrossRef]
- Ptashne, M. Lambda’s Switch: Lessons from a Module Swap. Curr. Biol. 2006, 16, R459–R462. [Google Scholar] [CrossRef]
- Buchan, A.; LeCleir, G.R.; Gulvik, C.A.; González, J.M. Master Recyclers: Features and Functions of Bacteria Associated with Phytoplankton Blooms. Nat. Rev. Microbiol. 2014, 12, 686–698. [Google Scholar] [CrossRef]
- Worden, A.Z.; Follows, M.J.; Giovannoni, S.J.; Wilken, S.; Zimmerman, A.E.; Keeling, P.J. Environmental Science. Rethinking the Marine Carbon Cycle: Factoring in the Multifarious Lifestyles of Microbes. Science 2015, 347, 1257594. [Google Scholar] [CrossRef]
- Romaniuk, K.; Styczynski, M.; Decewicz, P.; Buraczewska, O.; Uhrynowski, W.; Fondi, M.; Wolosiewicz, M.; Szuplewska, M.; Dziewit, L. Diversity and Horizontal Transfer of Antarctic Pseudomonas spp. Plasmids. Genes 2019, 10, 850. [Google Scholar] [CrossRef]
- Wietz, M.; Bienhold, C.; Metfies, K.; Torres-Valdés, S.; von Appen, W.J.; Salter, I.; Boetius, A. The Polar Night Shift: Seasonal Dynamics and Drivers of Arctic Ocean Microbiomes Revealed by Autonomous Sampling. ISME Commun. 2021, 1, 76. [Google Scholar] [CrossRef]
- Liu, Z.; Jiang, W.; Kim, C.; Peng, X.; Fan, C.; Wu, Y.; Xie, Z.; Peng, F. A Pseudomonas Lysogenic Bacteriophage Crossing the Antarctic and Arctic, Representing a New Genus of Autographiviridae. Int. J. Mol. Sci. 2023, 24, 7662. [Google Scholar] [CrossRef]
- Venger, M.P.; Kopylov, A.I.; Zabotkina, E.A.; Makarevich, P.R. The Influence of Viruses on Bacterioplankton of the Offshore and Coastal Parts of the Barents Sea. Russ. J. Mar. Biol. 2016, 42, 26–35. [Google Scholar] [CrossRef]
- Shirokolobova, T.I.; Zhichkin, A.P.; Venger, M.P.; Vodopyanova, V.V.; Moiseev, D.V. Bacteria and Viruses of the Ice-Free Aquatic Area of the Barents Sea at the Beginning of Polar Night. Dokl. Biol. Sci. 2016, 469, 182–186. [Google Scholar] [CrossRef]
- Payne, C.M.; Roesler, C.S. Characterizing the Influence of Atlantic Water Intrusion on Water Mass Formation and Phytoplankton Distribution in Kongsfjorden, Svalbard. Contin. Shelf Res. 2019, 191, 104005. [Google Scholar] [CrossRef]
- Kim, K.E.; Joo, H.M.; Lee, T.K.; Kim, H.J.; Kim, Y.J.; Kim, B.K.; Ha, S.Y.; Jung, S.W. Covariance of Marine Nucleocytoplasmic Large DNA Viruses with Eukaryotic Plankton Communities in the sub-Arctic Kongsfjorden Ecosystem: A Metagenomic Analysis of Marine Microbial Ecosystems. Microorganisms 2023, 11, 169. [Google Scholar] [CrossRef]
- Kim, K.E.; Jung, S.W.; Park, J.S.; Kim, H.-J.; Lee, C.; Ha, S.-Y.; Lee, T.-K. Optimized Metavirome Analysis of Marine DNA Virus Communities for Taxonomic Profiling. Ocean Sci. J. 2022, 57, 259–268. [Google Scholar] [CrossRef]
- Nurk, S.; Meleshko, D.; Korobeynikov, A.; Pevzner, P.A. metaSPAdes: A New Versatile Metagenomic Assembler. Genome Res. 2017, 27, 824–834. [Google Scholar] [CrossRef]
- Nayfach, S.; Camargo, A.P.; Schulz, F.; Eloe-Fadrosh, E.; Roux, S.; Kyrpides, N.C. CheckV Assesses the Quality and Completeness of Metagenome-Assembled Viral Genomes. Nat. Biotechnol. 2021, 39, 578–585. [Google Scholar] [CrossRef]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A Versatile Open Source Tool for Metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef]
- Roux, S.; Adriaenssens, E.M.; Dutilh, B.E.; Koonin, E.V.; Kropinski, A.M.; Krupovic, M.; Kuhn, J.H.; Lavigne, R.; Brister, J.R.; Varsani, A.; et al. Minimum Information About an Uncultivated Virus Genome (MIUViG). Nat. Biotechnol. 2019, 37, 29–37. [Google Scholar] [CrossRef]
- Bushnell, B. BBMap: A Fast, Accurate, Splice-Aware Aligner; No. LBNL-7065E; Ernest Orlando Lawrence Berkeley National Laboratory: Berkeley, CA, USA, 2014; Available online: https://sourceforge.net/projects/bbmap (accessed on 22 October 2023).
- Jeong, G.; Kim, H.J.; Kim, K.E.; Kim, Y.J.; Lee, T.K.; Shim, W.J.; Jung, S.W. Selective Attachment of Prokaryotes and Emergence of Potentially Pathogenic Prokaryotes on Four Plastic Surfaces: Adhesion Study in a Natural Marine Environment. Mar. Pollut. Bull. 2023, 193, 115149. [Google Scholar] [CrossRef]
- Li, R.W.; Connor, E.E.; Li, C.; Baldwin Vi, R.L.; Sparks, M.E. Characterization of the Rumen Microbiota of Pre-ruminant Calves Using Metagenomic Tools. Environ. Microbiol. 2012, 14, 129–139. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2. WIREs Comput. Stats. 2011, 3, 180–185. [Google Scholar] [CrossRef]
- Clarke, K.; Gorley, R. Getting Started with PRIMER v7; PRIMER-E; Plymouth Marine Laboratory: Plymouth, UK, 2015; Available online: https://www.primer-e.com/our-software/primer-version-7 (accessed on 22 March 2023).
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’Hara, R.; Simpson, G.L.; Solymos, P. Package ‘Vegan’. Community Ecology Package, Version 2. 2019. Available online: https://cran.r-project.org/package=vegan (accessed on 15 October 2023).
- Xia, L.C.; Steele, J.A.; Cram, J.A.; Cardon, Z.G.; Simmons, S.L.; Vallino, J.J.; Fuhrman, J.A.; Sun, F. Extended Local Similarity Analysis (eLSA) of Microbial Community and Other Time Series Data with Replicates. BMC Syst. Biol. 2011, 5 (Suppl. S2), S15. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Gilbert, J.A.; Steele, J.A.; Caporaso, J.G.; Steinbrück, L.; Reeder, J.; Temperton, B.; Huse, S.; McHardy, A.C.; Knight, R.; Joint, I.; et al. Defining Seasonal Marine Microbial Community Dynamics. ISME J. 2012, 6, 298–308. [Google Scholar] [CrossRef]
- Ghiglione, J.F.; Murray, A.E. Pronounced Summer to Winter Differences and Higher Wintertime Richness in Coastal Antarctic Marine Bacterioplankton. Environ. Microbiol. 2012, 14, 617–629. [Google Scholar] [CrossRef]
- Chen, X.; Wei, W.; Wang, J.; Li, H.; Sun, J.; Ma, R.; Jiao, N.; Zhang, R. Tide Driven Microbial Dynamics Through Virus-Host Interactions in the Estuarine Ecosystem. Water Res. 2019, 160, 118–129. [Google Scholar] [CrossRef]
- Jiao, N.; Herndl, G.J.; Hansell, D.A.; Benner, R.; Kattner, G.; Wilhelm, S.W.; Kirchman, D.L.; Weinbauer, M.G.; Luo, T.; Chen, F.; et al. Microbial Production of Recalcitrant Dissolved Organic Matter: Long-Term Carbon Storage in the Global Ocean. Nat. Rev. Microbiol. 2010, 8, 593–599. [Google Scholar] [CrossRef]
- Yau, S.; Seth-Pasricha, M. Viruses of Polar Aquatic Environments. Viruses 2019, 11, 189. [Google Scholar] [CrossRef]
- Azam, F.; Malfatti, F. Microbial structuring of marine ecosystems. Nat. Rev. Microbiol. 2007, 5, 782–791. [Google Scholar] [CrossRef]
- Young, R. Phage Lysis: Three Steps, Three Choices, One Outcome. J. Microbiol. 2014, 52, 243–258. [Google Scholar] [CrossRef]
- Weitz, J.S.; Beckett, S.J.; Brum, J.R.; Cael, B.B.; Dushoff, J. Lysis, Lysogeny and Virus–Microbe Ratios. Nature 2017, 549, E1–E3. [Google Scholar] [CrossRef]
- Howard-Varona, C.; Hargreaves, K.R.; Abedon, S.T.; Sullivan, M.B. Lysogeny in Nature: Mechanisms, Impact and Ecology of Temperate Phages. ISME J. 2017, 11, 1511–1520. [Google Scholar] [CrossRef]
- Williamson, S.J.; Houchin, L.A.; McDaniel, L.; Paul, J.H. Seasonal Variation in Lysogeny as Depicted by Prophage Induction in Tampa Bay, Florida. Appl. Environ. Microbiol. 2002, 68, 4307–4314. [Google Scholar] [CrossRef]
- Payet, J.P.; Suttle, C.A. To Kill or Not to Kill: The Balance Between Lytic and Lysogenic Viral Infection Is Driven by Trophic Status. Limnol. Oceanogr. 2013, 58, 465–474. [Google Scholar] [CrossRef]
- Brum, J.R.; Hurwitz, B.L.; Schofield, O.; Ducklow, H.W.; Sullivan, M.B. Seasonal Time Bombs: Dominant Temperate Viruses Affect Southern Ocean Microbial Dynamics. ISME J. 2016, 10, 437–449. [Google Scholar] [CrossRef]
- Cao, S.; Zhang, W.; Ding, W.; Wang, M.; Fan, S.; Yang, B.; McMinn, A.; Wang, M.; Xie, B.B.; Qin, Q.L.; et al. Structure and Function of the Arctic and Antarctic Marine Microbiota as Revealed by Metagenomics. Microbiome 2020, 8, 47. [Google Scholar] [CrossRef]
- Urios, L.; Intertaglia, L.; Lesongeur, F.; Lebaron, P. Eionea nigra gen. nov., sp. nov., a Gammaproteobacterium from the Mediterranean Sea. Int. J. Syst. Evol. Microbiol. 2011, 61, 1677–1681. [Google Scholar] [CrossRef]
- Winder, J.C.; Boulton, W.; Salamov, A.; Eggers, S.L.; Metfies, K.; Moulton, V.; Mock, T. Genetic and Structural Diversity of Prokaryotic Ice-Binding Proteins from the Central Arctic Ocean. Genes 2023, 14, 363. [Google Scholar] [CrossRef]
- Barak-Gavish, N.; Frada, M.J.; Ku, C.; Lee, P.A.; DiTullio, G.R.; Malitsky, S.; Aharoni, A.; Green, S.J.; Rotkopf, R.; Kartvelishvily, E.; et al. Bacterial Virulence Against an Oceanic Bloom-Forming Phytoplankter Is Mediated by Algal DMSP. Sci. Adv. 2018, 4, eaau5716. [Google Scholar] [CrossRef]
- Aalto, N.J.; Schweitzer, H.D.; Krsmanovic, S.; Campbell, K.; Bernstein, H.C. Diversity and Selection of Surface Marine Microbiomes in the Atlantic-Influenced Arctic. Front. Microbiol. 2022, 13, 892634. [Google Scholar] [CrossRef]
- Nguyen, H.T.; Lee, Y.M.; Hong, J.K.; Hong, S.; Chen, M.; Hur, J. Climate Warming-Driven Changes in the Flux of Dissolved Organic Matter and Its Effects on Bacterial Communities in the Arctic Ocean: A Review. Front. Mar. Sci. 2022, 9, 968583. [Google Scholar] [CrossRef]
- Sorokin, D.Y. Sulfitobacter pontiacus gen. nov., sp. nov.—A New Heterotrophic Bacterium from the Black Sea, Specialized on Sulfite Oxidation. Microbiology 1995, 64, 295–305. [Google Scholar]
- Kim, J.G.; Park, S.J.; Quan, Z.X.; Jung, M.Y.; Cha, I.T.; Kim, S.J.; Kim, K.H.; Yang, E.J.; Kim, Y.N.; Lee, S.H.; et al. Unveiling Abundance and Distribution of Planktonic Bacteria and Archaea in a Polynya in Amundsen Sea, Antarctica. Environ. Microbiol. 2014, 16, 1566–1578. [Google Scholar] [CrossRef]
- West, N.J.; Obernosterer, I.; Zemb, O.; Lebaron, P. Major Differences of Bacterial Diversity and Activity Inside and Outside of a Natural Iron-Fertilized Phytoplankton Bloom in the Southern Ocean. Environ. Microbiol. 2008, 10, 738–756. [Google Scholar] [CrossRef]
- Teeling, H.; Fuchs, B.M.; Becher, D.; Klockow, C.; Gardebrecht, A.; Bennke, C.M.; Kassabgy, M.; Huang, S.; Mann, A.J.; Waldmann, J.; et al. Substrate-Controlled Succession of Marine Bacterioplankton Populations Induced by a Phytoplankton Bloom. Science 2012, 336, 608–611. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, H.; Jose, L. Phage and Nucleocytoplasmic Large Viral Sequences Dominate Coral Viromes from the Arabian Gulf. Front. Microbiol. 2017, 8, 2063. [Google Scholar] [CrossRef]
- Gao, C.; Xia, J.; Zhou, X.; Liang, Y.; Jiang, Y.; Wang, M.; Shao, H.; Shi, X.; Guo, C.; He, H.; et al. Viral Characteristics of the Warm Atlantic and Cold Arctic Water Masses in the Nordic Seas. Appl. Environ. Microbiol. 2021, 87, e0116021. [Google Scholar] [CrossRef] [PubMed]
- Buchholz, H.H.; Bolaños, L.M.; Bell, A.G.; Michelsen, M.L.; Allen, M.J.; Temperton, B. Novel Pelagiphage Isolate Polarivirus Skadi Is a Polar Specialist That Dominates SAR11-Associated Bacteriophage Communities at High Latitudes. ISME J. 2023, 17, 1660–1670. [Google Scholar] [CrossRef]
- Qin, F.; Du, S.; Zhang, Z.; Ying, H.; Wu, Y.; Zhao, G.; Yang, M.; Zhao, Y. Newly Identified HMO-2011-type Phages Reveal Genomic Diversity and Biogeographic Distributions of This Marine Viral Group. ISME J. 2022, 16, 1363–1375. [Google Scholar] [CrossRef] [PubMed]
- Du, S.; Qin, F.; Zhang, Z.; Tian, Z.; Yang, M.; Liu, X.; Zhao, G.; Xia, Q.; Zhao, Y. Genomic Diversity, Life Strategies and Ecology of Marine HTVC010P-Type Pelagiphages. Microb. Genom. 2021, 7, 000596. [Google Scholar] [CrossRef] [PubMed]
- Williams, K.P. Integration Sites for Genetic Elements in Prokaryotic tRNA and tmRNA Genes: Sublocation Preference of Integrase Subfamilies. Nucleic Acids Res. 2002, 30, 866–875. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Ha, C.; Lee, S.; Kwon, J.; Cho, H.; Gorham, T.; Lee, J. Characterization of Cyanophages in Lake Erie: Interaction Mechanisms and Structural Damage of Toxic Cyanobacteria. Toxins 2019, 11, 444. [Google Scholar] [CrossRef]
- Šabacká, M.; Elster, J. Response of Cyanobacteria and Algae from Antarctic Wetland Habitats to Freezing and Desiccation Stress. Polar Biol. 2006, 30, 31–37. [Google Scholar] [CrossRef]
- Gong, Z.; Liang, Y.; Wang, M.; Jiang, Y.; Yang, Q.; Xia, J.; Zhou, X.; You, S.; Gao, C.; Wang, J.; et al. Viral Diversity and Its Relationship with Environmental Factors at the Surface and Deep Sea of Prydz Bay, Antarctica. Front. Microbiol. 2018, 9, 2981. [Google Scholar] [CrossRef]
- John, S.G.; Mendez, C.B.; Deng, L.; Poulos, B.; Kauffman, A.K.; Kern, S.; Brum, J.; Polz, M.F.; Boyle, E.A.; Sullivan, M.B. A Simple and Efficient Method for Concentration of Ocean Viruses by Chemical Flocculation. Environ. Microbiol. Rep. 2011, 3, 195–202. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. In Babraham Bioinformatics; Babraham Institute: Cambridge, UK, 2010; Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 22 October 2023).
- Magoč, T.; Salzberg, S.L. FLASH: Fast Length Adjustment of Short Reads to Improve Genome Assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing Mothur: Open-Source, Platform-Independent, Community-Supported Software for Describing and Comparing Microbial Communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Source | T | p (perm) |
|---|---|---|---|
| Bacteria | Surface × Bottom | 0.225 | 0.452 |
| April × June | 5.288 | 0.002 | |
| Bacteriophage | Surface × Bottom | 0.869 | 0.605 |
| April × June | 2.598 | 0.002 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, K.E.; Joo, H.M.; Kim, Y.J.; Kang, D.; Lee, T.-K.; Jung, S.W.; Ha, S.-Y. Ecological Interaction between Bacteriophages and Bacteria in Sub-Arctic Kongsfjorden Bay, Svalbard, Norway. Microorganisms 2024, 12, 276. https://doi.org/10.3390/microorganisms12020276
Kim KE, Joo HM, Kim YJ, Kang D, Lee T-K, Jung SW, Ha S-Y. Ecological Interaction between Bacteriophages and Bacteria in Sub-Arctic Kongsfjorden Bay, Svalbard, Norway. Microorganisms. 2024; 12(2):276. https://doi.org/10.3390/microorganisms12020276
Chicago/Turabian StyleKim, Kang Eun, Hyoung Min Joo, Yu Jin Kim, Donhyug Kang, Taek-Kyun Lee, Seung Won Jung, and Sun-Yong Ha. 2024. "Ecological Interaction between Bacteriophages and Bacteria in Sub-Arctic Kongsfjorden Bay, Svalbard, Norway" Microorganisms 12, no. 2: 276. https://doi.org/10.3390/microorganisms12020276
APA StyleKim, K. E., Joo, H. M., Kim, Y. J., Kang, D., Lee, T.-K., Jung, S. W., & Ha, S.-Y. (2024). Ecological Interaction between Bacteriophages and Bacteria in Sub-Arctic Kongsfjorden Bay, Svalbard, Norway. Microorganisms, 12(2), 276. https://doi.org/10.3390/microorganisms12020276