
Genomic Diversity and Virulence Factors of Clostridium perfringens Isolated from Healthy and Necrotic Enteritis-Affected Broiler Chicken Farms in Quebec Province
 ,
,  , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and Isolation of C. perfringens Strains
2.2. C. perfringens Culture, DNA Extraction, Sequencing Library Preparation, and WGS
2.3. Bioinformatics Workflow for Genome Assembly and Annotation
2.4. In Silico Profiling of Virulence Factors
2.5. Pangenome Analysis and Prediction of Prophages, Antibiotic Resistance Genes, and Plasmids
3. Results
3.1. Sample Collection and Isolation of C. perfringens Strains
3.2. Genome Assembly and Annotation
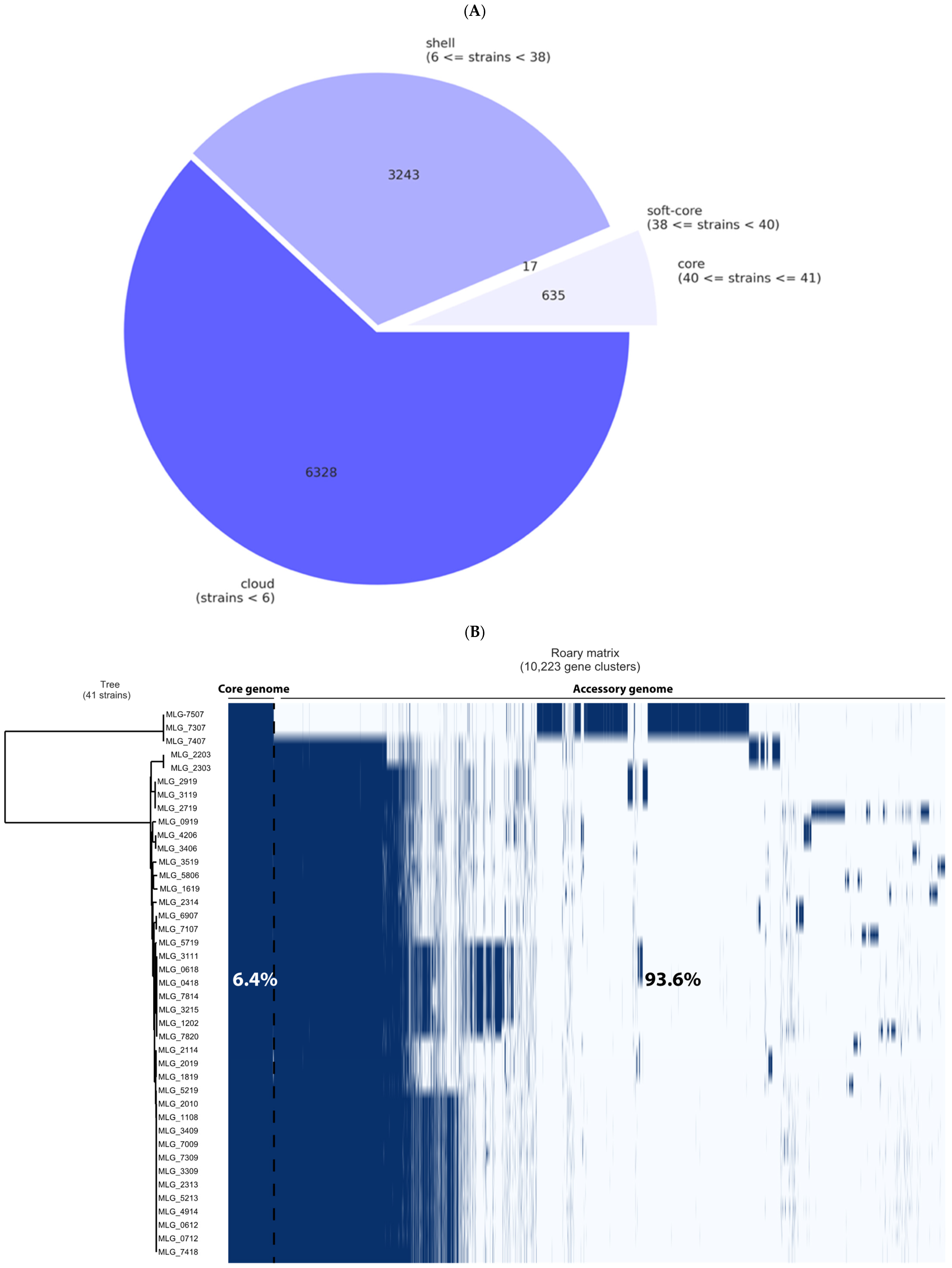
3.3. Pangenome Analysis and Genetic Relationships Between Isolates
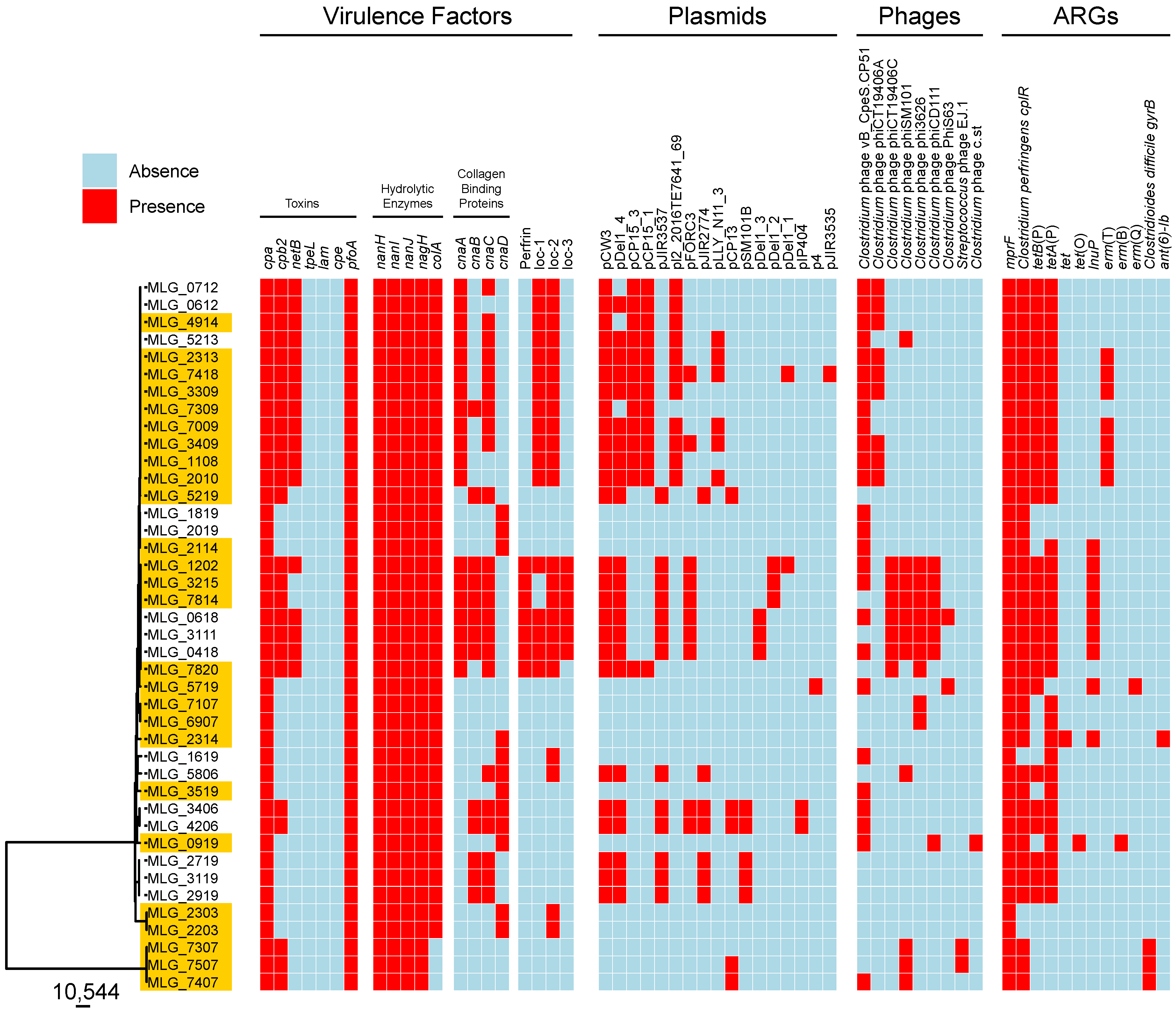
3.4. In Silico Profiling of Virulence Traits
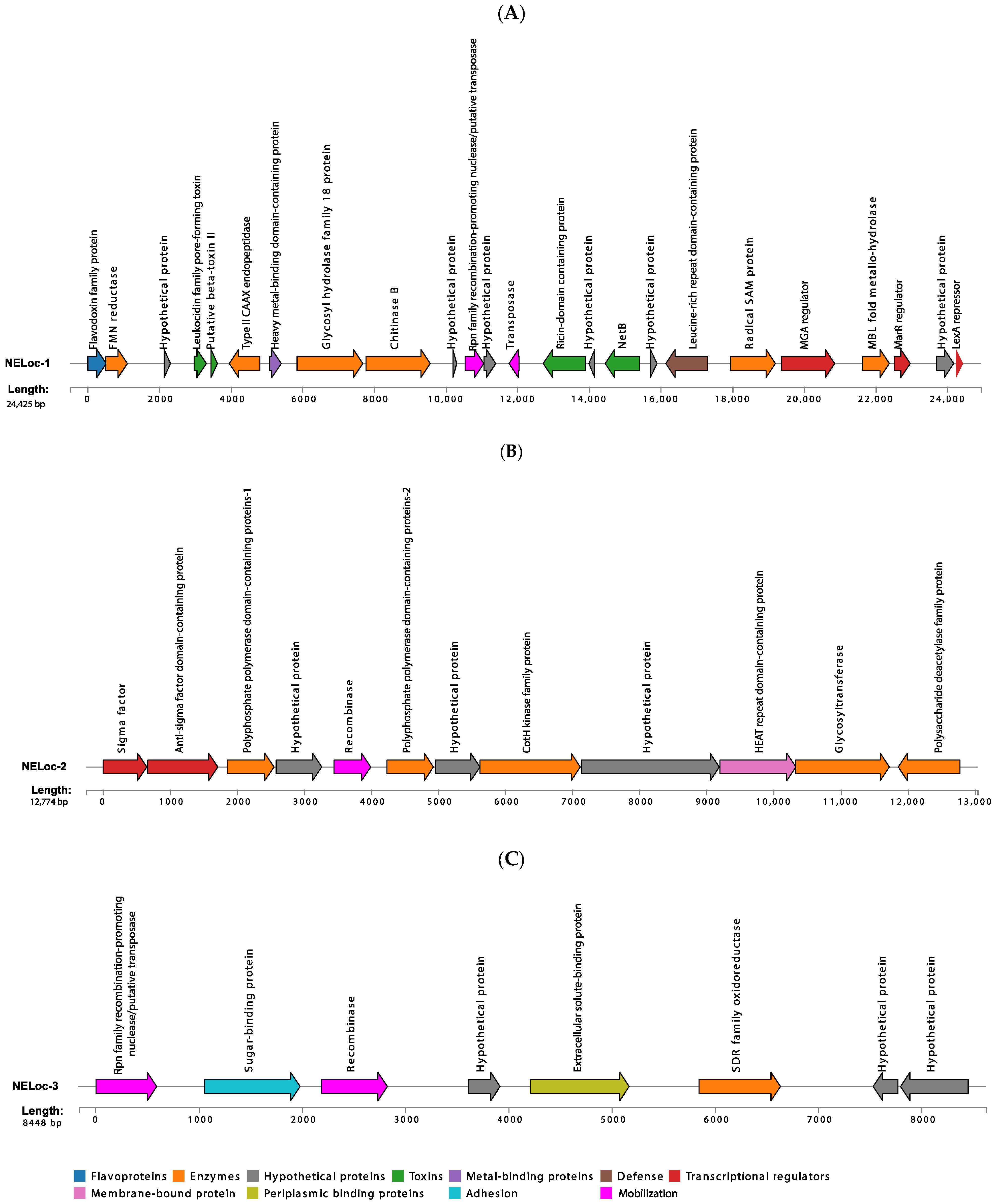
3.5. Characterization of NELoc-1–3
3.6. ARG Prediction
3.7. Prophage Content of C. perfringens Isolates
3.8. Presence of Plasmids in C. perfringens Isolates
4. Discussion
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Kiu, R.; Hall, L.J. An update on the human and animal enteric pathogen Clostridium perfringens. Emerg. Microbes Infect. 2018, 7, 141. [Google Scholar] [CrossRef] [PubMed]
- Mehdizadeh Gohari, I.; Navarro, M.A.; Li, J.; Shrestha, A.; Uzal, F.; McClane, B.A. Pathogenicity and virulence of Clostridium perfringens. Virulence 2021, 12, 723–753. [Google Scholar] [CrossRef] [PubMed]
- Fathima, S.; Hakeem, W.G.A.; Shanmugasundaram, R.; Selvaraj, R.K. Necrotic Enteritis in Broiler Chickens: A Review on the Pathogen, Pathogenesis, and Prevention. Microorganisms 2022, 10, 1958. [Google Scholar] [CrossRef] [PubMed]
- Alizadeh, M.; Shojadoost, B.; Boodhoo, N.; Astill, J.; Taha-Abdelaziz, K.; Hodgins, D.C.; Kulkarni, R.R.; Sharif, S. Necrotic enteritis in chickens: A review of pathogenesis, immune responses and prevention, focusing on probiotics and vaccination. Anim. Health Res. Rev. 2021, 22, 147–162. [Google Scholar] [CrossRef]
- Xu, W.; Wang, H.; Liu, L.; Miao, Z.; Huo, Y.; Zhong, Z. Prevalence and characterization of Clostridium perfringens isolated from different chicken farms in China. Anaerobe 2021, 72, 102467. [Google Scholar] [CrossRef]
- Mwangi, S.; Timmons, J.; Fitz-Coy, S.; Parveen, S. Characterization of Clostridium perfringens recovered from broiler chicken affected by necrotic enteritis. Poult. Sci. 2019, 98, 128–135. [Google Scholar] [CrossRef]
- Chalmers, G.; Martin, S.W.; Prescott, J.F.; Boerlin, P. Typing of Clostridium perfringens by multiple-locus variable number of tandem repeats analysis. Vet. Microbiol. 2008, 128, 126–135. [Google Scholar] [CrossRef]
- De Cesare, A.; Borilova, G.; Svobodova, I.; Bondioli, V.; Manfreda, G. Clostridium perfringens occurrence and ribotypes in healthy broilers reared in different European countries. Poult. Sci. 2009, 88, 1850–1857. [Google Scholar] [CrossRef]
- Lacey, J.A.; Allnutt, T.R.; Vezina, B.; Van, T.T.H.; Stent, T.; Han, X.; Rood, J.I.; Wade, B.; Keyburn, A.L.; Seemann, T.; et al. Whole genome analysis reveals the diversity and evolutionary relationships between necrotic enteritis-causing strains of Clostridium perfringens. BMC Genom. 2018, 19, 379. [Google Scholar] [CrossRef]
- Lepp, D.; Gong, J.; Songer, J.G.; Boerlin, P.; Parreira, V.R.; Prescott, J.F. Identification of accessory genome regions in poultry Clostridium perfringens isolates carrying the netB plasmid. J. Bacteriol. 2013, 195, 1152–1166. [Google Scholar] [CrossRef]
- Gaucher, M.L.; Perron, G.G.; Arsenault, J.; Letellier, A.; Boulianne, M.; Quessy, S. Recurring Necrotic Enteritis Outbreaks in Commercial Broiler Chicken Flocks Strongly Influence Toxin Gene Carriage and Species Richness in the Resident Clostridium perfringens Population. Front. Microbiol. 2017, 8, 881. [Google Scholar] [CrossRef] [PubMed]
- Park, M.; Deck, J.; Foley, S.L.; Nayak, R.; Songer, J.G.; Seibel, J.R.; Khan, S.A.; Rooney, A.P.; Hecht, D.W.; Rafii, F. Diversity of Clostridium perfringens isolates from various sources and prevalence of conjugative plasmids. Anaerobe 2016, 38, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Parreira, V.R.; Costa, M.; Eikmeyer, F.; Blom, J.; Prescott, J.F. Sequence of two plasmids from Clostridium perfringens chicken necrotic enteritis isolates and comparison with C. perfringens conjugative plasmids. PLoS ONE 2012, 7, e49753. [Google Scholar] [CrossRef] [PubMed]
- Lepp, D.; Roxas, B.; Parreira, V.R.; Marri, P.R.; Rosey, E.L.; Gong, J.; Songer, J.G.; Vedantam, G.; Prescott, J.F. Identification of novel pathogenicity loci in Clostridium perfringens strains that cause avian necrotic enteritis. PLoS ONE 2010, 5, e10795. [Google Scholar] [CrossRef]
- Zhou, H.; Lepp, D.; Pei, Y.; Liu, M.; Yin, X.; Ma, R.; Prescott, J.F.; Gong, J. Influence of pCP1NetB ancillary genes on the virulence of Clostridium perfringens poultry necrotic enteritis strain CP1. Gut Pathog. 2017, 9, 6. [Google Scholar] [CrossRef]
- Barbara, A.J.; Trinh, H.T.; Glock, R.D.; Glenn Songer, J. Necrotic enteritis-producing strains of Clostridium perfringens displace non-necrotic enteritis strains from the gut of chicks. Vet. Microbiol. 2008, 126, 377–382. [Google Scholar] [CrossRef]
- Myers, G.S.; Rasko, D.A.; Cheung, J.K.; Ravel, J.; Seshadri, R.; DeBoy, R.T.; Ren, Q.; Varga, J.; Awad, M.M.; Brinkac, L.M.; et al. Skewed genomic variability in strains of the toxigenic bacterial pathogen, Clostridium perfringens. Genome Res. 2006, 16, 1031–1040. [Google Scholar] [CrossRef]
- Kiu, R.; Caim, S.; Alexander, S.; Pachori, P.; Hall, L.J. Probing Genomic Aspects of the Multi-Host Pathogen Clostridium perfringens Reveals Significant Pangenome Diversity, and a Diverse Array of Virulence Factors. Front. Microbiol. 2017, 8, 2485. [Google Scholar] [CrossRef]
- Keyburn, A.L.; Sheedy, S.A.; Ford, M.E.; Williamson, M.M.; Awad, M.M.; Rood, J.I.; Moore, R.J. Alpha-toxin of Clostridium perfringens is not an essential virulence factor in necrotic enteritis in chickens. Infect. Immun. 2006, 74, 6496–6500. [Google Scholar] [CrossRef]
- Meniaï, I.; Thibodeau, A.; Quessy, S.; Parreira, V.R.; Fravalo, P.; Beauchamp, G.; Gaucher, M.L. Putative antigenic proteins identified by comparative and subtractive reverse vaccinology in necrotic enteritis-causing Clostridium perfringens isolated from broiler chickens. BMC Genom. 2021, 22, 890. [Google Scholar] [CrossRef]
- Coil, D.; Jospin, G.; Darling, A.E. A5-miseq: An updated pipeline to assemble microbial genomes from Illumina MiSeq data. Bioinformatics 2015, 31, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef] [PubMed]
- Page, A.J.; Cummins, C.A.; Hunt, M.; Wong, V.K.; Reuter, S.; Holden, M.T.; Fookes, M.; Falush, D.; Keane, J.A.; Parkhill, J. Roary: Rapid large-scale prokaryote pan genome analysis. Bioinformatics 2015, 31, 3691–3693. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Xu, S.; Li, L.; Luo, X.; Chen, M.; Tang, W.; Zhan, L.; Dai, Z.; Lam, T.T.; Guan, Y.; Yu, G. Ggtree: A serialized data object for visualization of a phylogenetic tree and annotation data. Imeta 2022, 1, e56. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing. (R Foundation for Statistical Computing, 2013). Available online: https://www.r-project.org/ (accessed on 30 September 2024).
- Wishart, D.S.; Han, S.; Saha, S.; Oler, E.; Peters, H.; Grant, J.R.; Stothard, P.; Gautam, V. PHASTEST: Faster than PHASTER, better than PHAST. Nucleic Acids Res. 2023, 51, W443–W450. [Google Scholar] [CrossRef]
- Roosaare, M.; Puustusmaa, M.; Möls, M.; Vaher, M.; Remm, M. PlasmidSeeker: Identification of known plasmids from bacterial whole genome sequencing reads. PeerJ 2018, 6, e4588. [Google Scholar] [CrossRef]
- Inouye, M.; Dashnow, H.; Raven, L.A.; Schultz, M.B.; Pope, B.J.; Tomita, T.; Zobel, J.; Holt, K.E. SRST2: Rapid genomic surveillance for public health and hospital microbiology labs. Genome Med. 2014, 6, 90. [Google Scholar] [CrossRef]
- Jia, B.; Raphenya, A.R.; Alcock, B.; Waglechner, N.; Guo, P.; Tsang, K.K.; Lago, B.A.; Dave, B.M.; Pereira, S.; Sharma, A.N.; et al. CARD 2017: Expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res. 2017, 45, D566–D573. [Google Scholar] [CrossRef] [PubMed]
- Camargo, A.; Guerrero-Araya, E.; Castañeda, S.; Vega, L.; Cardenas-Alvarez, M.X.; Rodríguez, C.; Paredes-Sabja, D.; Ramírez, J.D.; Muñoz, M. Intra-species diversity of Clostridium perfringens: A diverse genetic repertoire reveals its pathogenic potential. Front. Microbiol. 2022, 13, 952081. [Google Scholar] [CrossRef] [PubMed]
- Adams, V.; Han, X.; Lyras, D.; Rood, J.I. Antibiotic resistance plasmids and mobile genetic elements of Clostridium perfringens. Plasmid 2018, 99, 32–39. [Google Scholar] [CrossRef]
- Lacey, J.A.; Johanesen, P.A.; Lyras, D.; Moore, R.J. In silico Identification of Novel Toxin Homologs and Associated Mobile Genetic Elements in Clostridium perfringens. Pathogens 2019, 8, 16. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, J.A.; Rood, J.I. The Tcp conjugation system of Clostridium perfringens. Plasmid 2017, 91, 28–36. [Google Scholar] [CrossRef]
- Yang, W.Y.; Chou, C.H.; Wang, C. Characterization of toxin genes and quantitative analysis of netB in necrotic enteritis (NE)-producing and non-NE-producing Clostridium perfringens isolated from chickens. Anaerobe 2018, 54, 115–120. [Google Scholar] [CrossRef]
- Yin, J.; Meng, Q.; Cheng, D.; Fu, J.; Luo, Q.; Liu, Y.; Yu, Z. Mechanisms of bactericidal action and resistance of polymyxins for Gram-positive bacteria. Appl. Microbiol. Biotechnol. 2020, 104, 3771–3780. [Google Scholar] [CrossRef]
- Ernst, C.M.; Peschel, A. Broad-spectrum antimicrobial peptide resistance by MprF-mediated aminoacylation and flipping of phospholipids. Mol. Microbiol. 2011, 80, 290–299. [Google Scholar] [CrossRef]
- Feßler, A.T.; Wang, Y.; Wu, C.; Schwarz, S. Mobile macrolide resistance genes in staphylococci. Plasmid 2018, 99, 2–10. [Google Scholar] [CrossRef]
- Yu, R.; Xu, Y.; Schwarz, S.; Shang, Y.; Yuan, X.; Zhang, Y.; Li, D.; Du, X.D. erm(T)-Mediated Macrolide-Lincosamide Resistance in Streptococcus suis. Microbiol. Spectr. 2022, 10, e0165721. [Google Scholar] [CrossRef]
- Woodbury, R.L.; Klammer, K.A.; Xiong, Y.; Bailiff, T.; Glennen, A.; Bartkus, J.M.; Lynfield, R.; Van Beneden, C.; Beall, B.W. Plasmid-Borne erm(T) from invasive, macrolide-resistant Streptococcus pyogenes strains. Antimicrob. Agents Chemother. 2008, 52, 1140–1143. [Google Scholar] [CrossRef] [PubMed]
- Varaldo, P.E.; Montanari, M.P.; Giovanetti, E. Genetic elements responsible for erythromycin resistance in streptococci. Antimicrob. Agents Chemother. 2009, 53, 343–353. [Google Scholar] [CrossRef] [PubMed]
- Abril, C.; Brodard, I.; Perreten, V. Two novel antibiotic resistance genes, tet(44) and ant(6)-Ib, are located within a transferable pathogenicity island in Campylobacter fetus subsp. fetus. Antimicrob. Agents Chemother. 2010, 54, 3052–3055. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Fan, X.; Zhu, L.; Yang, X.; Liu, Y.; Gao, S.; Jin, X.; Liu, D.; Ding, J.; Guo, Y.; et al. Phylogenetic and genomic analysis reveals high genomic openness and genetic diversity of Clostridium perfringens. Microb. Genom. 2020, 6, mgen000441. [Google Scholar] [CrossRef] [PubMed]
- Gervasi, T.; Curto, R.L.; Narbad, A.; Mayer, M.J. Complete genome sequence of ΦCP51, a temperate bacteriophage of Clostridium perfringens. Arch. Virol. 2013, 158, 2015–2017. [Google Scholar] [CrossRef]
- Nariya, H.; Miyata, S.; Tamai, E.; Sekiya, H.; Maki, J.; Okabe, A. Identification and characterization of a putative endolysin encoded by episomal phage phiSM101 of Clostridium perfringens. Appl. Microbiol. Biotechnol. 2011, 90, 1973–1979. [Google Scholar] [CrossRef]
- Afshari, A.; Jamshidi, A.; Razmyar, J.; Rad, M. Genotyping of Clostridium perfringens isolated from broiler meat in northeastern of Iran. Vet. Res. Forum 2015, 6, 279–284. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
| Strain | Year of Isolation | Isolate Origin | Type IV Pilus Profile | PFGE Profile | Genome | No of CDSs | BioProject_ID | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Length (bp) | No of Contigs | Coverage (x) | GC (%) | N50 | |||||||
| MLG_7309 | 2010–2011 | NE-affected | 1 | V1 | 3,603,633 | 204 | 85 | 28 | 51,385 | 3403 | PRJNA1188553 |
| MLG_7009 | 2010–2011 | NE-affected | 1 | V2 | 3,577,078 | 100 | 93 | 28 | 197,277 | 3347 | PRJNA1188553 |
| MLG_1108 | 2010–2011 | NE-affected | 1 | V3 | 3,546,273 | 101 | 88 | 28 | 190,425 | 3300 | PRJNA1188553 |
| MLG_3309 | 2010–2011 | NE-affected | 2 | V3 | 3,538,464 | 144 | 77 | 28 | 58,373 | 3298 | PRJNA1188553 |
| MLG_3409 | 2010–2011 | NE-affected | 1 | V3 | 3,551,654 | 100 | 87 | 28 | 189,683 | 3297 | PRJNA1188553 |
| MLG_2010 | 2010–2011 | NE-affected | 2 | V3 | 3,551,275 | 104 | 92 | 28 | 152,768 | 3306 | PRJNA1188553 |
| MLG_2313 | 2010–2011 | NE-affected | 9 | V3 | 3,545,125 | 88 | 84 | 28 | 177,971 | 3290 | PRJNA734442 |
| MLG_2114 | 2010–2011 | NE-affected | 4 | V3 | 3,307,721 | 49 | 72 | 28.1 | 219,784 | 2940 | PRJNA1188553 |
| MLG_7418 | 2010–2011 | NE-affected | 2 | V3 | 3,544,119 | 82 | 87 | 28 | 219,477 | 3286 | PRJNA1188553 |
| MLG_1202 | 2010–2011 | NE-affected | 8 | V5 | 3,747,051 | 129 | 67 | 27.9 | 114,628 | 3572 | PRJNA734442 |
| MLG_3215 | 2010–2011 | NE-affected | 6 | V5 | 3,680,156 | 96 | 83 | 28 | 170,870 | 3498 | PRJNA1188553 |
| MLG_7814 | 2010–2011 | NE-affected | 5 | V6 | 3,687,085 | 124 | 60 | 28 | 80,413 | 3511 | PRJNA734442 |
| MLG_7820 | 2010–2011 | NE-affected | 5 | V7 | 3,730,847 | 209 | 91 | 27.9 | 48,863 | 3570 | PRJNA734442 |
| MLG_2314 | 2010–2011 | NE-affected | 12 | V10 | 3,291,552 | 43 | 75 | 28.1 | 291,371 | 2920 | PRJNA1188553 |
| MLG_4914 | 2010–2011 | NE-affected | 6 | V12 | 3,579,960 | 152 | 77 | 28 | 82,578 | 3345 | PRJNA1188553 |
| MLG_5806 | 2010–2011 | Healthy | 6 | C1 | 3,324,484 | 32 | 87 | 28.1 | 474,093 | 2963 | PRJNA734442 |
| MLG_4206 | 2010–2011 | Healthy | 2 | C2 | 3,400,900 | 48 | 64 | 28 | 141,651 | 3040 | PRJNA734442 |
| MLG_3406 | 2010–2011 | Healthy | 7 | C2 | 3,400,562 | 55 | 68 | 28 | 126,010 | 3039 | PRJNA734442 |
| MLG_5213 | 2010–2011 | Healthy | 6 | C3 | 3,527,345 | 92 | 64 | 28 | 158,351 | 3269 | PRJNA734442 |
| MLG_0612 | 2010–2011 | Healthy | 6 | C3 | 3,537,182 | 94 | 104 | 28 | 217,561 | 3283 | PRJNA734442 |
| MLG_0712 | 2010–2011 | Healthy | 6 | C3 | 3,527,154 | 171 | 72 | 28 | 49,443 | 3287 | PRJNA1188553 |
| MLG_7307 | 2010–2011 | NE-affected | 13 | V1 | 3,660,280 | 90 | 74 | 28.5 | 97,597 | 3296 | PRJNA1188553 |
| MLG_7407 | 2010–2011 | NE-affected | 14 | V1 | 3,661,751 | 58 | 76 | 28.5 | 172,371 | 3293 | PRJNA1188553 |
| MLG_7507 | 2010–2011 | NE-affected | 15 | V1 | 3,665,112 | 49 | 90 | 28.5 | 233,259 | 3292 | PRJNA1188553 |
| MLG_6907 | 2010–2011 | NE-affected | 1 | V2 | 3,281,089 | 30 | 97 | 28.1 | 699,417 | 2921 | PRJNA1188553 |
| MLG_7107 | 2010–2011 | NE-affected | 1 | V2 | 3,281,533 | 36 | 89 | 28.1 | 222,145 | 2920 | PRJNA1188553 |
| MLG_2203 | 2010–2011 | NE-affected | 16 | V3 | 3,281,091 | 36 | 83 | 28.1 | 219,382 | 2880 | PRJNA1188553 |
| MLG_2303 | 2010–2011 | NE-affected | 17 | V3 | 3,283,967 | 29 | 66 | 28.1 | 721,013 | 2881 | PRJNA1188553 |
| MLG_3519 | 2010–2011 | NE-affected | 4 | V5 | 3,227,573 | 33 | 64 | 28.2 | 255,420 | 2867 | PRJNA1188553 |
| MLG_0919 | 2010–2011 | NE-affected | 18 | V6 | 3,892,868 | 148 | 77 | 28.5 | 62,235 | 3661 | PRJNA1188553 |
| MLG_5719 | 2010–2011 | NE-affected | 1 | V7 | 3,379,598 | 58 | 70 | 28.2 | 133,605 | 3023 | PRJNA1188553 |
| MLG_5219 | 2010–2011 | NE-affected | 19 | V8 | 3,291,656 | 99 | 81 | 28.1 | 62,796 | 2953 | PRJNA1188553 |
| MLG_3119 | 2010–2011 | Healthy | 7 | C1 | 3,435,369 | 42 | 86 | 28 | 265,064 | 3053 | PRJNA734442 |
| MLG_2919 | 2010–2011 | Healthy | 20 | C1 | 3,434,769 | 42 | 113 | 28 | 397,745 | 3051 | PRJNA734442 |
| MLG_2719 | 2010–2011 | Healthy | 21 | C1 | 3,434,532 | 39 | 108 | 28 | 413,284 | 3049 | PRJNA734442 |
| MLG_2019 | 2010–2011 | Healthy | 3 | C2 | 3,233,362 | 60 | 82 | 28.2 | 127,551 | 2880 | PRJNA734442 |
| MLG_1819 | 2010–2011 | Healthy | 3 | C3 | 3,234,658 | 48 | 65 | 28.2 | 198,347 | 2884 | PRJNA1188553 |
| MLG_1619 | 2010–2011 | Healthy | 22 | C3 | 3,269,123 | 39 | 94 | 28.1 | 232,490 | 2913 | PRJNA734442 |
| MLG_0418 | 2010–2011 | Healthy | 5 | C4 | 3,788,295 | 115 | 94 | 28 | 114,406 | 3622 | PRJNA1188553 |
| MLG_0618 | 2010–2011 | Healthy | 8 | C4 | 3,782,263 | 143 | 126 | 28 | 71,277 | 3624 | PRJNA1188553 |
| MLG_3111 | 2010–2011 | Healthy | 5 | C5 | 3,790,123 | 124 | 70 | 28 | 102,785 | 3633 | PRJNA1188553 |
| AVERAGE | 3,500,210 | 86 | 83 | 28 | 200,369 | 3206 | |||||
| Description | Description | Nb of Strains | Nb of Genes | Pangenome (%) |
|---|---|---|---|---|
| Core genome | Core genes | (99% ≤ strains ≤ 100%) | 635 | (n = 652), 6.4% |
| Soft-core genes | (95% ≤ strains < 99%) | 17 | ||
| Accessory genome | Shell genes | (15% ≤ strains < 95%) | 3243 | (n = 9571), 93.6% |
| Cloud genes | (0% ≤ strains < 15%) | 6328 | ||
| Pangenome | Pangenome | (0% ≤ strains ≤ 100%) | 10,223 | 100% |
| Toxin | Enzyme | Enzyme | Enzyme | Enzyme | Toxin | Enzyme | Collagen Binding Protein | Collagen Binding Protein | Collagen Binding Protein | Collagen Binding Protein | Bacteriocin | Toxin | Toxin | NE Loci | NE Loci | NE Loci | Toxin | Toxin | Toxin | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Isolate origin | cpa | nanH | nanI | nanJ | nagH | pfoA | colA | cnaA | cnaB | cnaC | cnaD | perfrin | netB | cpb2 | NE Loc-1 | NE Loc-2 | NE Loc-3 | tpeL | cpe | lam |
| NE-affected (n = 26) | 100% | 100% | 100% | 100% | 100% | 100% | 88% | 50% | 19% | 46% | 23% | 15% | 42% | 65% | 42% | 57% | 11% | 0% | 0% | 0% |
| Healthy (n = 15) | 100% | 100% | 100% | 100% | 100% | 100% | 100% | 40% | 53% | 73% | 40% | 20% | 40% | 53% | 40% | 53% | 20% | 0% | 0% | 0% |
| Total (n = 41) | 100% | 100% | 100% | 100% | 100% | 100% | 92% | 46% | 31% | 56% | 29% | 17% | 41% | 60% | 41% | 56% | 14% | 0% | 0% | 0% |
| ARGs | Description | Occurrence | ||
|---|---|---|---|---|
| NE-Affected Isolates | Healthy Isolates | Total | ||
| mprF | Resistance to cationic peptides | 100% | 100% | 100% |
| C. perfringens cplR | Resistance to pleuromutilin, lincosamide, and streptogramin A | 92% | 93% | 92% |
| tetA(P) | Resistance to tetracycline antibiotic | 76% | 86% | 80% |
| tetB(P) | Resistance to tetracycline antibiotic | 57% | 80% | 65% |
| tet | Resistance to tetracycline antibiotic | 3% | 0% | 2% |
| tet(O) | Resistance to tetracycline antibiotic | 3% | 0% | 2% |
| erm(B) | Resistance to macrolide, lincosamide, and streptogramin | 3% | 0% | 2% |
| erm(Q) | Resistance to macrolide, lincosamide, and streptogramin | 3% | 0% | 2% |
| erm(T) | Resistance to macrolide, lincosamide, and streptogramin | 27% | 0% | 17% |
| lnuP | Lincosamide nucleotidyltransferase (LNU) | 23% | 20% | 22% |
| C. difficile gyrB | Resistance to fluoroquinolones | 11% | 0% | 7% |
| ant(6)-Ib | Aminoglycoside nucleotidyltransferase | 3% | 0% | 2% |
| Prophage | Description | Occurrence | ||
|---|---|---|---|---|
| NE-Affected Isolates | Healthy Isolates | Total | ||
| phiCP51 | Holin, amidase, endolysin, stage III sporulation protein D, and putative sporulation sigma factor production | 61% | 66% | 63% |
| phiCT19406A | Endolysin production | 27% | 13% | 22% |
| phiCT19406C | Endolysin production | 15% | 20% | 17% |
| phiSM101 | Endolysin production | 23% | 33% | 26% |
| phi3626 | Endolysin production, stage III sporulation protein D | 23% | 20% | 22% |
| phiCD111 | Endolysin production | 15% | 20% | 17% |
| PhiS63 | Endolysin; glycosyl-hydrolase production | 3% | 6% | 4% |
| Streptococcus phage EJ-1 | Endolysin production | 7% | 0% | 4% |
| Clostridium phage c-st | Endolysin production | 3% | 0% | 2% |
| Name and NCBI Reference Sequence | Size (bp) | Description | Occurrence | ||
|---|---|---|---|---|---|
| NE-Affected Isolates | Healthy Isolates | Total | |||
| pCW3-like plasmid (NC_010937.1) | 47,263 | tcp conjugation locus Tetracycline-resistance determinant Putative transposase | 53% | 80% | 63% |
| pDel1_4-like plasmid (NZ_CP019580.1) | 49,728 | tcp conjugation locus Tetracycline-resistance determinant Putative transposase | 46% | 73% | 56% |
| pCP15_3-like plasmid (NZ_CP019570.1) | 3843 | - | 38% | 20% | 31% |
| pCP15_1-like plasmid (NZ_CP019568.1) | 3202 | - | 38% | 20% | 31% |
| pJIR3537-like plasmid (NZ_CP025504.1) | 48,779 | Tetracycline-resistance determinant tcp conjugation locus Putative transposase | 15% | 60% | 31% |
| pl2_2016TE7641_69-like plasmid (NZ_MN503253.1) | 82,669 | tcp conjugation locus Tn3 family transposase Collagen binding domain-containing protein | 30% | 20% | 26% |
| pFORC3-like plasmid (NZ_CP009558.1) | 56,577 | tcp conjugation locus Tetracycline-resistance determinant Putative transposase | 19% | 33% | 24% |
| pJIR2774-like plasmid (DQ338473.1) | 14,640 | tcp conjugation locus | 3% | 40% | 17% |
| pLLY_N11_3-like plasmid (NZ_CP023413.1) | 72,060 | tcp conjugation locus Tetracycline-resistance determinant Putative transposase | 19% | 6% | 14% |
| pCP13-like plasmid (AP003515.1) | 54,310 | Probable collagen adhesin Probable transposase Resolvase | 11% | 13% | 12% |
| pSM101B-like plasmid (NC_008264.1) | 12,206 | BhlA/UviB family holin-like peptide | 0% | 33% | 12% |
| pDel1_3-like plasmid (NZ_CP019579.1) | 49,582 | Holin family protein IS630 transposase-related protein Chloramphenicol resistance protein | 0% | 20% | 7% |
| pDel1_2-like plasmid (NZ_CP019578.1) | 69,827 | tcp conjugation locus Putative transposase | 11% | 0% | 7% |
| pDel1_1-like plasmid (NZ_CP019577.1) | 82,596 | tcp conjugation locus Putative transposase Collagen binding domain-containing protein | 7% | 0% | 4% |
| pIP404-like plasmid (NC_001388.1) | 10,206 | Bacteriocin UviB | 0% | 13% | 4% |
| p4-like plasmid (NZ_MK275622.1) | 40,125 | IS1595 family transposase | 3% | 0% | 2% |
| pJIR3535-like plasmid (NZ_CP025502.1) | 81,826 | tcp conjugation locus Tn3 family transposase Collagen binding domain-containing protein | 3% | 0% | 2% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heidarpanah, S.; Li, K.; Thibodeau, A.; Meniaï, I.; Parreira, V.R.; Quessy, S.; Segura, M.; Fittipaldi, N.; Gaucher, M.-L. Genomic Diversity and Virulence Factors of Clostridium perfringens Isolated from Healthy and Necrotic Enteritis-Affected Broiler Chicken Farms in Quebec Province. Microorganisms 2024, 12, 2624. https://doi.org/10.3390/microorganisms12122624
Heidarpanah S, Li K, Thibodeau A, Meniaï I, Parreira VR, Quessy S, Segura M, Fittipaldi N, Gaucher M-L. Genomic Diversity and Virulence Factors of Clostridium perfringens Isolated from Healthy and Necrotic Enteritis-Affected Broiler Chicken Farms in Quebec Province. Microorganisms. 2024; 12(12):2624. https://doi.org/10.3390/microorganisms12122624
Chicago/Turabian StyleHeidarpanah, Sara, Kevin Li, Alexandre Thibodeau, Ilhem Meniaï, Valeria R. Parreira, Sylvain Quessy, Mariela Segura, Nahuel Fittipaldi, and Marie-Lou Gaucher. 2024. "Genomic Diversity and Virulence Factors of Clostridium perfringens Isolated from Healthy and Necrotic Enteritis-Affected Broiler Chicken Farms in Quebec Province" Microorganisms 12, no. 12: 2624. https://doi.org/10.3390/microorganisms12122624
APA StyleHeidarpanah, S., Li, K., Thibodeau, A., Meniaï, I., Parreira, V. R., Quessy, S., Segura, M., Fittipaldi, N., & Gaucher, M.-L. (2024). Genomic Diversity and Virulence Factors of Clostridium perfringens Isolated from Healthy and Necrotic Enteritis-Affected Broiler Chicken Farms in Quebec Province. Microorganisms, 12(12), 2624. https://doi.org/10.3390/microorganisms12122624