
Comparative Genomics Reveal Distinct Environment Preference and Functional Adaptation Among Lineages of Gemmatimonadota

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Data Acquisition
2.2. Genome Dereplication and Taxonomic and Phylogenomic Analysis
2.3. Functional Annotation and Metabolic Reconstruction
2.4. Comparison on Functional Profiles
2.5. Identification of Functional Genes Related to Adaptation to Different Environments
3. Results and Discussion
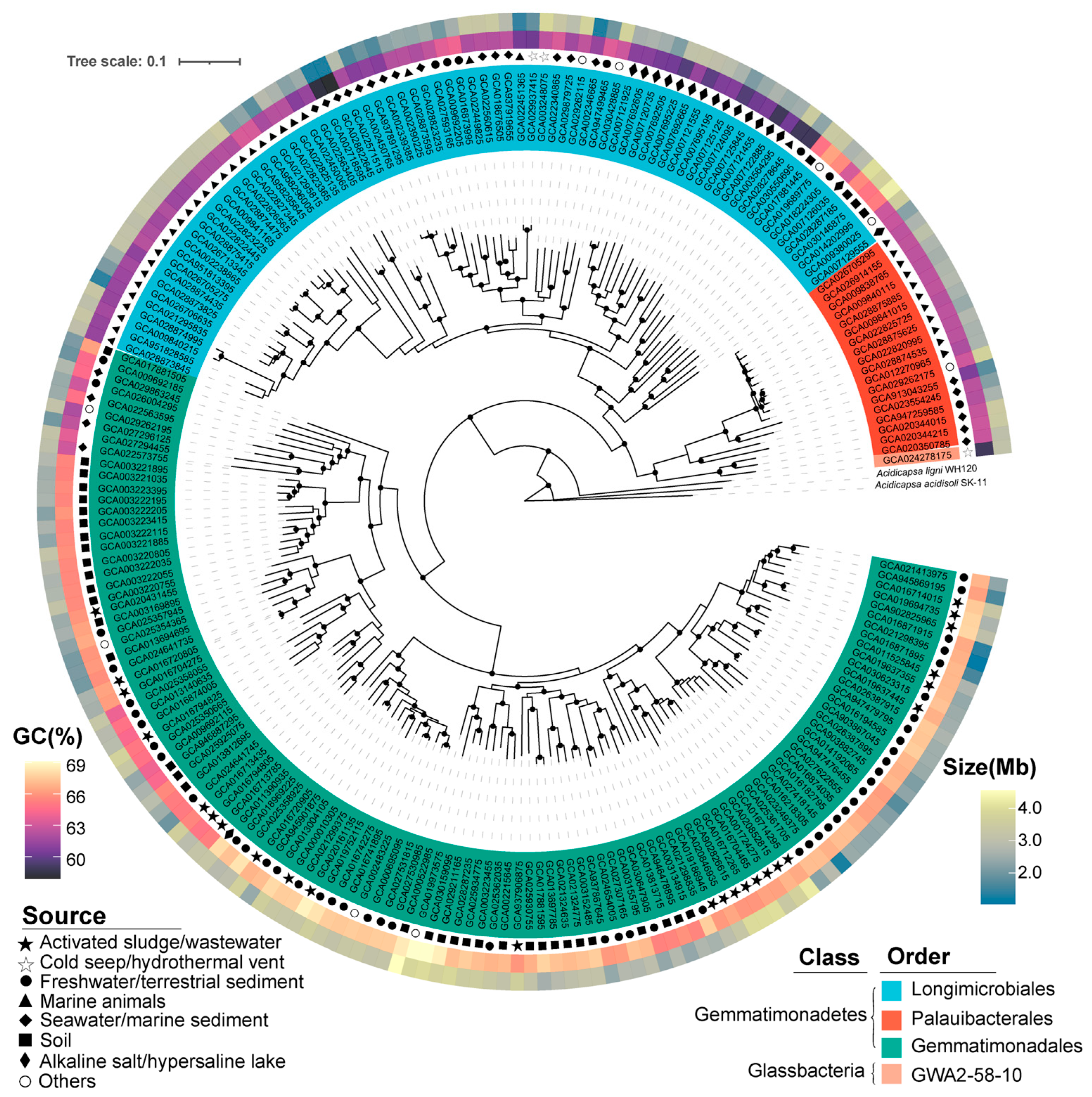
3.1. Biogeography and Taxonomy Analysis of Gemmatimonadota
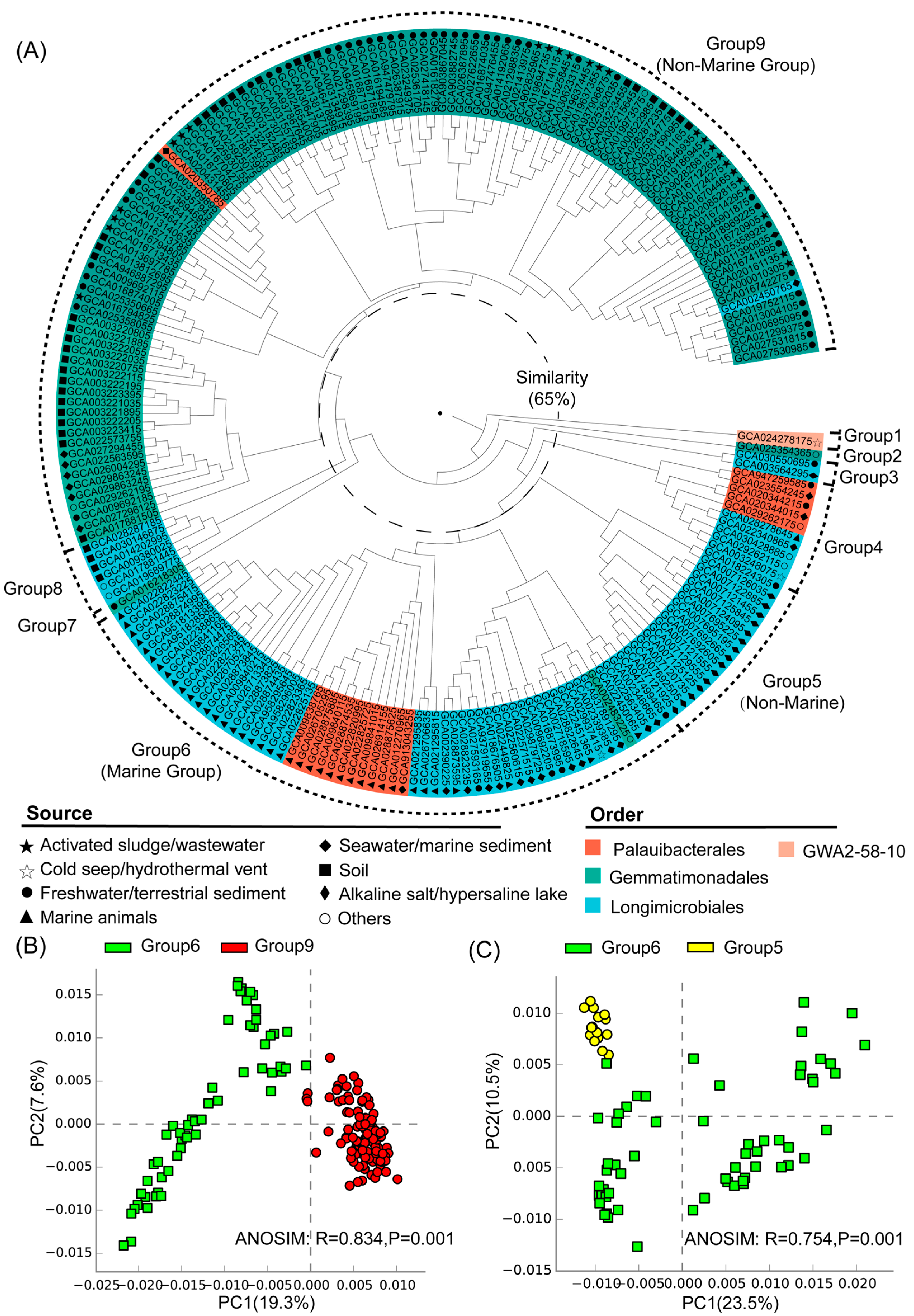
3.2. Environmental Preference of Gemmatimonadota Based on Functional Profiles
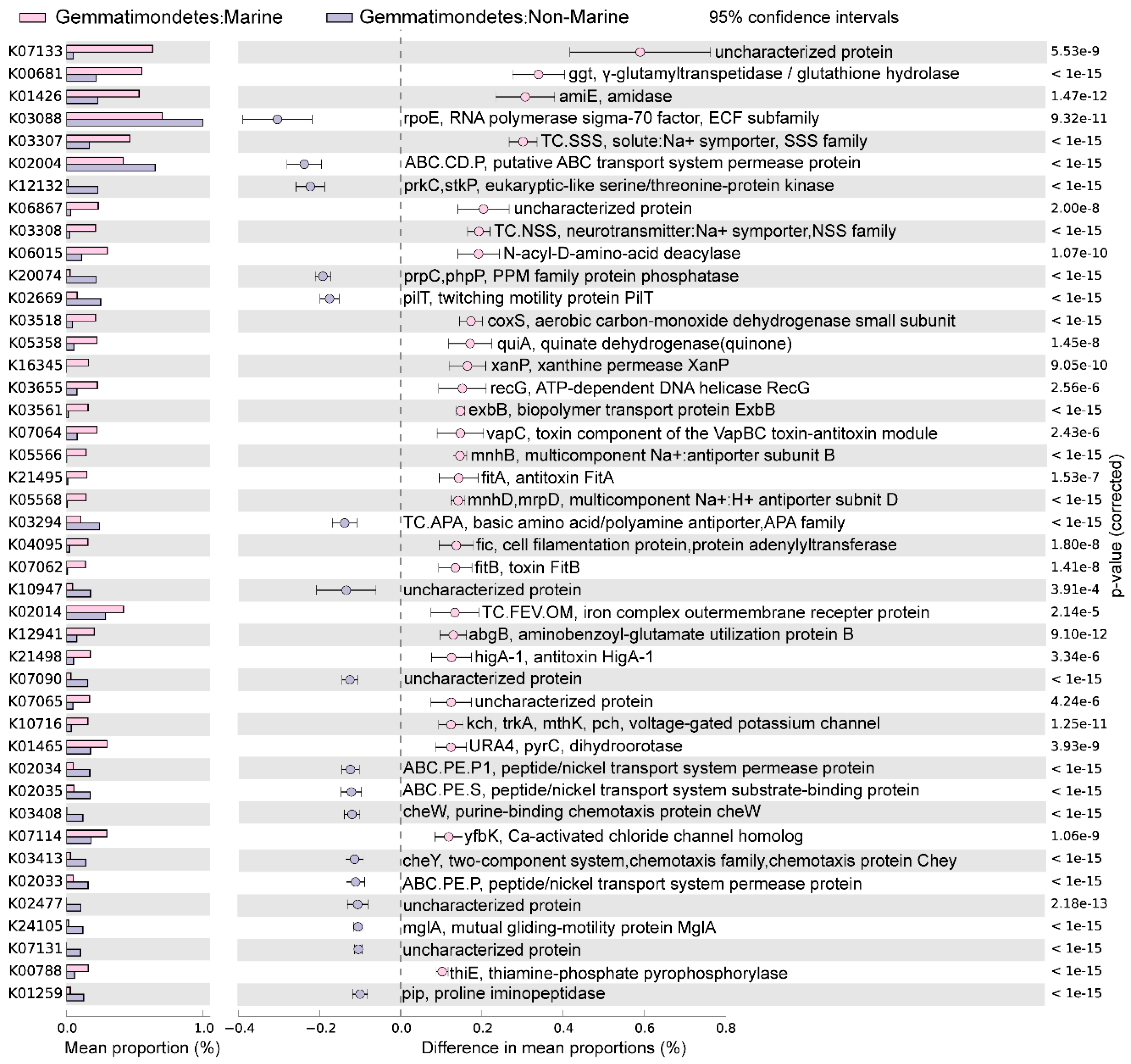
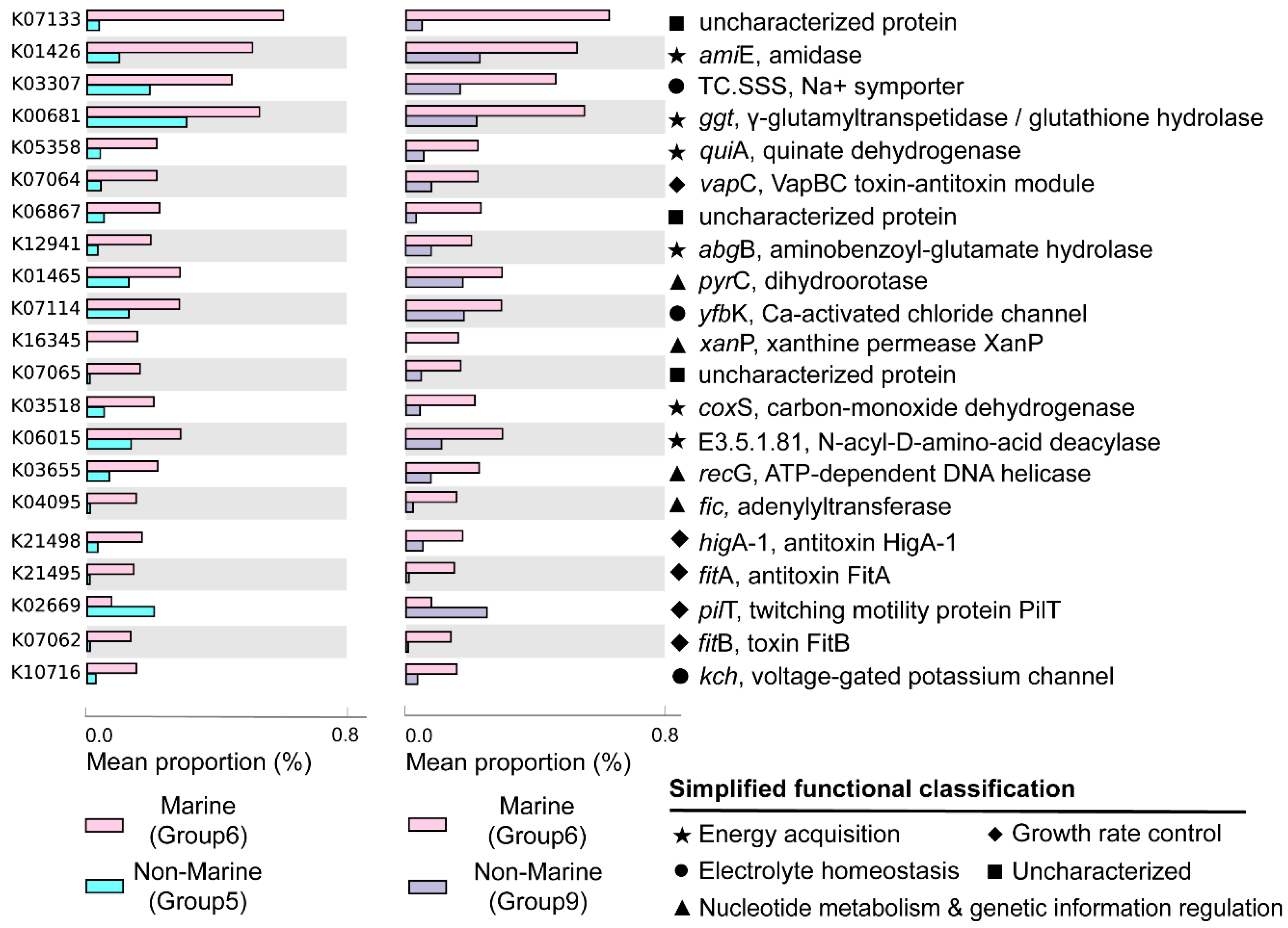
3.3. Statistical Comparison of Genomic and Functional Features Related to Adaptations to Marine Environment
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Zeng, Y.; Baumbach, J.; Barbosa, E.G.V.; Azevedo, V.; Zhang, C.; Koblížek, M. Metagenomic evidence for the presence of phototrophic Gemmatimonadetes bacteria in diverse environments. Environ. Microbiol. Rep. 2016, 8, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Dai, X.; Zhu, Y.; Yang, J.; Jiang, H.; Dong, H.; Huang, L. (Meta)Genomic Analysis Reveals Diverse Energy Conservation Strategies Employed by Globally Distributed Gemmatimonadota. mSystems 2022, 7, e0022822. [Google Scholar] [CrossRef] [PubMed]
- Mujakić, I.; Andrei, A.; Shabarova, T.; Fecskeová, L.K.; Salcher, M.M.; Piwosz, K.; Ghai, R.; Koblížek, M. Common Presence of Phototrophic Gemmatimonadota in Temperate Freshwater Lakes. mSystems 2021, 6, 10–128. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.; Wang, D.; Ruan, Y.; Xue, C.; Zhang, J.; Li, R.; Shen, Q. Deep 16S rRNA Pyrosequencing Reveals a Bacterial Community Associated with Banana Fusarium Wilt Disease Suppression Induced by Bio-Organic Fertilizer Application. PLoS ONE 2014, 9, e98420. [Google Scholar] [CrossRef] [PubMed]
- Carbonetto, B.; Rascovan, N.; Álvarez, R.; Mentaberry, A.; Vázquez, M.P. Structure, Composition and Metagenomic Profile of Soil Microbiomes Associated to Agricultural Land Use and Tillage Systems in Argentine Pampas. PLoS ONE 2014, 9, e99949. [Google Scholar] [CrossRef]
- DeBruyn, J.M.; Nixon, L.T.; Fawaz, M.N.; Johnson, A.M.; Radosevich, M. Global Biogeography and Quantitative Seasonal Dynamics of Gemmatimonadetes in Soil. Appl. Environ. Microbiol. 2011, 77, 6295–6300. [Google Scholar] [CrossRef]
- Janssen, P.H. Identifying the Dominant Soil Bacterial Taxa in Libraries of 16S rRNA and 16S rRNA Genes. Appl. Environ. Microbiol. 2006, 72, 1719–1728. [Google Scholar] [CrossRef]
- Delgado-Baquerizo, M.; Oliverio, A.M.; Brewer, T.E.; Benavent-González, A.; Eldridge, D.J.; Bardgett, R.D.; Maestre, F.T.; Singh, B.K.; Fierer, N. A global atlas of the dominant bacteria found in soil. Science 2018, 359, 320–325. [Google Scholar] [CrossRef]
- Mujakić, I.; Piwosz, K.; Koblížek, M. Phylum Gemmatimonadota and Its Role in the Environment. Microorganisms 2022, 10, 151. [Google Scholar] [CrossRef]
- Liu, R.; Wang, Z.; Wang, L.; Li, Z.; Fang, J.; Wei, X.; Wei, W.; Cao, J.; Wei, Y.; Xie, Z. Bulk and Active Sediment Prokaryotic Communities in the Mariana and Mussau Trenches. Front. Microbiol. 2020, 11, 1521. [Google Scholar] [CrossRef]
- Mujakić, I.; Cabello-Yeves, P.J.; Villena-Alemany, C.; Piwosz, K.; Rodriguez-Valera, F.; Picazo, A.; Camacho, A.; Koblížek, M.; Orlic, S. Multi-environment ecogenomics analysis of the cosmopolitan phylum Gemmatimonadota. Microbiol. Spectr. 2023, 11, e0111223. [Google Scholar] [CrossRef] [PubMed]
- Bay, S.K.; Dong, X.; Bradley, J.A.; Leung, P.M.; Grinter, R.; Jirapanjawat, T.; Arndt, S.K.; Cook, P.L.M.; LaRowe, D.E.; Nauer, P.A.; et al. Trace gas oxidizers are widespread and active members of soil microbial communities. Nat. Microbiol. 2021, 6, 246–256. [Google Scholar] [CrossRef] [PubMed]
- Chee-Sanford, J.; Tian, D.; Sanford, R. Consumption of N2O and other N-cycle intermediates by Gemmatimonas aurantiaca strain T-27. Microbiology 2019, 165, 1345–1354. [Google Scholar] [CrossRef] [PubMed]
- Park, D.; Kim, H.; Yoon, S. Nitrous Oxide Reduction by an Obligate Aerobic Bacterium, Gemmatimonas aurantiaca Strain T-27. Appl. Environ. Microbiol. 2017, 83, e00502-17. [Google Scholar] [CrossRef]
- Deng, J.; Bai, X.; Zhou, Y.; Zhu, W.; Yin, Y. Variations of soil microbial communities accompanied by different vegetation restoration in an open-cut iron mining area. Sci. Total. Environ. 2020, 704, 135243. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, H.; Chen, Z.; Hu, H.; Deng, C.; Wang, X. The benefits of autotrophic nitrogen removal from high concentration of urea wastewater through a process of urea hydrolysis and partial nitritation in sequencing batch reactor. J. Environ. Manag. 2021, 292, 112762. [Google Scholar] [CrossRef]
- Zeng, Y.; Selyanin, V.; Lukeš, M.; Dean, J.; Kaftan, D.; Feng, F.; Koblížek, M. Characterization of the microaerophilic, bacteriochlorophyll a-containing bacterium Gemmatimonas phototrophica sp. nov., and emended descriptions of the genus Gemmatimonas and Gemmatimonas aurantiaca. Int. J. Syst. Evol. Microbiol. 2015, 65, 2410–2419. [Google Scholar] [CrossRef]
- Zeng, Y.; Nupur; Wu, N.; Madsen, A.M.; Chen, X.; Gardiner, A.T.; Koblížek, M. Gemmatimonas groenlandica sp. nov. Is an Aerobic Anoxygenic Phototroph in the Phylum Gemmatimonadetes. Front. Microbiol. 2021, 11, 606612. [Google Scholar] [CrossRef]
- DeBruyn, J.M.; Fawaz, M.N.; Peacock, A.D.; Dunlap, J.R.; Nixon, L.T.; Cooper, K.E.; Radosevich, M. Gemmatirosa kalamazoonesis gen. nov., sp. nov., a member of the rarely-cultivated bacterial phylum Gemmatimonadetes. J. Gen. Appl. Microbiol. 2013, 59, 305–312. [Google Scholar] [CrossRef]
- Koblížek, M.; Dachev, M.; Bína, D.; Nupur; Piwosz, K.; Kaftan, D. Utilization of light energy in phototrophic Gemmatimonadetes. J. Photochem. Photobiol. B Biol. 2020, 213, 112085. [Google Scholar] [CrossRef]
- Beccati, A.; Gerken, J.; Quast, C.; Yilmaz, P.; Glöckner, F.O. SILVA tree viewer: Interactive web browsing of the SILVA phylogenetic guide trees. BMC Bioinform. 2017, 18, 433. [Google Scholar] [CrossRef] [PubMed]
- Aldeguer-Riquelme, B.; Antón, J.; Santos, F. Distribution, abundance, and ecogenomics of the Palauibacterales , a new cosmopolitan thiamine-producing order within the Gemmatimonadota phylum. mSystems 2023, 8, e0021523. [Google Scholar] [CrossRef] [PubMed]
- Olm, M.R.; Brown, C.T.; Brooks, B.; Banfield, J.F. dRep: A tool for fast and accurate genomic comparisons that enables improved genome recovery from metagenomes through de-replication. ISME J. 2017, 11, 2864–2868. [Google Scholar] [CrossRef] [PubMed]
- Chaumeil, P.-A.; Mussig, A.J.; Hugenholtz, P.; Parks, D.H. GTDB-Tk: A toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics 2019, 36, 1925–1927. [Google Scholar] [CrossRef] [PubMed]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef] [PubMed]
- Weizhi, S. Songweizhi/BioSAK: First Release of BioSAK, version 1.25.4; Zenodo: Geneva, Switzerland, 2020. [Google Scholar] [CrossRef]
- Hyatt, D.; Chen, G.-L.; Locascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef]
- Kanehisa, M. The KEGG database. In ‘In Silico’ Simulation of Biological Processes: Novartis Foundation Symposium 247; John Wiley & Sons Ltd.: Chichester, UK, 2002; Volume 247, pp. 91–103. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Morishima, K. BlastKOALA and GhostKOALA: KEGG Tools for Functional Characterization of Genome and Metagenome Sequences. J. Mol. Biol. 2016, 428, 726–731. [Google Scholar] [CrossRef]
- Clarke, K.R.; Gorley, R.N. PRIMER: Getting Started with v6; PRIMER-E Ltd.: Plymouth, UK, 2005; pp. 931–932. [Google Scholar]
- Parks, D.H.; Tyson, G.W.; Hugenholtz, P.; Beiko, R.G. STAMP: Statistical analysis of taxonomic and functional profiles. Bioinformatics 2014, 30, 3123–3124. [Google Scholar] [CrossRef]
- Minami, H.; Suzuki, H.; Kumagai, H. γ-Glutamyltranspeptidase, but Not YwrD, Is Important in Utilization of Extracellular Glutathione as a Sulfur Source in Bacillus subtilis. J. Bacteriol. 2004, 186, 1213–1214. [Google Scholar] [CrossRef]
- Oelgeschläger, E.; Rother, M. Carbon monoxide-dependent energy metabolism in anaerobic bacteria and archaea. Arch. Microbiol. 2008, 190, 257–269. [Google Scholar] [CrossRef]
- Diender, M.; Stams, A.J.M.; Sousa, D.Z. Pathways and Bioenergetics of Anaerobic Carbon Monoxide Fermentation. Front. Microbiol. 2015, 6, 1275. [Google Scholar] [CrossRef] [PubMed]
- Wakayama, M.; Ashika, T.; Miyamoto, Y.; Yoshikawa, T.; Sonoda, Y.; Sakai, K.; Moriguchi, M. Primary Structure of N-Acyl-D-Glutamate Amidohydrolase from Alcaligenes xylosoxydans subsp. xylosoxydans A-61. J. Biochem. 1995, 118, 204–209. [Google Scholar] [CrossRef] [PubMed]
- Hussein, M.J.; Green, J.M.; Nichols, B.P. Characterization of Mutations That Allow p-Aminobenzoyl-Glutamate Utilization by Escherichia coli. J. Bacteriol. 1998, 180, 6260–6268. [Google Scholar] [CrossRef] [PubMed]
- Pollmann, S.; Neu, D.; Weiler, E.W. Molecular cloning and characterization of an amidase from Arabidopsis thaliana capable of converting indole-3-acetamide into the plant growth hormone, indole-3-acetic acid. Phytochemistry 2003, 62, 293–300. [Google Scholar] [CrossRef]
- Herrero, A.; Muro-Pastor, A.M.; Flores, E. Nitrogen Control in Cyanobacteria. J. Bacteriol. 2001, 183, 411–425. [Google Scholar] [CrossRef]
- Chen, W.; Yang, F.; Zhang, L.; Wang, J. Organic Acid Secretion and Phosphate Solubilizing Efficiency ofPseudomonas sp. PSB12: Effects of Phosphorus Forms and Carbon Sources. Geomicrobiol. J. 2016, 33, 870–877. [Google Scholar] [CrossRef]
- Wu, Z.; Liu, C.; Zhang, Z.; Zheng, R.; Zheng, Y. Amidase as a versatile tool in amide-bond cleavage: From molecular features to biotechnological applications. Biotechnol. Adv. 2020, 43, 107574. [Google Scholar] [CrossRef]
- Whitby, M.; Vincent, S.; Lloyd, R. Branch migration of Holliday junctions: Identification of RecG protein as a junction specific DNA helicase. EMBO J. 1994, 13, 5220–5228. [Google Scholar] [CrossRef]
- Mattoo, S.; Durrant, E.; Chen, M.J.; Xiao, J.; Lazar, C.S.; Manning, G.; Dixon, J.E.; Worby, C.A. Comparative Analysis of Histophilus somni Immunoglobulin-binding Protein A (IbpA) with Other Fic Domain-containing Enzymes Reveals Differences in Substrate and Nucleotide Specificities. J. Biol. Chem. 2011, 286, 32834–32842. [Google Scholar] [CrossRef]
- Palanivelu, D.V.; Goepfert, A.; Meury, M.; Guye, P.; Dehio, C.; Schirmer, T. Fic domain-catalyzed adenylylation: Insight provided by the structural analysis of the type IV secretion system effector BepA. Protein Sci. 2011, 20, 492–499. [Google Scholar] [CrossRef]
- Karatza, P.; Frillingos, S. Cloning and functional characterization of two bacterial members of the NAT/NCS2 family inEscherichia coli. Mol. Membr. Biol. 2005, 22, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Porter, T.N.; Li, Y.; Raushel, F.M. Mechanism of the Dihydroorotase Reaction. Biochemistry 2004, 43, 16285–16292. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.K.; Ahn, B.; Bohr, V.A. Roles of RECQ helicases in recombination based DNA repair, genomic stability and aging. Biogerontology 2009, 10, 235–252. [Google Scholar] [CrossRef] [PubMed]
- McDaniel, L.D.; Young, E.; Delaney, J.; Ruhnau, F.; Ritchie, K.B.; Paul, J.H. High Frequency of Horizontal Gene Transfer in the Oceans. Science 2010, 330, 50. [Google Scholar] [CrossRef] [PubMed]
- Riva, F.; Riva, V.; Eckert, E.M.; Colinas, N.; Di Cesare, A.; Borin, S.; Mapelli, F.; Crotti, E. An Environmental Escherichia coli Strain Is Naturally Competent to Acquire Exogenous DNA. Front. Microbiol. 2020, 11, 574301. [Google Scholar] [CrossRef]
- Von Blohn, C.; Kempf, B.; Kappes, R.M.; Bremer, E. Osmostress response in Bacillus subtilis: Characterization of a proline uptake system (OpuE) regulated by high osmolarity and the alternative transcription factor sigma B. Mol. Microbiol. 1997, 25, 175–187. [Google Scholar] [CrossRef]
- Hiramatsu, T.; Kodama, K.; Kuroda, T.; Mizushima, T.; Tsuchiya, T. A Putative Multisubunit Na+/H+ Antiporter from Staphylococcus aureus. J. Bacteriol. 1998, 180, 6642–6648. [Google Scholar] [CrossRef]
- Parfenova, L.V.; Abarca-Heidemann, K.; Crane, B.M.; Rothberg, B.S. Molecular Architecture and Divalent Cation Activation of TvoK, a Prokaryotic Potassium Channel. J. Biol. Chem. 2007, 282, 24302–24309. [Google Scholar] [CrossRef]
- Wilbur, J.S.; Chivers, P.T.; Mattison, K.; Potter, L.; Brennan, R.G.; So, M. Neisseria gonorrhoeae FitA Interacts with FitB To Bind DNA through Its Ribbon−Helix−Helix Motif. Biochemistry 2005, 44, 12515–12524. [Google Scholar] [CrossRef]
- Budde, P.P.; Davis, B.M.; Yuan, J.; Waldor, M.K. Characterization of a higBA Toxin-Antitoxin Locus in Vibrio cholerae. J. Bacteriol. 2007, 189, 491–500. [Google Scholar] [CrossRef]
- Sharrock, A.; Ruthe, A.; Andrews, E.S.V.; Arcus, V.A.; Hicks, J.L. VapC proteins from Mycobacterium tuberculosis share ribonuclease sequence specificity but differ in regulation and toxicity. PLoS ONE 2018, 13, e0203412. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Du, J.; Wang, Z.; Hu, L.; Wang, L.; Fang, J.; Liu, R. Comparative Genomics Reveal Distinct Environment Preference and Functional Adaptation Among Lineages of Gemmatimonadota. Microorganisms 2024, 12, 2198. https://doi.org/10.3390/microorganisms12112198
Du J, Wang Z, Hu L, Wang L, Fang J, Liu R. Comparative Genomics Reveal Distinct Environment Preference and Functional Adaptation Among Lineages of Gemmatimonadota. Microorganisms. 2024; 12(11):2198. https://doi.org/10.3390/microorganisms12112198
Chicago/Turabian StyleDu, Jiangtao, Zhixuan Wang, Lin Hu, Li Wang, Jiasong Fang, and Rulong Liu. 2024. "Comparative Genomics Reveal Distinct Environment Preference and Functional Adaptation Among Lineages of Gemmatimonadota" Microorganisms 12, no. 11: 2198. https://doi.org/10.3390/microorganisms12112198
APA StyleDu, J., Wang, Z., Hu, L., Wang, L., Fang, J., & Liu, R. (2024). Comparative Genomics Reveal Distinct Environment Preference and Functional Adaptation Among Lineages of Gemmatimonadota. Microorganisms, 12(11), 2198. https://doi.org/10.3390/microorganisms12112198