
Evaluating the Effects of Sugar Shift® Symbiotic on Microbiome Composition and LPS Regulation: A Double-Blind, Placebo-Controlled Study

 , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethical Considerations
2.2. Study Design
2.3. Sample Collection
2.4. Lipopolysaccharide (LPS) Determinations
2.5. HOMA-IR Index Calculation
2.6. Metagenome Analysis
2.6.1. 16S rRNA Gene Metagenomic Sequencing
2.6.2. 16S Metagenomic Taxonomic Profiling
2.6.3. Metagenomic Shotgun Sequencing
2.6.4. Metagenomic Functional Profiling
2.6.5. Metagenomic Taxonomic Profiling
2.6.6. Alpha Diversity Assessment
3. Results
3.1. Clinical Results
3.2. Metagenome Quality
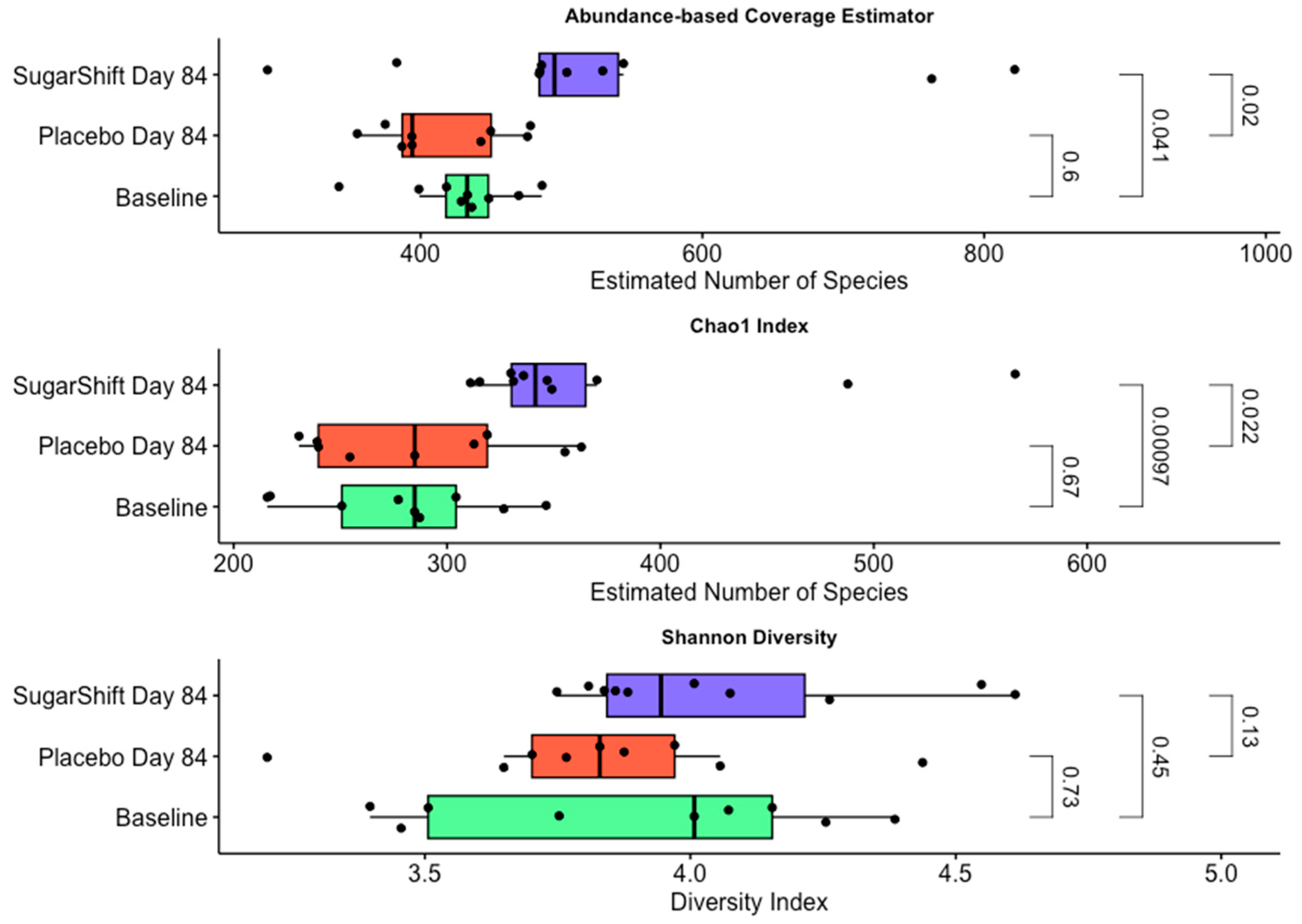
3.3. Changes in Alpha Diversity
3.4. Functional Annotation and Pathway Analysis of Metagenomic Data
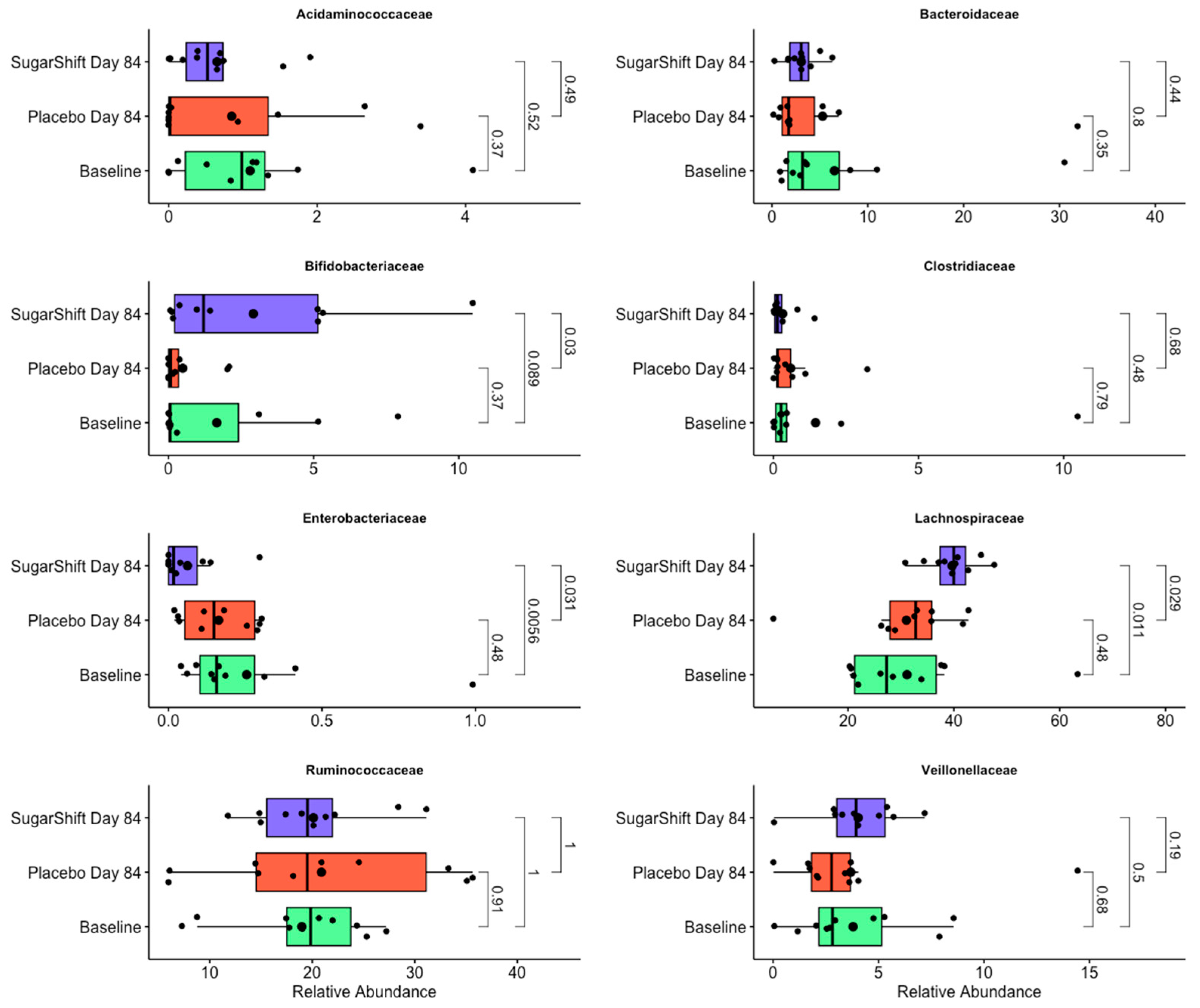
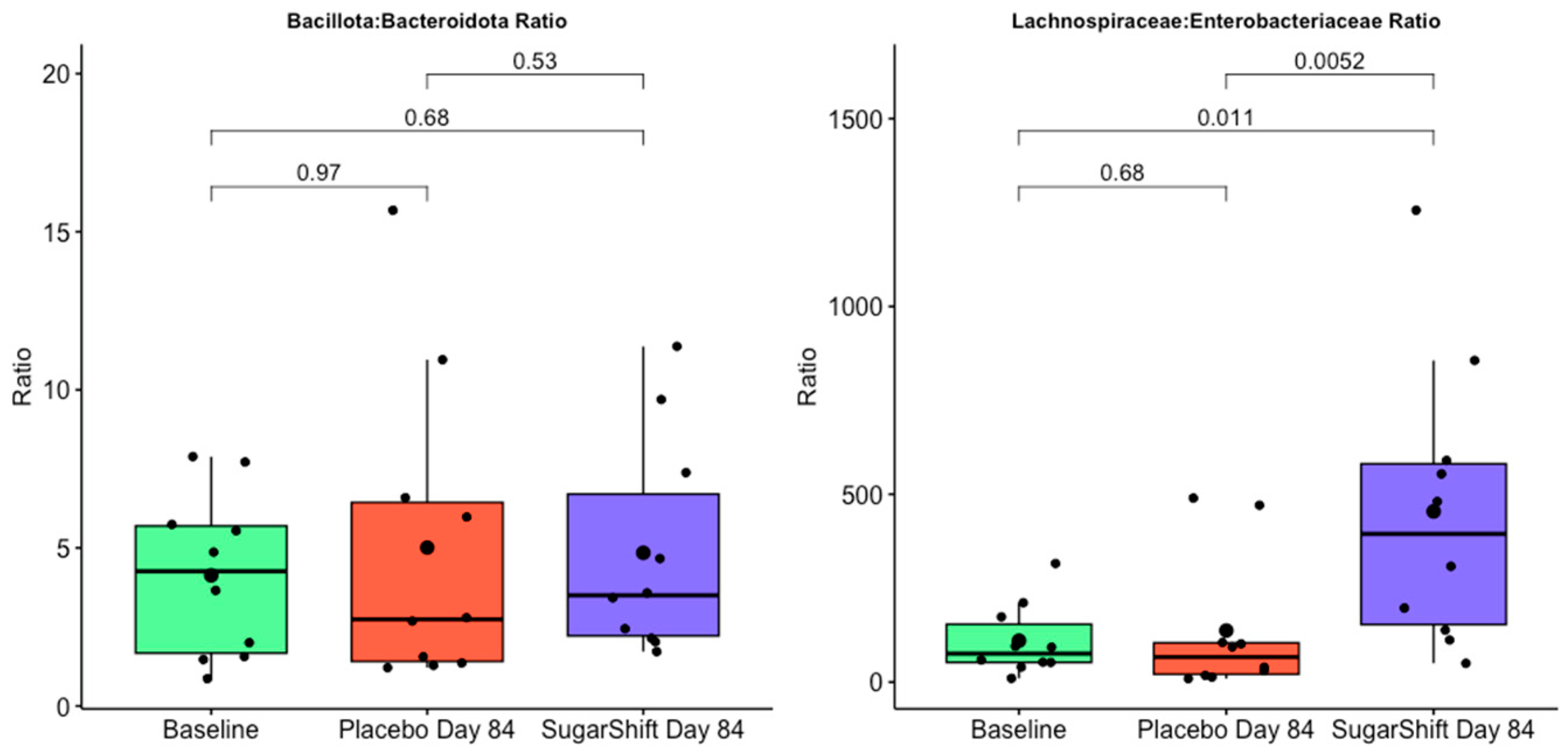
3.5. Taxonomic Composition and Findings
4. Discussion
4.1. Clinical Parameters
4.2. Metagenomic Sequences
4.3. Alpha Diversity Trends
4.4. Functional Analysis Trends
4.5. Taxonomic Diversity Analysis
4.6. Biomarkers for Gut Health
4.7. Mechanisms of Gut Microbiome Modulation by the Probiotic Sugar Shift in Type 2 Diabetes Management
4.8. Limitation of the Study
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hameed, I.; Masoodi, S.R.; Mir, S.A.; Nabi, M.; Ghazanfar, K.; Ganai, B.A. Type 2 diabetes mellitus: From a metabolic disorder to an inflammatory condition. World J. Diabetes 2015, 6, 598. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Tripathi, P. Gut microbiome and type 2 diabetes: Where we are and where to go? J. Nutr. Biochem. 2019, 63, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Saeedi, P.; Karuranga, S.; Pinkepank, M.; Ogurtsova, K.; Duncan, B.B.; Stein, C.; Basit, A.; Chan, J.C.; Mbanya, J.C.; et al. IDF Diabetes Atlas: Global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045. Diabetes Res. Clin. Pract. 2022, 183, 109119. [Google Scholar] [CrossRef] [PubMed]
- Singer-Englar, T.; Barlow, G.; Mathur, R. Obesity, diabetes, and the gut microbiome: An updated review. Expert Rev. Gastroenterol. Hepatol. 2019, 13, 3–15. [Google Scholar] [CrossRef]
- Ramachandran, A.; Snehalatha, C.; Raghavan, A.; Nanditha, A. Classification and diagnosis of diabetes. In Textbook of Diabetes; Wiley: Hoboken, NJ, USA, 2024; pp. 22–27. [Google Scholar]
- Dendup, T.; Feng, X.; Clingan, S.; Astell-Burt, T. Environmental risk factors for developing type 2 diabetes mellitus: A systematic review. Int. J. Environ. Res. Public Health 2018, 15, 78. [Google Scholar] [CrossRef]
- Gnauck, A.; Lentle, R.G.; Kruger, M.C. The characteristics and function of bacterial lipopolysaccharides and their endotoxic potential in humans. Int. Rev. Immunol. 2016, 35, 189–218. [Google Scholar] [CrossRef]
- Merkevičius, K.; Kundelis, R.; Maleckas, A.; Veličkienė, D. Microbiome changes after type 2 diabetes treatment: A systematic review. Medicina 2021, 57, 1084. [Google Scholar] [CrossRef]
- Rorato, R.; Borges, B.d.C.; Uchoa, E.T.; Antunes-Rodrigues, J.; Elias, C.F.; Elias, L.L.K. LPS-induced low-grade inflammation increases hypothalamic JNK expression and causes central insulin resistance irrespective of body weight changes. Int. J. Mol. Sci. 2017, 18, 1431. [Google Scholar] [CrossRef]
- Di Lorenzo, F.; De Castro, C.; Silipo, A.; Molinaro, A. Lipopolysaccharide structures of Gram-negative populations in the gut microbiota and effects on host interactions. FEMS Microbiol. Rev. 2019, 43, 257–272. [Google Scholar] [CrossRef]
- Massier, L.; Blüher, M.; Kovacs, P.; Chakaroun, R.M. Impaired intestinal barrier and tissue bacteria: Pathomechanisms for metabolic diseases. Front. Endocrinol. 2021, 12, 616506. [Google Scholar] [CrossRef]
- Moreira de Gouveia, M.I.; Bernalier-Donadille, A.; Jubelin, G. Enterobacteriaceae in the Human Gut: Dynamics and Ecological Roles in Health and Disease. Biology 2024, 13, 142. [Google Scholar] [CrossRef] [PubMed]
- Al Bander, Z.; Nitert, M.A.-O.; Mousa, A.A.-O.; Naderpoor, N.A.-O. The Gut Microbiota and Inflammation: An Overview. Int. J. Environ. Res. Public Health 2020, 17, 7618. [Google Scholar] [CrossRef]
- Yang, G.; Wei, J.; Liu, P.; Zhang, Q.; Tian, Y.; Hou, G.; Meng, L.; Xin, Y.; Jiang, X. Role of the gut microbiota in type 2 diabetes and related diseases. Metabolism 2021, 117, 154712. [Google Scholar] [CrossRef]
- Roshanravan, N.; Bastani, S.; Tutunchi, H.; Kafil, B.; Nikpayam, O.; Alamdari, N.M.; Hadi, A.; Sotoudeh, S.; Ghaffari, S.; Ostadrahimi, A. A comprehensive systematic review of the effectiveness of Akkermansia muciniphila, a member of the gut microbiome, for the management of obesity and associated metabolic disorders. Arch. Physiol. Biochem. 2023, 129, 741–751. [Google Scholar] [CrossRef]
- Liu, E.; Ji, X.; Zhou, K. Akkermansia muciniphila for the prevention of type 2 diabetes and obesity: A meta-analysis of animal studies. Nutrients 2024, 16, 3440. [Google Scholar] [CrossRef]
- Zeng, Z.; Chen, M.; Liu, Y.; Zhou, Y.; Liu, H.; Wang, S.; Ji, Y. Role of Akkermansia muciniphila in insulin resistance. J. Gastroenterol. Hepatol. 2024. [Google Scholar] [CrossRef] [PubMed]
- García, G.; Soto, J.; Rodríguez, L.; Nuez, M.; Domínguez, N.; Buchaca, E.F.; Martínez, D.; Gómez, R.J.; Ávila, Y.; Carlin, M.R.; et al. Metabolic Shifting Probiotic in Type 2 Diabetes Mellitus Management: Randomized Clinical Trial. J. Biotechnol. Biomed. 2023, 6, 270–280. [Google Scholar] [CrossRef]
- Shen, X.; Ma, C.; Yang, Y.; Liu, X.; Wang, B.; Wang, Y.; Zhang, G.; Bian, X.; Zhang, N. The Role and Mechanism of Probiotics Supplementation in Blood Glucose Regulation: A Review. Foods 2024, 13, 2719. [Google Scholar] [CrossRef]
- Carlin, M.R.; Kazemi, S.K.; Sangwan, N.; Cano, R.D.J. Probiotics and Methods of Use. U.S. Patent 11,850,270, 26 December 2023. [Google Scholar]
- Lewis, N.E.; Nagarajan, H.; Palsson, B.O. Constraining the metabolic genotype-phenotype relationship using a phylogeny of in silico methods. Nat. Rev. Microbiol. 2012, 10, 291–305. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, B.; Dunn, L. The declaration of Helsinki on medical research involving human subjects: A review of seventh revision. J. Nepal Health Res. Counc. 2019, 17, 548–552. [Google Scholar] [CrossRef]
- Moher, D.; Hopewell, S.; Schulz, K.F.; Montori, V.; Gøtzsche, P.C.; Devereaux, P.; Elbourne, D.; Egger, M.; Altman, D.G. CONSORT 2010 explanation and elaboration: Updated guidelines for reporting parallel group randomised trials. Int. J. Surg. 2012, 10, 28–55. [Google Scholar] [CrossRef] [PubMed]
- Friedman, M. The use of ranks to avoid the assumption of normality implicit in the analysis of variance. J. Am. Stat. Assoc. 1937, 32, 675–701. [Google Scholar] [CrossRef]
- Rosner, B.; Glynn, R.J.; Lee, M.L.T. The Wilcoxon signed rank test for paired comparisons of clustered data. Biometrics 2006, 62, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Christiansen, T.; Lauritsen, J. EpiData-Comprehensive Data Management and Basic Statistical Analysis System; EpiData Association: Odense, Denmark, 2010. [Google Scholar]
- Petersmann, A.; Müller-Wieland, D.; Müller, U.A.; Landgraf, R.; Nauck, M.; Freckmann, G.; Heinemann, L.; Schleicher, E. Definition, Classification and Diagnosis of Diabetes Mellitus. Exp. Clin. Endocrinol. Diabetes 2019, 127, S1–S7. [Google Scholar] [CrossRef]
- Trivedi, C.B.; Keuschnig, C.; Larose, C.; Rissi, D.V.; Mourot, R.; Bradley, J.A.; Winkel, M.; Benning, L.G. DNA/RNA preservation in glacial snow and ice samples. Front. Microbiol. 2022, 13, 894893. [Google Scholar] [CrossRef]
- Matthews, D.R.; Hosker, J.P.; Rudenski, A.S.; Naylor, B.A.; Treacher, D.F.; Turner, R.C. Homeostasis model assessment: Insulin resistance and β-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 1985, 28, 412–419. [Google Scholar] [CrossRef]
- Wallace, T.M.; Levy, J.C.; Matthews, D.R. Use and abuse of HOMA modeling. Diabetes Care 2004, 27, 1487–1495. [Google Scholar] [CrossRef]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef]
- Yoon, S.-H.; Ha, S.-M.; Kwon, S.; Lim, J.; Kim, Y.; Seo, H.; Chun, J. Introducing EzBioCloud: A taxonomically united database of 16S rRNA gene sequences and whole-genome assemblies. Int. J. Syst. Evol. Microbiol. 2017, 67, 1613. [Google Scholar] [CrossRef]
- Prasad, D.V.; Madhusudanan, S.; Jaganathan, S. uCLUST-a new algorithm for clustering unstructured data. ARPN J. Eng. Appl. Sci. 2015, 10, 2108–2117. [Google Scholar]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Doak, T.G. A parsimony approach to biological pathway reconstruction/inference for genomes and metagenomes. PLoS Comput. Biol. 2009, 5, e1000465. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wood, D.E.; Lu, J.; Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef]
- Chalita, M.; Ha, S.-M.; Kim, Y.O.; Oh, H.-S.; Yoon, S.-H.; Chun, J. Improved metagenomic taxonomic profiling using a curated core gene-based bacterial database reveals unrecognized species in the genus Streptococcus. Pathogens 2020, 9, 204. [Google Scholar] [CrossRef]
- Yoon, H.; Lee, D.H.; Lee, J.H.; Kwon, J.E.; Shin, C.M.; Yang, S.-J.; Park, S.-H.; Lee, J.H.; Kang, S.W.; Lee, J.-S.; et al. Characteristics of the Gut Microbiome of Healthy Young Male Soldiers in South Korea: The Effects of Smoking. Gut Liver 2020, 15, 243–252. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef]
- Daniel, W.W. Kruskal–Wallis one-way analysis of variance by ranks. In Applied Nonparametric Statistics; PWS-Kent: Boston, MA, USA, 1990; pp. 226–234. [Google Scholar]
- Chao, A.; Lee, S.-M. Estimating the number of classes via sample coverage. J. Am. Stat. Assoc. 1992, 87, 210–217. [Google Scholar] [CrossRef]
- Chao, A. Nonparametric estimation of the number of classes in a population. Scand. J. Stat. 1984, 11, 265–270. [Google Scholar]
- Vavrek, M.J. Fossil: Palaeoecological and palaeogeographical analysis tools. Palaeontol. Electron. 2011, 14, 16. [Google Scholar]
- Schloss, P.D.; Handelsman, J. Introducing DOTUR, a computer program for defining operational taxonomic units and estimating species richness. Appl. Environ. Microbiol. 2005, 71, 1501–1506. [Google Scholar] [CrossRef] [PubMed]
- Shannon, C.E. A mathematical theory of communication. ACM SIGMOBILE Mob. Comput. Commun. Rev. 2001, 5, 3–55. [Google Scholar] [CrossRef]
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; et al. Vegan Community Ecology Package Version 2. 5–7 November 2020. Available online: https://CRAN.R-project.org/package=vegan (accessed on 22 October 2024).
- Faith, D.P. The role of the phylogenetic diversity measure, PD, in bio-informatics: Getting the definition right. Evol. Bioinform. Online 2007, 2, 277–283. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Fisher, R.A. Statistical methods for research workers. In Breakthroughs in Statistics: Methodology and Distribution; Springer: Berlin/Heidelberg, Germany, 1970; pp. 66–70. [Google Scholar]
- Chao, A.; Li, P.C.; Agatha, S.; Foissner, W. A statistical approach to estimate soil ciliate diversity and distribution based on data from five continents. Oikos 2006, 114, 479–493. [Google Scholar] [CrossRef]
- Student. The probable error of a mean. Biometrika 1908, 6, 1–25. [Google Scholar] [CrossRef]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef]
- Kassambara, A. ggpubr:‘ggplot2’based Publication Ready Plots. R Package Version. 2018. Available online: https://cran.r-project.org/package=ggpubr (accessed on 22 October 2024).
- Alkhammash, A.M.; Alshehri, W.A.; Alhozali, A.M.; Bahieldin, A. Link of Gut Microbiome with Risk of Type 2 Diabetes Mellitus. J. Contemp. Med. Sci. 2022, 8, 363–369. [Google Scholar] [CrossRef]
- El-Zayat, S.R.; Sibaii, H.; Mannaa, F.A. Toll-like receptors activation, signaling, and targeting: An overview. Bull. Natl. Res. Cent. 2019, 43, 187. [Google Scholar] [CrossRef]
- Zhao, L.; Zhang, F.; Ding, X.; Wu, G.; Lam, Y.Y.; Wang, X.; Fu, H.; Xue, X.; Lu, C.; Ma, J.; et al. Gut bacteria selectively promoted by dietary fibers alleviate type 2 diabetes. Science 2018, 359, 1151–1156. [Google Scholar] [CrossRef] [PubMed]
- Koh, A.; De Vadder, F.; Kovatcheva-Datchary, P.; Bäckhed, F. From dietary fiber to host physiology: Short-chain fatty acids as key bacterial metabolites. Cell 2016, 165, 1332–1345. [Google Scholar] [CrossRef]
- Cani, P.D.; Amar, J.; Iglesias, M.A.; Poggi, M.; Knauf, C.; Bastelica, D.; Neyrinck, A.M.; Fava, F.; Tuohy, K.M.; Chabo, C.; et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007, 56, 1761–1772. [Google Scholar] [CrossRef] [PubMed]
- Sechovcová, H.; Mahayri, T.M.; Mrázek, J.; Jarošíková, R.; Husáková, J.; Wosková, V.; Fejfarová, V. Gut microbiota in relationship to diabetes mellitus and its late complications with a focus on diabetic foot syndrome: A review. Folia Microbiol. 2024, 69, 259–282. [Google Scholar] [CrossRef] [PubMed]
- Facchin, S.; Bertin, L.; Bonazzi, E.; Lorenzon, G.; De Barba, C.; Barberio, B.; Zingone, F.; Maniero, D.; Scarpa, M.; Ruffolo, C.; et al. Short-Chain Fatty Acids and Human Health: From Metabolic Pathways to Current Therapeutic Implications. Life 2024, 14, 559. [Google Scholar] [CrossRef]
- Ranneh, Y.; Fadel, A.; Md Akim, A.; Idris, I.; Ilesanmi-Oyelere, B.L.; Ismail, L.C. Effect of Dietary Fiber Supplementation on Metabolic Endotoxemia: A Protocol for Systematic Review and Meta-Analysis of Randomized Clinical Trials. Methods Protoc. 2023, 6, 84. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Gonzalez Peña, A.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hugerth, L.W.; Andersson, A.F. Analysing microbial community composition through amplicon sequencing: From sampling to hypothesis testing. Front. Microbiol. 2017, 8, 1561. [Google Scholar] [CrossRef]
- Knight, R.; Vrbanac, A.; Taylor, B.C.; Aksenov, A.; Callewaert, C.; Debelius, J.; Gonzalez, A.; Kosciolek, T.; McCall, L.-I.; McDonald, D.; et al. Best practices for analysing microbiomes. Nat. Rev. Microbiol. 2018, 16, 410–422. [Google Scholar] [CrossRef]
- Zhou, J.; He, Z.; Yang, Y.; Deng, Y.; Tringe, S.G.; Alvarez-Cohen, L. High-throughput metagenomic technologies for complex microbial community analysis: Open and closed formats. mBio 2015, 6, e02288-14. [Google Scholar] [CrossRef] [PubMed]
- Samal, A.; Ghosh, T.S. Meta-Analytic Investigation of Gut Microbial Community Structure Identifies a Panel of Stability-Promoting Microbiome Members Consistently Reduced with Gut Inflammation. Ph.D. Thesis, IIIT-Delhi, Delhi, India, 2023. [Google Scholar]
- Gil Sorribes, M. Predictive gut microbiome analysis for health assessment. Procedia Comput. Sci. 2024, 239, 1452–1459. [Google Scholar] [CrossRef]
- Moya, A.; Ferrer, M. Functional redundancy-induced stability of gut microbiota subjected to disturbance. Trends Microbiol. 2016, 24, 402–413. [Google Scholar] [CrossRef] [PubMed]
- Fassarella, M.; Blaak, E.E.; Penders, J.; Nauta, A.; Smidt, H.; Zoetendal, E.G. Gut microbiome stability and resilience: Elucidating the response to perturbations in order to modulate gut health. Gut 2021, 70, 595–605. [Google Scholar] [CrossRef]
- Cipelli, M.; da Silva, E.M.; Câmara, N.O.S. Gut Microbiota Resilience Mechanisms Against Pathogen Infection and its Role in Inflammatory Bowel Disease. Curr. Clin. Microbiol. Rep. 2023, 10, 187–197. [Google Scholar] [CrossRef]
- Peterson, C.T.; Perez Santiago, J.; Iablokov, S.N.; Chopra, D.; Rodionov, D.A.; Peterson, S.N. Short-chain fatty acids modulate healthy gut microbiota composition and functional potential. Curr. Microbiol. 2022, 79, 128. [Google Scholar] [CrossRef]
- Mohammad, S.; Thiemermann, C. Role of metabolic endotoxemia in systemic inflammation and potential interventions. Front. Immunol. 2021, 11, 594150. [Google Scholar] [CrossRef]
- Lin, X.; Han, H.; Wang, N.; Wang, C.; Qi, M.; Wang, J.; Liu, G. The Gut Microbial Regulation of Epigenetic Modification from a Metabolic Perspective. Int. J. Mol. Sci. 2024, 25, 7175. [Google Scholar] [CrossRef]
- Singh, V.; Lee, G.; Son, H.; Koh, H.; Kim, E.S.; Unno, T.; Shin, J.H. Butyrate producers,“The Sentinel of Gut”: Their intestinal significance with and beyond butyrate, and prospective use as microbial therapeutics. Front. Microbiol. 2023, 13, 1103836. [Google Scholar] [CrossRef]
- Fu, Y.; Lyu, J.; Wang, S. The role of intestinal microbes on intestinal barrier function and host immunity from a metabolite perspective. Front. Immunol. 2023, 14, 1277102. [Google Scholar] [CrossRef]
- Archana, A.K.; Gupta, A.K.; Noumani, A.; Panday, D.K.; Zaidi, F.; Sahu, G.K.; Joshi, G.; Yadav, M.; Borah, S.J.; Susmitha, V.; et al. Gut microbiota derived short-chain fatty acids in physiology and pathology: An update. Cell Biochem. Funct. 2024, 42, e4108. [Google Scholar] [CrossRef] [PubMed]
- Markowiak-Kopeć, P.; Śliżewska, K. The effect of probiotics on the production of short-chain fatty acids by human intestinal microbiome. Nutrients 2020, 12, 1107. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Zhu, Y.; Li, X.; Sun, B. Dynamic balancing of intestinal short-chain fatty acids: The crucial role of bacterial metabolism. Trends Food Sci. Technol. 2020, 100, 118–130. [Google Scholar] [CrossRef]
- Liu, X.; Mao, B.; Gu, J.; Wu, J.; Cui, S.; Wang, G.; Zhao, J.; Zhang, H.; Chen, W. Blautia—A new functional genus with potential probiotic properties? Gut Microbes 2021, 13, 1875796. [Google Scholar] [CrossRef]
- Gryaznova, M.; Dvoretskaya, Y.; Burakova, I.; Syromyatnikov, M.; Popov, E.; Kokina, A.; Mikhaylov, E.; Popov, V. Dynamics of changes in the gut microbiota of healthy mice fed with lactic acid bacteria and bifidobacteria. Microorganisms 2022, 10, 1020. [Google Scholar] [CrossRef]
- Gao, Y.; Zhao, T.; Lv, N.; Liu, S.; Yuan, T.; Fu, Y.; Zhao, W.; Zhu, B. Metformin-induced changes of the gut microbiota in patients with type 2 diabetes mellitus: Results from a prospective cohort study. Endocrine 2024, 85, 1178–1192. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D.; Osto, M.; Geurts, L.; Everard, A. Involvement of gut microbiota in the development of low-grade inflammation and type 2 diabetes associated with obesity. Gut Microbes 2012, 3, 279–288. [Google Scholar] [CrossRef]
- Snelson, M.; de Pasquale, C.; Ekinci, E.I.; Coughlan, M.T. Gut microbiome, prebiotics, intestinal permeability and diabetes complications. Best Pract. Res. Clin. Endocrinol. Metab. 2021, 35, 101507. [Google Scholar] [CrossRef]
- Candelli, M.; Franza, L.; Pignataro, G.; Ojetti, V.; Covino, M.; Piccioni, A.; Gasbarrini, A.; Franceschi, F. Interaction between Lipopolysaccharide and Gut Microbiota in Inflammatory Bowel Diseases. Int. J. Mol. Sci. 2021, 22, 6242. [Google Scholar] [CrossRef]
- Salguero, M.V.; Al-Obaide, M.A.I.; Singh, R.; Siepmann, T.; Vasylyeva, T.L. Dysbiosis of Gram-negative gut microbiota and the associated serum lipopolysaccharide exacerbates inflammation in type 2 diabetic patients with chronic kidney disease. Exp. Ther. Med. 2019, 18, 3461–3469. [Google Scholar] [CrossRef]
- Baldelli, V.; Scaldaferri, F.; Putignani, L.; Del Chierico, F. The role of Enterobacteriaceae in gut microbiota dysbiosis in inflammatory bowel diseases. Microorganisms 2021, 9, 697. [Google Scholar] [CrossRef] [PubMed]
- Rigottier-Gois, L. Dysbiosis in inflammatory bowel diseases: The oxygen hypothesis. ISME J. 2013, 7, 1256–1261. [Google Scholar] [CrossRef] [PubMed]
- Litvak, Y.; Byndloss, M.X.; Tsolis, R.M.; Bäumler, A.J. Dysbiotic Proteobacteria expansion: A microbial signature of epithelial dysfunction. Curr. Opin. Microbiol. 2017, 39, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Winter, S.E.; Bäumler, A.J. Gut dysbiosis: Ecological causes and causative effects on human disease. Proc. Natl. Acad. Sci. USA 2023, 120, e2316579120. [Google Scholar] [CrossRef]
- Rivera-Chávez, F.; Lopez, C.A.; Bäumler, A.J. Oxygen as a driver of gut dysbiosis. Free Radic. Biol. Med. 2017, 105, 93–101. [Google Scholar] [CrossRef]
- Lay, C.; Chu, C.W.; Purbojati, R.W.; Acerbi, E.; Drautz-Moses, D.I.; de Sessions, P.F.; Jie, S.; Ho, E.; Kok, Y.J.; Bi, X.; et al. A synbiotic intervention modulates meta-omics signatures of gut redox potential and acidity in elective caesarean born infants. BMC Microbiol. 2021, 21, 191. [Google Scholar] [CrossRef]
- Rath, E.; Haller, D. Intestinal epithelial cell metabolism at the interface of microbial dysbiosis and tissue injury. Mucosal Immunol. 2022, 15, 595–604. [Google Scholar] [CrossRef]
- André, A.C.; Debande, L.; Marteyn, B.S. The selective advantage of facultative anaerobes relies on their unique ability to cope with changing oxygen levels during infection. Cell. Microbiol. 2021, 23, e13338. [Google Scholar] [CrossRef] [PubMed]
- Gueddouri, D.; Caüzac, M.; Fauveau, V.; Benhamed, F.; Charifi, W.; Beaudoin, L.; Rouland, M.; Sicherre, F.; Lehuen, A.; Postic, C.; et al. Insulin resistance per se drives early and reversible dysbiosis-mediated gut barrier impairment and bactericidal dysfunction. Mol. Metab. 2022, 57, 101438. [Google Scholar] [CrossRef]
- Fusco, W.; Lorenzo, M.B.; Cintoni, M.; Porcari, S.; Rinninella, E.; Kaitsas, F.; Lener, E.; Mele, M.C.; Gasbarrini, A.; Collado, M.C.; et al. Short-chain fatty-acid-producing bacteria: Key components of the human gut microbiota. Nutrients 2023, 15, 2211. [Google Scholar] [CrossRef]
- Caricilli, A.M.; Saad, M.J. The role of gut microbiota on insulin resistance. Nutrients 2013, 5, 829–851. [Google Scholar] [CrossRef] [PubMed]
- Gurung, M.; Li, Z.; You, H.; Rodrigues, R.; Jump, D.B.; Morgun, A.; Shulzhenko, N. Role of gut microbiota in type 2 diabetes pathophysiology. EBioMedicine 2020, 51, 102590. [Google Scholar] [CrossRef] [PubMed]
- Qian, X.; Si, Q.; Lin, G.; Zhu, M.; Lu, J.; Zhang, H.; Wang, G.; Chen, W. Bifidobacterium adolescentis Is Effective in Relieving Type 2 Diabetes and May Be Related to Its Dominant Core Genome and Gut Microbiota Modulation Capacity. Nutrients 2022, 14, 2479. [Google Scholar] [CrossRef]
- Louis, P.; Flint, H.J. Formation of propionate and butyrate by the human colonic microbiota. Environ. Microbiol. 2017, 19, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Kamada, N.; Seo, S.-U.; Chen, G.Y.; Núñez, G. Role of the gut microbiota in immunity and inflammatory disease. Nat. Rev. Immunol. 2013, 13, 321–335. [Google Scholar] [CrossRef]
- O’Callaghan, A.; Van Sinderen, D. Bifidobacteria and their role as members of the human gut microbiota. Front. Microbiol. 2016, 7, 925. [Google Scholar] [CrossRef]
- Rivière, A.; Selak, M.; Lantin, D.; Leroy, F.; De Vuyst, L. Bifidobacteria and butyrate-producing colon bacteria: Importance and strategies for their stimulation in the human gut. Front. Microbiol. 2016, 7, 979. [Google Scholar] [CrossRef]
- Scott, K.P.; Martin, J.C.; Duncan, S.H.; Flint, H.J. Prebiotic stimulation of human colonic butyrate-producing bacteria and bifidobacteria, in vitro. FEMS Microbiol. Ecol. 2014, 87, 30–40. [Google Scholar] [CrossRef]
- Laursen, M.F.; Laursen, R.P.; Larnkjær, A.; Mølgaard, C.; Michaelsen, K.F.; Frøkiær, H.; Bahl, M.I.; Licht, T.R. Faecalibacterium Gut Colonization Is Accelerated by Presence of Older Siblings. mSphere 2017, 2, e00448-17. [Google Scholar] [CrossRef]
- Parsaei, M.; Sarafraz, N.; Moaddab, S.Y.; Ebrahimzadeh Leylabadlo, H. The importance of Faecalibacterium prausnitzii in human health and diseases. New Microbes New Infect. 2021, 43, 100928. [Google Scholar] [CrossRef]
- Sokol, H.; Pigneur, B.; Watterlot, L.; Lakhdari, O.; Bermúdez-Humaran, L.G.; Gratadoux, J.-J.; Blugeon, S.; Bridonneau, C.; Furet, J.-P.; Corthier, G.; et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc. Natl. Acad. Sci. USA 2008, 105, 16731–16736. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, Y.; Yang, L.; Feng, J.; Tian, S.; Chen, L.; Huang, W.; Liu, J.; Wang, X. Integrated 16S rRNA sequencing and metagenomics insights into microbial dysbiosis and distinct virulence factors in inflammatory bowel disease. Front. Microbiol. 2024, 15, 1375804. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, D.; Jia, H.; Feng, Q.; Wang, D.; Liang, D.; Wu, X.; Li, J.; Tang, L.; Li, Y.; et al. The oral and gut microbiomes are perturbed in rheumatoid arthritis and partly normalized after treatment. Nat. Med. 2015, 21, 895–905. [Google Scholar] [CrossRef]
- Jayashree, B.; Bibin, Y.S.; Prabhu, D.; Shanthirani, C.S.; Gokulakrishnan, K.; Lakshmi, B.S.; Mohan, V.; Balasubramanyam, M. Increased circulatory levels of lipopolysaccharide (LPS) and zonulin signify novel biomarkers of proinflammation in patients with type 2 diabetes. Mol. Cell. Biochem. 2014, 388, 203–210. [Google Scholar] [CrossRef]
- Ishikawa, D.; Zhang, X.; Nomura, K.; Shibuya, T.; Hojo, M.; Yamashita, M.; Koizumi, S.; Yamazaki, F.; Iwamoto, S.; Saito, M.; et al. Anti-inflammatory Effects of Bacteroidota Strains Derived From Outstanding Donors of Fecal Microbiota Transplantation for the Treatment of Ulcerative Colitis. Inflammatory Bowel Diseases. Inflamm. Bowel Dis. 2024, 30, 2136–2145. [Google Scholar] [CrossRef] [PubMed]
- Czarnowski, P.; Mikula, M.; Ostrowski, J.; Żeber-Lubecka, N. Gas Chromatography–Mass Spectrometry-Based Analyses of Fecal Short-Chain Fatty Acids (SCFAs): A Summary Review and Own Experience. Biomedicines 2024, 12, 1904. [Google Scholar] [CrossRef]
- Kozhakhmetov, S.; Kaiyrlykyzy, A.; Jarmukhanov, Z.; Vinogradova, E.; Zholdasbekova, G.; Alzhanova, D.; Kunz, J.; Kushugulova, A.; Askarova, S. Inflammatory Manifestations Associated With Gut Dysbiosis in Alzheimer’s Disease. Int. J. Alzheimer’s Dis. 2024, 2024, 9741811. [Google Scholar] [CrossRef]
- Huda, M.N.; Kim, M.; Bennett, B.J. Modulating the microbiota as a therapeutic intervention for type 2 diabetes. Front. Endocrinol. 2021, 12, 632335. [Google Scholar] [CrossRef]
- Hernandez-Sanabria, E.; Heiremans, E.; Arroyo, M.C.; Props, R.; Leclercq, L.; Snoeys, J.; Van de Wiele, T. Short-term supplementation of celecoxib-shifted butyrate production on a simulated model of the gut microbial ecosystem and ameliorated in vitro inflammation. NPJ Biofilms Microbiomes 2020, 6, 9. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, B.S.; Profir, M.; Rosu, O.A.; Ionescu, R.F.; Cretoiu, S.M. The Intestinal Microbiome in Humans: Its Role for a Healthy Life and in the Onset of Diseases. In Human Physiology Annual Volume 2024; IntechOpen: London, UK, 2024. [Google Scholar]
- Kusnadi, Y.; Saleh, M.I.; Ali, Z.; Hermansyah, H.; Murti, K.; Hafy, Z.; Yuristo, E. Firmicutes/Bacteroidetes ratio of gut microbiota and its relationships with clinical parameters of type 2 diabetes mellitus: A systematic review. Open Access Maced. J. Med Sci. 2023, 11, 67–72. [Google Scholar] [CrossRef]
- Ahmed, K.; Choi, H.-N.; Cho, S.-R.; Yim, J.-E. Association of Firmicutes/Bacteroidetes Ratio with Body Mass Index in Korean Type 2 Diabetes Mellitus Patients. Metabolites 2024, 14, 518. [Google Scholar] [CrossRef] [PubMed]
- Frolova, M.S.; Suvorova, I.A.; Iablokov, S.N.; Petrov, S.N.; Rodionov, D.A. Genomic reconstruction of short-chain fatty acid production by the human gut microbiota. Front. Mol. Biosci. 2022, 9, 949563. [Google Scholar] [CrossRef] [PubMed]
- Malesza, I.J.; Malesza, M.; Walkowiak, J.; Mussin, N.; Walkowiak, D.; Aringazina, R.; Bartkowiak-Wieczorek, J.; Mądry, E. High-fat, western-style diet, systemic inflammation, and gut microbiota: A narrative review. Cells 2021, 10, 3164. [Google Scholar] [CrossRef] [PubMed]
- Caballero-Flores, G.; Pickard, J.M.; Núñez, G. Microbiota-mediated colonization resistance: Mechanisms and regulation. Nat. Rev. Microbiol. 2023, 21, 347–360. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D.; Bibiloni, R.; Knauf, C.; Waget, A.; Neyrinck, A.M.; Delzenne, N.M.; Burcelin, R. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet–induced obesity and diabetes in mice. Diabetes 2008, 57, 1470–1481. [Google Scholar] [CrossRef]
- Wu, S.; Yang, L.; Fu, Y.; Liao, Z.; Cai, D.; Liu, Z. Intestinal barrier function and neurodegenerative disease. CNS Neurol. Disord.-Drug Targets 2024, 23, 1134–1142. [Google Scholar] [CrossRef]
- Gheorghe, A.S.; Negru, M.; Preda, M.; Mihăilă, R.I.; Komporaly, I.A.; Dumitrescu, E.A.; Lungulescu, C.V.; Kajanto, L.A.; Georgescu, B.; Radu, E.A.; et al. Biochemical and Metabolical Pathways Associated with Microbiota-Derived Butyrate in Colorectal Cancer and Omega-3 Fatty Acids Implications: A Narrative Review. Nutrients 2022, 14, 1152. [Google Scholar] [CrossRef]
- Mazhar, S.; Khokhlova, E.; Colom, J.; Simon, A.; Deaton, J.; Rea, K. In vitro and in silico assessment of probiotic and functional properties of Bacillus subtilis DE111®. Front. Microbiol. 2023, 13, 1101144. [Google Scholar] [CrossRef]
- Freedman, K.E.; Hill, J.L.; Wei, Y.; Vazquez, A.R.; Grubb, D.S.; Trotter, R.E.; Wrigley, S.D.; Johnson, S.A.; Foster, M.T.; Weir, T.L. Examining the gastrointestinal and immunomodulatory effects of the novel probiotic Bacillus subtilis DE111. Int. J. Mol. Sci. 2021, 22, 2453. [Google Scholar] [CrossRef]
- Fukuda, S.; Toh, H.; Hase, K.; Oshima, K.; Nakanishi, Y.; Yoshimura, K.; Tobe, T.; Clarke, J.M.; Topping, D.L.; Suzuki, T.; et al. Bifidobacteria can protect from enteropathogenic infection through production of acetate. Nature 2011, 469, 543. [Google Scholar] [CrossRef]
- De Vries, M.C.; Vaughan, E.E.; Kleerebezem, M.; de Vos, W.M. Lactobacillus plantarum—Survival, functional and potential probiotic properties in the human intestinal tract. Int. Dairy J. 2006, 16, 1018–1028. [Google Scholar] [CrossRef]
- Adıgüzel, E.; Çiçek, B. (Eds.) Dietary Approaches and Nutritional Supplements in the Management of Autism Spectrum Disorder. In Autismo: Uma Abordagem Multiprofissional; Editora Científica Digital: Juazeiro, BA, Brazil, 2023. [Google Scholar]
- Shin, S.-Y.; Han, N.S. Leuconostoc spp. as starters and their beneficial roles in fermented foods. In Beneficial Microorganisms in Food and Nutraceuticals; Springer: Berlin/Heidelberg, Germany, 2015; pp. 111–132. [Google Scholar]
- Mathur, S.; Singh, R. Antibiotic resistance in food lactic acid bacteria—A review. Int. J. Food Microbiol. 2005, 105, 281–295. [Google Scholar] [CrossRef] [PubMed]
- Anjana a Tiwari, S.K. Bacteriocin-producing probiotic lactic acid bacteria in controlling dysbiosis of the gut microbiota. Front. Cell. Infect. Microbiol. 2022, 12, 851140. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Demographic Variables | SS Cohort (n = 30) a | Placebo (n = 27) a | p-Value | |||
|---|---|---|---|---|---|---|
| No. | % | No. | % | |||
| Sex | Female | 18 | 60.0 | 14 | 51.9 | 0.725 b |
| Male | 12 | 40.0 | 13 | 48.1 | ||
| Age | Median ± SD | 56.3 ± 6.7 | 53.2 ± 7.6 | 0.120 c | ||
| Nutritional Assessment | Normal weight | 3 | 10.0 | 5 | 18.5 | 0.722 a |
| Overweight | 16 | 53.3 | 8 | 29.6 | ||
| Obesity | 11 | 36.7 | 14 | 51.9 | ||
| Kind of Treatment | Diet | 3 | 10.0 | 2 | 7.4 | 0.549 a |
| Diet plus oral hypoglycemic agents | 19 | 63.3 | 15 | 55.6 | ||
| Insulin | 0 | 0.0 | 2 | 7.4 | ||
| Combined treatment | 8 | 26.7 | 8 | 29.6 | ||
| Ortholog | Gene | Enzyme | Baseline | Sugar Shift Day 84 |
|---|---|---|---|---|
| Mean ± SD | Mean ± SD | |||
| LPS Biosynthesis Genes | ||||
| K02847 | waaL/kdsB | 3-deoxy-manno-octulosonate cytidylyltransferase [EC:2.7.7.38] | 59.45 ± 21.55 | 36.54 ± 5.71 |
| p = 0.006 * | ||||
| K02848 | waaP/kdsC | 3-deoxy-manno-octulosonate 8-phosphate phosphatase [EC:3.1.3.45] | 13.04 ± 6.31 | 3.93 ± 1.71 |
| p = 0.0008 | ||||
| K03760 | eptA/kdsA | 3-deoxy-manno-octulosonate 8-phosphate synthase [EC:2.5.1.55] | 99.61 ± 49.02 | 34.32 ± 5.65 |
| p = 0.005 | ||||
| K00677 | lpxA | UDP-N-acetylglucosamine acyltransferase [EC:2.3.1.129] | 694.20 ± 304.15 | 439.13 ± 142.52 |
| p = 0.005 | ||||
| K00748 | lpxB | lipid-A-disaccharide synthase [EC:2.4.1.182] | 895.65 ± 421.56 | 470.14 ± 152.40 |
| p = 0.0008 | ||||
| K02535 | lpxC | UDP-3-O-[3-hydroxymyristoyl] N-acetylglucosamine deacetylase [EC:3.5.1.108] | 1009.99 ± 459.61 | 400.88 ± 152.02 |
| p = 0.0006 | ||||
| K02536 | lpxD | UDP-3-O-[3-hydroxymyristoyl] glucosamine N-acyltransferase [EC:2.3.1.191] | 778.59 ± 276.12 | 371.92 ± 115.31 |
| p = 0.0007 | ||||
| K03269 | lpxH | UDP-2,3-diacylglucosamine hydrolase [EC:3.6.1.54] | 719.29 ± 233.54 | 279.04 ± 126.52 |
| p = 0.0005 | ||||
| K00912 | lphK | tetraacyldisaccharide 4′-kinase [EC:2.7.1.130] | 858.77 ± 272.11 | 425.71 ± 129.76 |
| p = 0.0003 | ||||
| K02515 | lphL | 3-deoxy-manno-octulosonate 8-phosphate phosphatase [EC:3.1.3.45] | 869.92 ± 240.07 | 446.64 ± 175.28 |
| p = 0.0005 | ||||
| SCFA Biosynthesis Genes | ||||
| K00925 | ackA | Acetate kinase [EC:2.7.2.1] | 1592.2 ± 969.99 | 3455.7 ± 1889.65 |
| p = 0.008 | ||||
| K00175 | crt | Enoyl-CoA hydratase [EC:4.2.1.17] | 365.3 ± 380.37 | 652.5 ± 396.11 |
| p = 0.037 | ||||
| K00823 | gabT | 4-aminobutyrate aminotransferase/(S)-3-amino-2-methylpropionate transaminase/5-aminovalerate transaminase [EC:2.6.1.19 2.6.1.22 2.6.1.48] | 164.1 ± 133.71 | 292.3 ± 162.1 |
| p = 0.008 | ||||
| K18566 | frdA | NADH-dependent fumarate reductase subunit A [EC:1.3.1.6] | 703.1 ± 642.5 | 1406.3 ± 297.61 |
| p = 0.016 | ||||
| Taxon Name | p-Value | Max Group | COHORT | ||
|---|---|---|---|---|---|
| Baseline | Sugar Shift Day 84 | Placebo Day 84 | |||
| Bacteroides eggerthii | 0.01974 | Baseline | 1.52822 | 0.13288 | 0.00000 |
| CP017245_s | 0.03314 | Sugar Shift Day 84 | 0.00000 | 0.85761 | 0.00000 |
| PAC001135_s | 0.04082 | Sugar Shift Day 84 | 0.10157 | 0.38603 | 0.04486 |
| LLKB_g | 0.04183 | Sugar Shift Day 84 | 0.14974 | 0.37675 | 0.13256 |
| PAC001206_s | 0.04590 | Sugar Shift Day 84 | 0.17914 | 0.18373 | 0.00000 |
| Oscillibacter_uc | 0.03216 | Baseline | 0.22961 | 0.22653 | 0.05031 |
| PAC001233_s | 0.02572 | Sugar Shift Day 84 | 0.02719 | 0.20221 | 0.04486 |
| Alistipes finegoldii | 0.03909 | Sugar Shift Day 84 | 0.06623 | 0.12565 | 0.00729 |
| PAC001036_s | 0.03175 | Sugar Shift Day 84 | 0.07542 | 0.10291 | 0.00000 |
| Lactococcus garvieae gp. | 0.01795 | Baseline | 0.07653 | 0.00000 | 0.00000 |
| Bacteroides xylanisolvens | 0.02666 | Baseline | 0.06002 | 0.00745 | 0.04993 |
| PAC001263_s | 0.02927 | Sugar Shift Day 84 | 0.00485 | 0.03417 | 0.00000 |
| PAC001458_s | 0.03314 | Placebo Day 84 | 0.00000 | 0.00000 | 0.02679 |
| PAC001458_g | 0.03314 | Placebo Day 84 | 0.00000 | 0.00000 | 0.02679 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
García, G.; Soto, J.; Netherland, M., Jr.; Hasan, N.A.; Buchaca, E.; Martínez, D.; Carlin, M.; de Jesus Cano, R. Evaluating the Effects of Sugar Shift® Symbiotic on Microbiome Composition and LPS Regulation: A Double-Blind, Placebo-Controlled Study. Microorganisms 2024, 12, 2525. https://doi.org/10.3390/microorganisms12122525
García G, Soto J, Netherland M Jr., Hasan NA, Buchaca E, Martínez D, Carlin M, de Jesus Cano R. Evaluating the Effects of Sugar Shift® Symbiotic on Microbiome Composition and LPS Regulation: A Double-Blind, Placebo-Controlled Study. Microorganisms. 2024; 12(12):2525. https://doi.org/10.3390/microorganisms12122525
Chicago/Turabian StyleGarcía, Gissel, Josanne Soto, Michael Netherland, Jr., Nur A. Hasan, Emilio Buchaca, Duniesky Martínez, Martha Carlin, and Raúl de Jesus Cano. 2024. "Evaluating the Effects of Sugar Shift® Symbiotic on Microbiome Composition and LPS Regulation: A Double-Blind, Placebo-Controlled Study" Microorganisms 12, no. 12: 2525. https://doi.org/10.3390/microorganisms12122525
APA StyleGarcía, G., Soto, J., Netherland, M., Jr., Hasan, N. A., Buchaca, E., Martínez, D., Carlin, M., & de Jesus Cano, R. (2024). Evaluating the Effects of Sugar Shift® Symbiotic on Microbiome Composition and LPS Regulation: A Double-Blind, Placebo-Controlled Study. Microorganisms, 12(12), 2525. https://doi.org/10.3390/microorganisms12122525