
Molecular Genomic Analyses of Enterococcus cecorum from Sepsis Outbreaks in Broilers



Abstract
1. Introduction
2. Materials and Methods
2.1. New Genome Assemblies
2.2. Genome Analyses
3. Results and Discussion
3.1. Genome Assemblies from Sepsis Surveys
3.2. Considerations Regarding Disease Trait Classifications
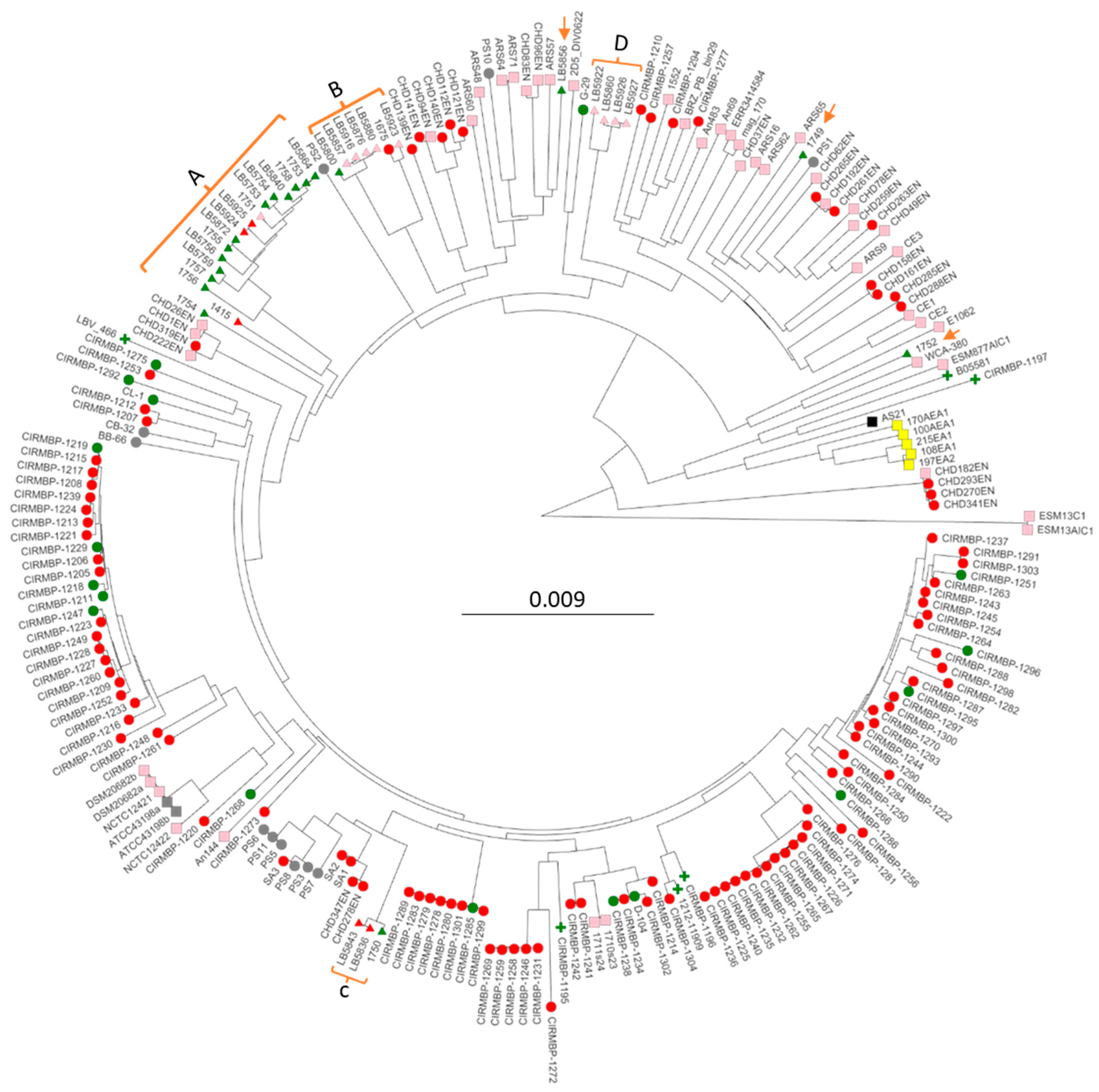
3.3. Phylogenomic Analysis of Sepsis and Osteomyelitis Isolates

3.4. Is Gene Acquisition Central to Sepsis or Disease Traits?
3.5. Are Core Genome Mutations Central to Sepsis or Disease Traits?
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Lebreton, F.; Willems, R.J.; Gilmore, M.S. Enterococcus diversity, origins in nature, and gut colonization. In Enterococci: From Commensals to Leading Causes of Drug Resistant Infection [Internet]; Gilmore, M.S., Clewell, D.B., Ike, Y., Shankar, N., Eds.; Massachusetts Eye and Ear Infirmary: Boston, MA, USA, 2014; pp. 1–46. [Google Scholar]
- Zhong, Z.; Zhang, W.; Song, Y.; Liu, W.; Xu, H.; Xi, X.; Menghe, B.; Zhang, H.; Sun, Z. Comparative genomic analysis of the genus Enterococcus. Microbiol. Res. 2017, 196, 95–105. [Google Scholar] [CrossRef]
- Naser, S.M.; Vancanneyt, M.; De Graef, E.; Devriese, L.A.; Snauwaert, C.; Lefebvre, K.; Hoste, B.; Švec, P.; Decostere, A.; Haesebrouck, F. Enterococcus canintestini sp. nov., from faecal samples of healthy dogs. Int. J. Syst. Evol. Microbiol. 2005, 55, 2177–2182. [Google Scholar] [CrossRef]
- Walker, G.K.; Suyemoto, M.M.; Gall, S.; Chen, L.; Thakur, S.; Borst, L.B. The role of Enterococcus faecalis during co-infection with avian pathogenic Escherichia coli in avian colibacillosis. Avian Pathol. 2020, 49, 589–599. [Google Scholar] [CrossRef]
- Sistek, V.; Maheux, A.F.; Boissinot, M.; Bernard, K.A.; Cantin, P.; Cleenwerck, I.; De Vos, P.; Bergeron, M.G. Enterococcus ureasiticus sp. nov. and Enterococcus quebecensis sp. nov., isolated from water. Int. J. Syst. Evol. Microbiol. 2012, 62, 1314–1320. [Google Scholar] [CrossRef] [PubMed]
- Moreno, M.F.; Sarantinopoulos, P.; Tsakalidou, E.; De Vuyst, L. The role and application of enterococci in food and health. Int. J. Food. Microbiol. 2006, 106, 1–24. [Google Scholar] [CrossRef] [PubMed]
- O’Driscoll, T.; Crank, C.W. Vancomycin-resistant enterococcal infections: Epidemiology, clinical manifestations, and optimal management. Infect. Drug Resist. 2015, 8, 217–230. [Google Scholar] [PubMed]
- Moellering Jr, R.C. Emergence of Enterococcus as a significant pathogen. Clin. Infect. Dis. 1992, 14, 1173–1176. [Google Scholar] [CrossRef] [PubMed]
- Jackson, C.R.; Fedorka-Cray, P.J.; Barrett, J.B. Use of a Genus- and Species-Specific Multiplex PCR for Identification of Enterococci. J. Clin. Microbiol. 2004, 42, 3558–3565. [Google Scholar] [CrossRef] [PubMed]
- Borst, L.B.; Suyemoto, M.M.; Sarsour, A.H.; Harris, M.C.; Martin, M.P.; Strickland, J.D.; Oviedo, E.O.; Barnes, H.J. Pathogenesis of Enterococcal Spondylitis Caused by Enterococcus cecorum in Broiler Chickens. Vet. Pathol. 2017, 54, 61–73. [Google Scholar] [CrossRef]
- Jung, A.; Chen, L.R.; Suyemoto, M.M.; Barnes, H.J.; Borst, L.B. A Review of Enterococcus cecorum Infection in Poultry. Avian Dis. 2018, 62, 261–271. [Google Scholar] [CrossRef] [PubMed]
- Logue, C.M.; Andreasen, C.B.; Borst, L.B.; Eriksson, H.; Hampson, D.J.; Sanchez, S.; Fulton, R.M. Other Bacterial Diseases. In Diseases of Poultry, 14th ed.; Swayne, D.E., Boulianne, M., Logue, C.M., McDougald, L.R., Nair, V., Suarez, D.L., Wit, S., Grimes, T., Johnson, D., Kromm, M., et al., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2020; pp. 995–1085. [Google Scholar]
- Borst, L.B.; Suyemoto, M.M.; Scholl, E.H.; Fuller, F.J.; Barnes, H.J. Comparative Genomic Analysis Identifies Divergent Genomic Features of Pathogenic Enterococcus cecorum Including a Type IC CRISPR-Cas System, a Capsule Locus, an epa-Like Locus, and Putative Host Tissue Binding Proteins. PLoS ONE 2015, 10, e0121294. [Google Scholar] [CrossRef]
- Paulsen, I.T.; Banerjei, L.; Myers, G.S.; Nelson, K.E.; Seshadri, R.; Read, T.D.; Fouts, D.E.; Eisen, J.A.; Gill, S.R.; Heidelberg, J.F.; et al. Role of mobile DNA in the evolution of vancomycin-resistant Enterococcus faecalis. Science 2003, 299, 2071–2074. [Google Scholar] [CrossRef]
- Palmer, K.L.; Godfrey, P.; Griggs, A.; Kos, V.N.; Zucker, J.; Desjardins, C.; Cerqueira, G.; Gevers, D.; Walker, S.; Wortman, J.; et al. Comparative Genomics of Enterococci: Variation in Enterococcus faecalis, Clade Structure in E. faecium, and Defining Characteristics of E. gallinarum and E. casseliflavus. mBio 2012, 3, 10-1128. [Google Scholar] [CrossRef]
- Laurentie, J.; Loux, V.; Hennequet-Antier, C.; Chambellon, E.; Deschamps, J.; Trotereau, A.; Furlan, S.; Darrigo, C.; Kempf, F.; Lao, J.; et al. Comparative Genome Analysis of Enterococcus cecorum Reveals Intercontinental Spread of a Lineage of Clinical Poultry Isolates. mSphere 2023, 8, e0049522. [Google Scholar] [CrossRef]
- Arango, M.; Forga, A.; Liu, J.; Zhang, G.; Gray, L.; Moore, R.; Coles, M.; Atencio, A.; Trujillo, C.; Latorre, J.D.; et al. Characterizing the impact of Enterococcus cecorum infection during late embryogenesis on disease progression, cecal microbiome composition, and early performance in broiler chickens. Poult. Sci. 2023, 102, 103059. [Google Scholar] [CrossRef]
- Kense, M.J.; Landman, W.J.M. Enterococcus cecorum infections in broiler breeders and their offspring: Molecular epidemiology. Avian Pathol. 2011, 40, 603–612. [Google Scholar] [CrossRef]
- Dunnam, G.; Thornton, J.; Pulido-Landinez, M. An Emerging Enterococcus cecorum Outbreak in a Broiler Integrator in the Southern US: Analysis of Antimicrobial resistance Trends. In Proceedings of the 71st Western Poultry Disease Conference, Vancouver, BC, Canada, 3–6 April 2022; Volume 2022, pp. 55–61. [Google Scholar]
- Hodak, C.R.; Bescucci, D.M.; Shamash, K.; Kelly, L.C.; Montina, T.; Savage, P.B.; Inglis, G.D. Antimicrobial Growth Promoters Altered the Function but Not the Structure of Enteric Bacterial Communities in Broiler Chicks ± Microbiota Transplantation. Animals 2023, 13, 997. [Google Scholar] [CrossRef]
- Sambrook, J.; Fritsch, E.F.; Maniatis, T. Molecular Cloning: A Laboratory Manual; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1989. [Google Scholar]
- Ekesi, N.S.; Hasan, A.; Alrubaye, A.; Rhoads, D. Analysis of Genomes of Bacterial Isolates from Lameness Outbreaks in Broilers. Poult. Sci. 2021, 100, 101148. [Google Scholar] [CrossRef] [PubMed]
- Olson, R.D.; Assaf, R.; Brettin, T.; Conrad, N.; Cucinell, C.; Davis, J.J.; Dempsey, D.M.; Dickerman, A.; Dietrich, E.M.; Kenyon, R.W.; et al. Introducing the Bacterial and Viral Bioinformatics Resource Center (BV-BRC): A resource combining PATRIC, IRD and ViPR. Nucleic Acid Res. 2022, 51, D678–D689. [Google Scholar] [CrossRef] [PubMed]
- Grant, J.R.; Stothard, P. The CGView Server: A comparative genomics tool for circular genomes. Nucleic Acid Res. 2008, 36, W181–W184. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.L.; Mullet, J.; Hindi, F.; Stoll, J.E.; Gupta, S.; Choi, M.; Keenum, I.; Vikesland, P.; Pruden, A.; Zhang, L. mobileOG-db: A Manually Curated Database of Protein Families Mediating the Life Cycle of Bacterial Mobile Genetic Elements. Appl. Environ. Microbiol. 2022, 88, e0099122. [Google Scholar] [CrossRef]
- Bertelli, C.; Laird, M.R.; Williams, K.P.; Lau, B.Y.; Hoad, G.; Winsor, G.L.; Brinkman, F.S.; Simon Fraser University Research Computing Group. IslandViewer 4: Expanded prediction of genomic islands for larger-scale datasets. Nucleic Acid Res. 2017, 45, W30–W35. [Google Scholar] [CrossRef]
- Pon, A.; Marcu, A.; Arndt, D.; Grant, J.R.; Sajed, T.; Liang, Y.; Wishart, D.S. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acid Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef]
- Lees, J.A.; Harris, S.R.; Tonkin-Hill, G.; Gladstone, R.A.; Lo, S.W.; Weiser, J.N.; Corander, J.; Bentley, S.D.; Croucher, N.J. Fast and flexible bacterial genomic epidemiology with PopPUNK. Genome Res. 2019, 29, 304–316. [Google Scholar] [CrossRef]
- Treangen, T.J.; Ondov, B.D.; Koren, S.; Phillippy, A.M. The Harvest suite for rapid core-genome alignment and visualization of thousands of intraspecific microbial genomes. Genome Biol. 2014, 15, 524. [Google Scholar] [CrossRef] [PubMed]
- Han, M.V.; Zmasek, C.M. phyloXML: XML for evolutionary biology and comparative genomics. BMC Bioinform. 2009, 10, 356. [Google Scholar] [CrossRef] [PubMed]
- Argimón, S.; Abudahab, K.; Goater, R.J.E.; Fedosejev, A.; Bhai, J.; Glasner, C.; Feil, E.J.; Holden, M.T.G.; Yeats, C.A.; Grundmann, H.; et al. Microreact: Visualizing and sharing data for genomic epidemiology and phylogeography. Microb. Genom. 2016, 2, e000093. [Google Scholar] [CrossRef] [PubMed]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Page, A.J.; Cummins, C.A.; Hunt, M.; Wong, V.K.; Reuter, S.; Holden, M.T.G.; Fookes, M.; Falush, D.; Keane, J.A.; Parkhill, J. Roary: Rapid large-scale prokaryote pan genome analysis. Bioinformatics 2015, 31, 3691–3693. [Google Scholar] [CrossRef]
- Brynildsrud, O.; Bohlin, J.; Scheffer, L.; Eldholm, V. Rapid scoring of genes in microbial pan-genome-wide association studies with Scoary. Genome Biol. 2016, 17, 238. [Google Scholar] [CrossRef]
- Shi, Z.J.; Nayfach, S.; Pollard, K.S. Maast: Genotyping thousands of microbial strains efficiently. Genome Biol. 2023, 24, 186. [Google Scholar] [CrossRef] [PubMed]
- Teufel, F.; Almagro Armenteros, J.J.; Johansen, A.R.; Gíslason, M.H.; Pihl, S.I.; Tsirigos, K.D.; Winther, O.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 6.0 predicts all five types of signal peptides using protein language models. Nat. Biotech. 2022, 40, 1023–1025. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Eeckhaut, V.; Goossens, E.; Rasschaert, G.; Van Erum, J.; Roovers, G.; Ducatelle, R.; Antonissen, G.; Van Immerseel, F. Bacterial chondronecrosis with osteomyelitis related Enterococcus cecorum isolates are genetically distinct from the commensal population and are more virulent in an embryo mortality model. Vet. Res. 2023, 54, 13. [Google Scholar] [CrossRef] [PubMed]
- Fiddaman, S.R.; Dimopoulos, E.A.; Lebrasseur, O.; du Plessis, L.; Vrancken, B.; Charlton, S.; Haruda, A.F.; Tabbada, K.; Flammer, P.G.; Dascalu, S.; et al. Ancient chicken remains reveal the origins of virulence in Marek’s disease virus. Science 2023, 382, 1276–1281. [Google Scholar] [CrossRef] [PubMed]
- Madan-Lala, R.; Sia, J.K.; King, R.; Adekambi, T.; Monin, L.; Khader, S.A.; Pulendran, B.; Rengarajan, J. Mycobacterium tuberculosis Impairs Dendritic Cell Functions through the Serine Hydrolase Hip1. J Immunol. 2014, 192, 4263–4272. [Google Scholar] [CrossRef]
- Liu, Y.; Yoo, B.B.; Hwang, C.-A.; Suo, Y.; Sheen, S.; Khosravi, P.; Huang, L. LMOf2365_0442 Encoding for a Fructose Specific PTS Permease IIA May Be Required for Virulence in L. monocytogenes Strain F2365. Front. Microbiol. 2017, 8, 1611. [Google Scholar] [CrossRef]
- Paganelli, F.L.; Huebner, J.; Singh, K.V.; Zhang, X.; van Schaik, W.; Wobser, D.; Braat, J.C.; Murray, B.E.; Bonten, M.J.M.; Willems, R.J.L.; et al. Genome-wide Screening Identifies Phosphotransferase System Permease BepA to Be Involved in Enterococcus faecium Endocarditis and Biofilm Formation. J. Infect. Dis. 2016, 214, 189–195. [Google Scholar] [CrossRef]
- Coleman, J.P.; Hudson, L.L.; McKnight, S.L.; Farrow, J.M.; Calfee, M.W.; Lindsey, C.A.; Pesci, E.C. Pseudomonas aeruginosa PqsA Is an Anthranilate-Coenzyme A Ligase. J. Bact. 2008, 190, 1247–1255. [Google Scholar] [CrossRef]
- Dash, A.; Modak, R. Protein Acetyltransferases Mediate Bacterial Adaptation to a Diverse Environment. J. Bact. 2021, 203, JB0023121. [Google Scholar] [CrossRef] [PubMed]
- Boël, G.; Smith, P.C.; Ning, W.; Englander, M.T.; Chen, B.; Hashem, Y.; Testa, A.J.; Fischer, J.J.; Wieden, H.-J.; Frank, J.; et al. The ABC-F protein EttA gates ribosome entry into the translation elongation cycle. Nat. Struct. Mol. Biol. 2014, 21, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Kazmierczak, M.J.; Wiedmann, M.; Boor, K.J. Alternative Sigma Factors and Their Roles in Bacterial Virulence. Microbiol. Mol. Biol. Rev. 2005, 69, 527–543. [Google Scholar] [CrossRef]
- Fukushima, S.; Yoshimura, M.; Chibazakura, T.; Sato, T.; Yoshikawa, H. The putative ABC transporter YheH/YheI is involved in the signalling pathway that activates KinA during sporulation initiation. FEMS Microbiol. Lett. 2006, 256, 90–97. [Google Scholar] [CrossRef][Green Version]
- Shwani, A.; Zuo, B.; Alrubaye, A.; Zhao, J.; Rhoads, D.D. A Simple, Inexpensive Alkaline Method for Bacterial DNA Extraction from Environmental Samples for PCR Surveillance and Microbiome Analyses. Appl. Sci. 2024, 14, 141. [Google Scholar] [CrossRef]
- Borst, L.B.; Suyemoto, M.M.; Keelara, S.; Dunningan, S.E.; Guy, J.S.; Barnes, H.J. A Chicken Embryo Lethality Assay for Pathogenic Enterococcus cecorum. Avian Dis. 2014, 58, 244–248. [Google Scholar] [CrossRef] [PubMed]
- Dolka, B.; Chrobak-Chmiel, D.; Czopowicz, M.; Szeleszczuk, P. Characterization of pathogenic Enterococcus cecorum from different poultry groups: Broiler chickens, layers, turkeys, and waterfowl. PLoS ONE 2017, 12, e0185199. [Google Scholar] [CrossRef] [PubMed]
- Ekesi, N.; Hasan, A.; Parveen, A.; Shwani, A.; Rhoads, D. Embryo Lethality Assay for Evaluating Virulence of Isolates from Bacterial Chondronecrosis with Osteomyelitis in Broilers. Poult. Sci. 2021. in review. [Google Scholar] [CrossRef] [PubMed]
- Garsin, D.A.; Frank, K.L.; Silanpää, J.; Ausubel, F.M.; Hartke, A.; Shankar, N.; Murray, B.E. Pathogenesis and Models of Enterococcal Infection. In Enterococci: From Commensals to Leading Causes of Drug Resistant Infection [Internet]; Gilmore, M.S., Clewell, D.B., Ike, Y., Shankar, N., Eds.; Massachusetts Eye and Ear Infirmary: Boston, MA, USA, 2014; pp. 139–192. [Google Scholar]

{kind=link}
| Strain | Source | Date Collected | Collect Location | Bird Age | Assembly Contigs | Total bp | BioSample | BLASTp | PopPUNK Cluster |
|---|---|---|---|---|---|---|---|---|---|
| 1415 | Tibial pus | 6/23/2016 | farm16 | 4.3 | 65 | 2,411,963 | SAMN38750234 | 100 | A |
| 1675 | Femoral head necrosis | 11/26/2020 | UAPRF | 8 | 56 | 2,188,186 | SAMN38750235 | 100 | B |
| 1749 | Liver | n/a | farm15 | 0.5 | 93 | 2,494,030 | SAMN38750236 | 15 | solo |
| 1750 | Heart | n/a | farm22 | 3 | 49 | 2,281,603 | SAMN38750237 | 100 | C |
| 1751 | n/a | n/a | farm19 | 2.5 | 49 | 2,300,397 | SAMN38750238 | 100 | A |
| 1752 | Liver | n/a | farm8 | 3.6 | 32 | 2,279,238 | SAMN38750239 | 17 | solo |
| 1753 | Heart | n/a | farm15 | 3.1 | 51 | 2,240,969 | SAMN38750240 | 100 | A |
| 1754 | n/a | n/a | farm2 | n/a | 47 | 2,180,738 | SAMN38750241 | 100 | A |
| 1755 | Heart | n/a | farm2 | n/a | 58 | 2,366,287 | SAMN38750242 | 100 | A |
| 1756 | Heart | n/a | farm8 | 3.2 | 62 | 2,288,864 | SAMN38750243 | 100 | A |
| 1757 | Heart | n/a | farm4 | 3.3 | 53 | 2,297,190 | SAMN38750244 | 100 | A |
| 1758 | Heart | n/a | farm22 | 3 | 53 | 2,249,959 | SAMN38750245 | 100 | A |
| LB5753 | Heart | 2/5/2021 | farm17 | 3 | 56 | 2,226,311 | SAMN38750246 | 100 | A |
| LB5754 | Heart | 2/12/2021 | farm9 | 5 | 62 | 2,234,879 | SAMN38750247 | 100 | A |
| LB5756 | Heart | 3/8/2021 | farm13 | 2.3 | 62 | 2,239,264 | SAMN38750248 | 100 | A |
| LB5759 | Heart | 3/8/2021 | farm11 | 2.6 | 75 | 2,392,627 | SAMN38750249 | 100 | A |
| LB5800 | Heart | 3/11/2021 | farm3 | 3 | 42 | 2,212,442 | SAMN38750250 | 100 | B |
| LB5836 | Vertebral osteomyelitis | 9/29/2020 | farm6 | 12 | 69 | 2,320,142 | SAMN38750251 | 100 | C |
| LB5840 | Heart | 10/13/2020 | farm5 | n/a | 57 | 2,258,228 | SAMN38750252 | 100 | A |
| LB5843 | Vertebral osteomyelitis | 10/20/2020 | farm7 | 8 | 70 | 2,319,059 | SAMN38750253 | 100 | C |
| LB5856 | Heart | 12/21/2020 | farm14 | n/a | 81 | 2,378,014 | SAMN38750254 | 40 | solo |
| LB5857 | Egg transfer residue | 3/26/2021 | hatchery 1 | Eggs | 47 | 2,180,187 | SAMN38750255 | 100 | B |
| LB5860 | Cull eggs | 3/24/2021 | hatchery 1 | Eggs | 102 | 2,230,151 | SAMN38750256 | 100 | D |
| LB5864 | Heart | 12/9/2020 | farm20 | 2.3 | 61 | 2,239,173 | SAMN38750257 | 100 | A |
| LB5872 | Heart | 4/21/2021 | farm18 | n/a | 65 | 2,365,322 | SAMN38750258 | 100 | A |
| LB5876 | Egg transfer residue | 5/10/2021 | hatchery 1 | Eggs | 37 | 2,183,317 | SAMN38750259 | 100 | B |
| LB5880 | Air sacculitis | 4/29/2021 | farm21 | 2 | 38 | 2,146,928 | SAMN38750260 | 100 | B |
| LB5916 | Egg transfer residue | 6/4/2021 | hatchery 1 | Eggs | 40 | 2,193,104 | SAMN38750261 | 100 | B |
| LB5922 | n/a | 6/16/2021 | farm12 | 1 | 88 | 2,169,308 | SAMN38750262 | 99 | D |
| LB5923 | intestinal tract | 6/21/2021 | farm 1 | 1.3 | 58 | 2,239,406 | SAMN38750263 | 100 | B |
| LB5924 | Air sacculitis | 4/2/2021 | farm10 | 2.4 | 61 | 2,296,440 | SAMN38750264 | 100 | A |
| LB5925 | Air sacculitis | 4/2/2021 | farm10 | 2.4 | 61 | 2,297,136 | SAMN38750265 | 94 | A |
| LB5926 | Egg transfer residue | 4/16/2021 | hatchery 2 | Eggs | 103 | 2,277,367 | SAMN38750266 | 100 | D |
| LB5927 | Egg transfer residue | 4/16/2021 | hatchery 2 | Eggs | 105 | 2,276,597 | SAMN38750267 | 97 | D |
| Phentotype | Count | Borst Gene Cluster Protein Encoded Gene | Average ± SEM | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 312 | 313 | 314 | 315 | 316 | 317 | 318 | 319 | 320 | 321 | 322 | 323 | |||
| None | 82 | 53 | 35 | 37 | 49 | 30 | 43 | 31 | 31 | 45 | 43 | 39 | 91 | 44 ± 5 |
| CD | 145 | 85 | 77 | 79 | 83 | 75 | 79 | 74 | 75 | 84 | 85 | 82 | 98 | 81 ± 2 |
| BCO | 113 | 84 | 76 | 77 | 82 | 73 | 77 | 72 | 73 | 81 | 83 | 79 | 97 | 80 ± 2 |
| SS | 34 | 88 | 80 | 83 | 86 | 81 | 83 | 79 | 79 | 89 | 88 | 88 | 98 | 85 ± 2 |
| Gene | Phenotypic Trait Group | ||||||
|---|---|---|---|---|---|---|---|
| Name | Function | AA Pos | Ref | Alt | BCO | Sepsis Strict | Sepsis Strict Reduced |
| PEG231 | Serine aminopeptidase S33 domain-containing protein | 261 | T | A | 35 | 66 | 50 |
| EIIABC | PTS fructose-specific EIIABC component | 555 | R | Q | 33 | 75 | 64 |
| 590 | A | V | 29 | 75 | 64 | ||
| CBCL1 | 4-chlorobenzoate--CoA ligase | 244 | G | H | 43 | 69 | 55 |
| 246 | I | L | 43 | 69 | 55 | ||
| 249 | H | Y | 43 | 69 | 55 | ||
| ElaA | Gcn5-related N-acetyltransferase (GNAT family) | 128 | N | K | 34 | 69 | 55 |
| EttA | Energy-dependent translational throttle protein | 106 | S | A | 40 | 69 | 55 |
| 534 | T | A | 39 | 66 | 50 | ||
| RpoN | RNA polymerase σ54 factor | 12–13 | TQ | -- | 25 | 38 | 50 |
| 22 | T | S | 26 | 38 | 50 | ||
| YheH | putative multidrug resistance ABC transporter ATP-binding/permease protein | 13 | I | L | 43 | 72 | 59 |
| 578 | D | N | 28 | 66 | 50 | ||
| 590 | S | I | 28 | 66 | 50 | ||
| 592–593 | EEI | GAD | 28 | 69 | 55 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rhoads, D.D.; Pummill, J.; Alrubaye, A.A.K. Molecular Genomic Analyses of Enterococcus cecorum from Sepsis Outbreaks in Broilers. Microorganisms 2024, 12, 250. https://doi.org/10.3390/microorganisms12020250
Rhoads DD, Pummill J, Alrubaye AAK. Molecular Genomic Analyses of Enterococcus cecorum from Sepsis Outbreaks in Broilers. Microorganisms. 2024; 12(2):250. https://doi.org/10.3390/microorganisms12020250
Chicago/Turabian StyleRhoads, Douglas D., Jeff Pummill, and Adnan Ali Khalaf Alrubaye. 2024. "Molecular Genomic Analyses of Enterococcus cecorum from Sepsis Outbreaks in Broilers" Microorganisms 12, no. 2: 250. https://doi.org/10.3390/microorganisms12020250
APA StyleRhoads, D. D., Pummill, J., & Alrubaye, A. A. K. (2024). Molecular Genomic Analyses of Enterococcus cecorum from Sepsis Outbreaks in Broilers. Microorganisms, 12(2), 250. https://doi.org/10.3390/microorganisms12020250