

An In Silico Bioremediation Study to Identify Essential Residues of Metallothionein Enhancing the Bioaccumulation of Heavy Metals in Pseudomonas aeruginosa
 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sequence Retrieval and Analysis
2.2. Comparative Genomic Analysis and Orthologs Identification
2.3. Three-Dimensional Structure Prediction and Validation
2.4. Visualization of the Structure
3. Results
3.1. Metallothionein Sequence Analysis
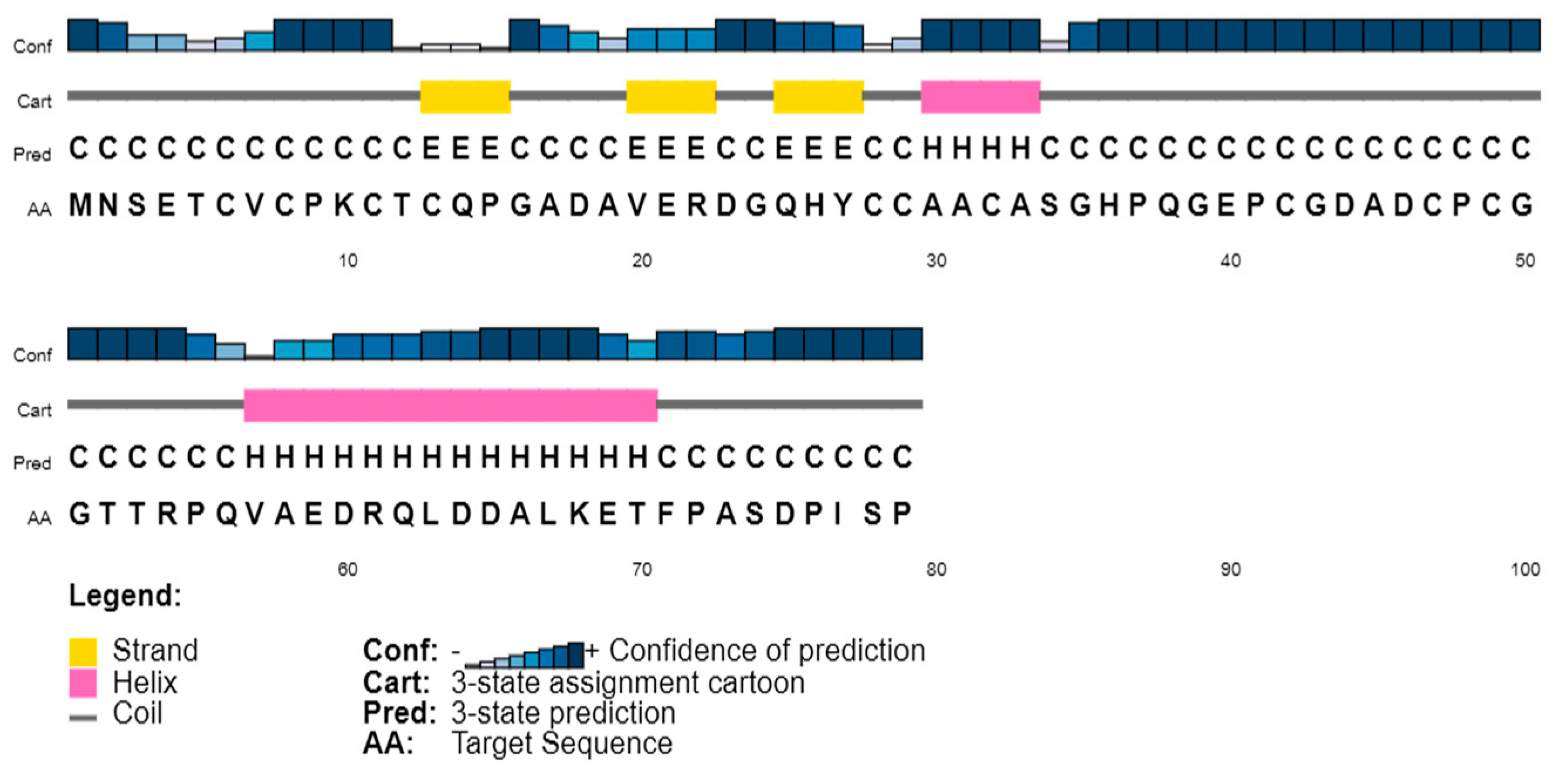
3.2. Secondary Structure Content in the Sequences
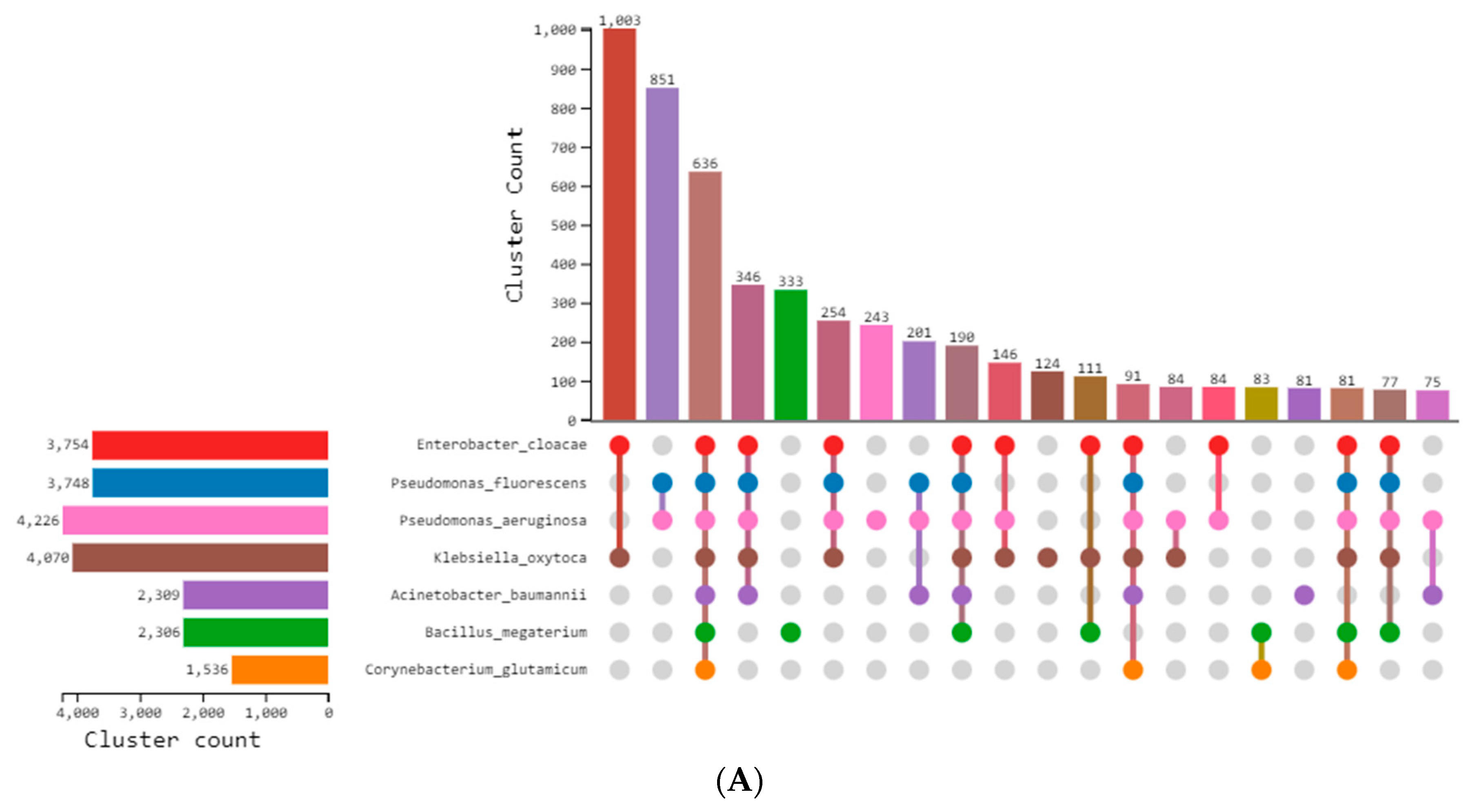
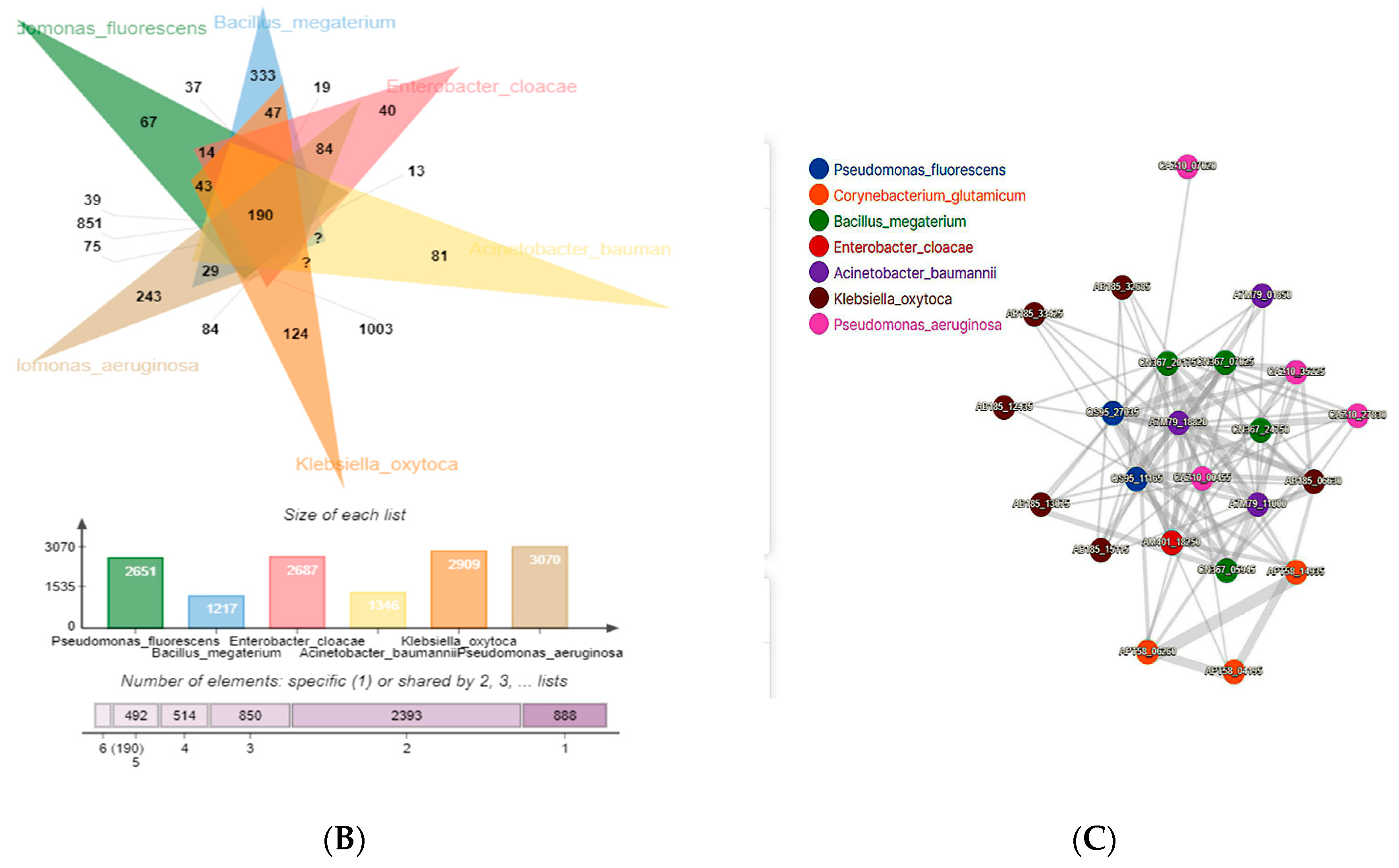
3.3. Comparative Genomic Analysis and Orthologs Identification
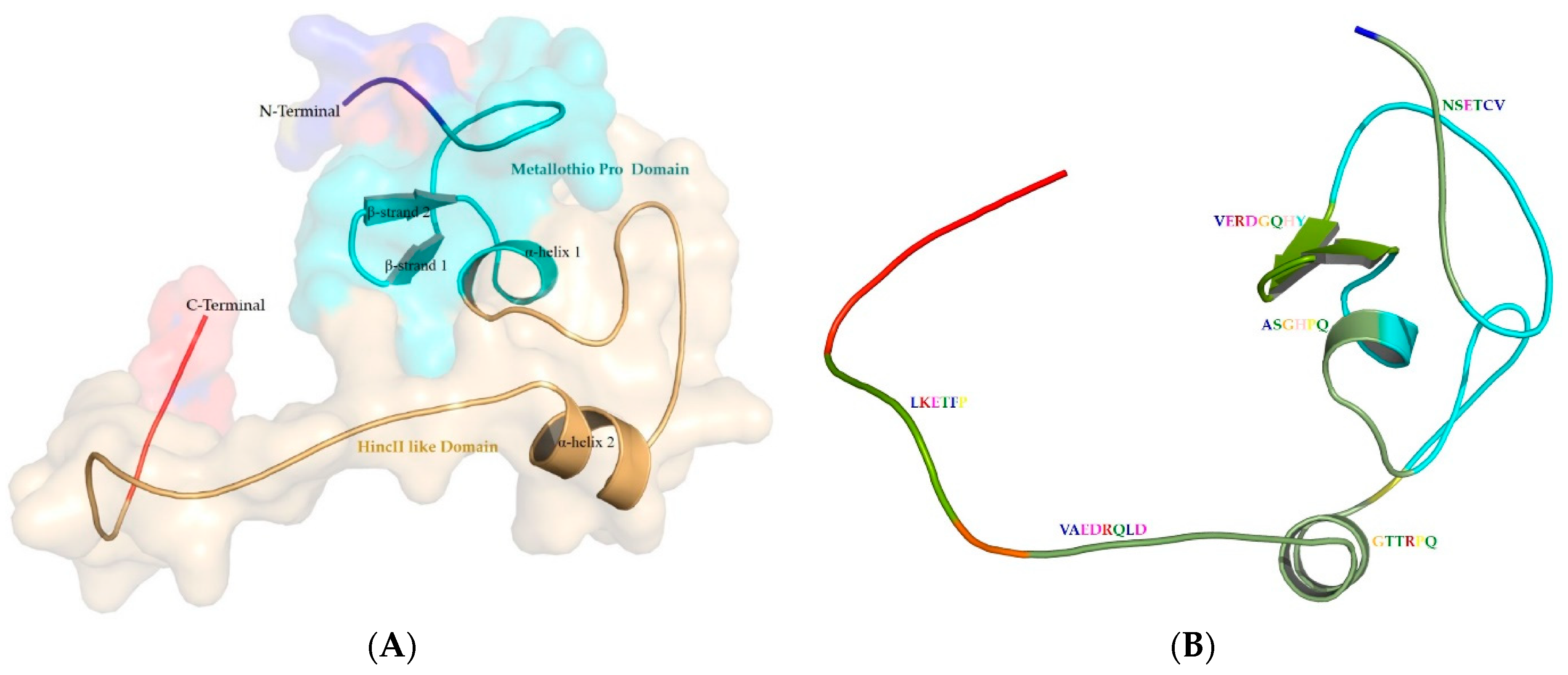
3.4. Structure of Metallothionein from P. aeruginosa
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Saeed, M.U.; Hussain, N.; Sumrin, A.; Shahbaz, A.; Noor, S.; Bilal, M.; Aleya, L.; Iqbal, H.M. Microbial bioremediation strategies with wastewater treatment potentialities—A review. Sci. Total. Environ. 2021, 818, 151754. [Google Scholar] [CrossRef]
- Kapahi, M.; Sachdeva, S. Bioremediation Options for Heavy Metal Pollution. J. Health Pollut. 2019, 9, 191203. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.V.; Lee, D.Y.; Li, B.; Quinlan, M.P.; Takahashi, F.; Maheswaran, S.; McDermott, U.; Azizian, N.; Zou, L.; Fischbach, M.A.; et al. A Chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell 2010, 141, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.; Xiao, W.; Zhou, C.; Pu, Q.; Deng, X.; Lan, L.; Liang, H.; Song, X.; Wu, M. Pseudomonas aeruginosa: Pathogenesis, virulence factors, antibiotic resistance, interaction with host, technology advances and emerging therapeutics. Signal Transduct. Target. Ther. 2022, 7, 199. [Google Scholar] [CrossRef] [PubMed]
- Jäger, A.V.; Arias, P.; Tribulatti, M.V.; Brocco, M.A.; Pepe, M.V.; Kierbel, A. The inflammatory response induced by Pseudomonas aeruginosa in macrophages enhances apoptotic cell removal. Sci. Rep. 2021, 11, 2393. [Google Scholar] [CrossRef]
- Zhang, B.-L.; Qiu, W.; Wang, P.-P.; Liu, Y.-L.; Zou, J.; Wang, L.; Ma, J. Mechanism study about the adsorption of Pb(II) and Cd(II) with iron-trimesic metal-organic frameworks. Chem. Eng. J. 2020, 385, 123507. [Google Scholar] [CrossRef]
- Maghraby, M.; Nasr, O.; Hamouda, M. Quality assessment of groundwater at south Al Madinah Al Munawarah area, Saudi Arabia. Environ. Earth Sci. 2013, 70, 1525–1538. [Google Scholar] [CrossRef]
- Ali, I.; Hasan, M.A.; Alharbi, O.M.L. Toxic metal ions contamination in the groundwater, Kingdom of Saudi Arabia. J. Taibah Univ. Sci. 2020, 14, 1571–1579. [Google Scholar] [CrossRef]
- Vélez, J.M.B.; Martínez, J.G.; Ospina, J.T.; Agudelo, S.O. Bioremediation potential of Pseudomonas genus isolates from residual water, capable of tolerating lead through mechanisms of exopolysaccharide production and biosorption. Biotechnol. Rep. 2021, 32, e00685. [Google Scholar] [CrossRef] [PubMed]
- Tiquia-Arashiro, S.M. Lead absorption mechanisms in bacteria as strategies for lead bioremediation. Appl. Microbiol. Biotechnol. 2018, 102, 5437–5444. [Google Scholar] [CrossRef]
- Taha, I.M.; El-Shafie, A.M. Leading causes and possible environmental contributors for end stage renal disease in al-madinah region in Saudi Arabia. In Proceedings of the 2nd Eurasian Multidisciplinary Forum, EMF 2014, Tbilisi, GA, USA, 23–26 October 2014. [Google Scholar]
- Al-Ghamdi, A.F. Electrochemical determination of Cd2+ in some Al-Madinah water samples and human plasma by cathodic stripping voltammetry in the presence of oxine as a chelating agent. J. Taibah Univ. Sci. 2014, 8, 19–25. [Google Scholar] [CrossRef]
- Alsehli, B.R. Evaluation and Comparison between a Conventional Acid Digestion Method and a Microwave Digestion System for Heavy Metals Determination in Mentha Samples by ICP-MS. Egypt. J. Chem. 2021, 64, 869–881. [Google Scholar] [CrossRef]
- Sayers, E.W.; Beck, J.; Bolton, E.E.; Bourexis, D.; Brister, J.R.; Canese, K.; Comeau, D.C.; Funk, K.; Kim, S.; Klimke, W.; et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2010, 38, D5–D16. [Google Scholar] [CrossRef]
- Tasleem, M.; Hussein, W.M.; El-Sayed, A.-A.A.A.; Alrehaily, A. Providencia alcalifaciens—Assisted Bioremediation of Chro-mium-Contaminated Groundwater: A Computational Study. Water 2023, 15, 1142. [Google Scholar] [CrossRef]
- Tasleem, M.; El-Sayed, A.-A.A.A.; Hussein, W.M.; Alrehaily, A. Bioremediation of Chromium-Contaminated Groundwater Using Chromate Reductase from Pseudomonas putida: An In silico Approach. Water 2023, 15, 150. [Google Scholar] [CrossRef]
- Saha, S.; Raghava, G. VICMpred: An SVM-based method for the prediction of functional proteins of Gram-negative bacteria using amino acid patterns and composition. Genom. Proteom. Bioinform. 2006, 4, 42–47. [Google Scholar] [CrossRef]
- Wang, Y.; Xiao, J.; Suzek, T.O.; Zhang, J.; Wang, J.; Bryant, S.H. PubChem: A public information system for analyzing bioac-tivities of small molecules. Nucleic Acids Res. 2009, 37 (Suppl. 2), W623–W633. [Google Scholar] [CrossRef]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.L.; Tosatto, S.C.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 2021, 49, D412–D419. [Google Scholar] [CrossRef]
- Blum, M.; Chang, H.-Y.; Chuguransky, S.; Grego, T.; Kandasaamy, S.; Mitchell, A.; Nuka, G.; Paysan-Lafosse, T.; Qureshi, M.; Raj, S.; et al. The InterPro protein families and domains database: 20 years on. Nucleic Acids Res. 2021, 49, D344–D354. [Google Scholar] [CrossRef]
- Bailey, T.L.; Johnson, J.; Grant, C.E.; Noble, W.S. The MEME suite. Nucleic Acids Res. 2015, 43, W39–W49. [Google Scholar] [CrossRef]
- McGuffin, L.J.; Bryson, K.; Jones, D.T.J.B. The PSIPRED protein structure prediction server. Bioinformatics 2000, 16, 404–405. [Google Scholar] [CrossRef]
- Montgomerie, S.; Cruz, J.A.; Shrivastava, S.; Arndt, D.; Berjanskii, M.; Wishart, D.S. PROTEUS2: A web server for compre-hensive protein structure prediction and structure-based annotation. Nucleic Acids Res. 2008, 36 (Suppl. 2), W202–W209. [Google Scholar] [CrossRef]
- Introduction to Program Evaluation for Public Health Programs: A Self-Study Guide. 2011; pp. 6–14. Available online: https://www.cdc.gov/evaluation/guide/introduction/index.htm (accessed on 15 March 2023).
- Fischer, S.; Brunk, B.P.; Chen, F.; Gao, X.; Harb, O.S.; Iodice, J.B.; Shanmugam, D.; Roos, D.S.; Stoeckert, C.J., Jr. Using OrthoMCL to Assign Proteins to OrthoMCL-DB Groups or to Cluster Proteomes into New Ortholog Groups. Curr. Protoc. Bioinform. 2011, 35, 6.12.1–6.12.19. [Google Scholar] [CrossRef]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J.E. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef]
- Zheng, W.; Wuyun, Q.; Zhou, X.; Li, Y.; Freddolino, P.L.; Zhang, Y. LOMETS3: Integrating deep learning and profile alignment for advanced protein template recognition and function annotation. Nucleic Acids Res. 2022, 50, W454–W464. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; De Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- Shin, W.-H.; Lee, G.R.; Heo, L.; Lee, H.; Seo, C. Prediction of protein structure and interaction by GALAXY protein modeling programs. Bio Des. 2014, 2, 1–11. [Google Scholar]
- Scott, W.R.P.; Hünenberger, P.H.; Tironi, I.G.; Mark, A.E.; Billeter, S.R.; Fennen, J.; Torda, A.E.; Huber, T.; Krüger, P.; van Gunsteren, W.F. The GROMOS biomolecular simulation program package. J. Phys. Chem. A 1999, 103, 3596–3607. [Google Scholar] [CrossRef]
- Wang, W.; Li, Z.; Wang, J.; Xu, D.; Shang, Y. PSICA: A fast and accurate web service for protein model quality analysis. Nucleic Acids Res. 2019, 47, W443–W450. [Google Scholar] [CrossRef] [PubMed]
- Dym, O.; Eisenberg, D.; Yeates, T.O. ERRAT. Int. J. Biol. 2012, 21, 678–679. [Google Scholar]
- Uziela, K.; Shu, N.; Wallner, B.; Elofsson, A. ProQ3: Improved model quality assessments using Rosetta energy terms. Sci. Rep. 2016, 6, 33509. [Google Scholar] [CrossRef]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Abeln, S.; Feenstra, K.A.; Heringa, J. Protein Three-Dimensional Structure Prediction. In Encyclopedia of Bioinformatics and Computational Biology; Ranganathan, S., Gribskov, M., Nakai, K., Schönbach, C., Eds.; Academic Press: Oxford, UK, 2019; pp. 497–511. [Google Scholar]
- Satyanarayana, S.D.; Krishna, M.; Kumar, P.P.; Jeereddy, S. In silico structural homology modeling of nif A protein of rhizobial strains in selective legume plants. J. Genet. Eng. Biotechnol. 2018, 16, 731–737. [Google Scholar] [CrossRef]
- Sun, J.; Lu, F.; Luo, Y.; Bie, L.; Xu, L.; Wang, Y. OrthoVenn3: An integrated platform for exploring and visualizing orthologous data across genomes. Nucleic Acids Res. 2023, 51, W397–W403. [Google Scholar] [CrossRef]
- Chellaiah, E.R. Cadmium (heavy metals) bioremediation by Pseudomonas aeruginosa: A minireview. Appl. Water Sci. 2018, 8, 154. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Jones, D.T.; Taylor, W.R.; Thornton, J.M. The rapid generation of mutation data matrices from protein sequences. Comput. Appl. Biosci. 1992, 8, 275–282. [Google Scholar] [CrossRef]
- Mendes, F.K.; Vanderpool, D.; Fulton, B.; Hahn, M.W. CAFE 5 models variation in evolutionary rates among gene families. Bioinformatics 2020, 36, 5516–5518. [Google Scholar] [CrossRef]
- Lespinet, O.; Wolf, Y.I.; Koonin, E.V.; Aravind, L. The role of lineage-specific gene family expansion in the evolution of eu-karyotes. Genome Res. 2002, 12, 1048–1059. [Google Scholar] [CrossRef]
- Johnson, M.S.; Srinivasan, N.; Sowdhamini, R.; Blundell, T.L. Knowledge-based protein modeling. Crit. Rev. Biochem. Mol. Biol. 1994, 29, 1–68. [Google Scholar] [CrossRef]
- McCarty, P.L.; Semprini, L. Ground-Water Treatment for Chlorinated Solvents. In Handbook of Bioremediation; CRC Press: Boca Raton, FL, USA, 1994. [Google Scholar]
- Matin, A. Starvation Promoters of Escherichia coli: Their Function, Regulation, and Use in Bioprocessing and Bioremediation. Ann. N. Y. Acad. Sci. 1994, 721, 277–291. [Google Scholar] [CrossRef]
- Michel, C.; Brugna, M.; Aubert, C.; Bernadac, A.; Bruschi, M. Enzymatic reduction of chromate: Comparative studies using sulfate-reducing bacteria. Key role of polyheme cytochromes c and hydrogenases. Appl. Microbiol. Biotechnol. 2001, 55, 95–100. [Google Scholar] [CrossRef]
- Ackerley, D.F.; Gonzalez, C.F.; Park, C.H.; Blake, R.; Keyhan, M.; Matin, A. Chromate-reducing properties of soluble flavoproteins from Pseudomonas putida and Escherichia coli. Appl. Environ. Microbiol. 2004, 70, 873–882. [Google Scholar] [CrossRef] [PubMed]
- Fowler, B.A.; Hildebrand, C.E.; Kojima, Y.; Webb, M. Nomenclature of metallothionein. Exp. Suppl. 1987, 52, 19–22. [Google Scholar]
- Blindauer, C.A.; Harrison, M.D.; Parkinson, J.A.; Robinson, A.K.; Cavet, J.S.; Robinson, N.J.; Sadler, P.J. A metallothionein containing a zinc finger within a four-metal cluster protects a bacterium from zinc toxicity. Proc. Natl. Acad. Sci. USA 2001, 98, 9593–9598. [Google Scholar] [CrossRef] [PubMed]
- Bjellqvist, B.; Sanchez, J.-C.; Pasquali, C.; Ravier, F.; Paquet, N.; Frutiger, S.; Hughes, G.J.; Hochstrasser, D. Micropreparative two-dimensional electrophoresis allowing the separation of samples containing milligram amounts of proteins. Electrophoresis 1993, 14, 1375–1378. [Google Scholar] [CrossRef]
- Ikai, A. Thermostability and aliphatic index of globular proteins. J. Biochem. 1980, 88, 1895–1898. [Google Scholar] [CrossRef]
- Kyte, J.; Doolittle, R.F. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982, 157, 105–132. [Google Scholar] [CrossRef]
- Gabaldón, T.; Koonin, E.V. Functional and evolutionary implications of gene orthology. Nat. Rev. Genet. 2013, 14, 360–366. [Google Scholar] [CrossRef]
- Fitch, W.M. Homology: A personal view on some of the problems. Trends Genet. 2000, 16, 227–231. [Google Scholar] [CrossRef]
- Mirny, L.A.; Gelfand, M.S. Non-orthologous gene displacement. Trends Genet. 1996, 12, 334–336. [Google Scholar]
- Mirny, L.A.; Gelfand, M. Using orthologous and paralogous proteins to identify specificity determining residues. Genome Biol. 2002, 3, preprint0002.1. [Google Scholar] [CrossRef]
- Engh, R.A.; Huber, R. Accurate bond and angle parameters for X-ray protein structure refinement. Acta Crystallogr. Sect. A Found. Crystallogr. 1991, 47, 392–400. [Google Scholar] [CrossRef]
- Senarathne, P.; Pathiratne, K. Accumulation of heavy metals in a food fish, Mystus gulio inhabiting Bolgoda Lake, Sri Lanka. Sri Lanka J. Aquat. Sci. 2007, 12, 61–75. [Google Scholar] [CrossRef]
- Naik, M.M.; Dubey, S.K. Lead-enhanced siderophore production and alteration in cell morphology in a Pb-resistant Pseudo-monas aeruginosa strain 4EA. Curr. Microbiol. 2011, 62, 409–414. [Google Scholar] [CrossRef]
- Liu, T.; Nakashima, S.; Hirose, K.; Uemura, Y.; Shibasaka, M.; Katsuhara, M.; Kasamo, K. A metallothionein and CPx-ATPase handle heavy-metal tolerance in the filamentous cyanobacterium Oscillatoria brevis. FEBS Lett. 2003, 542, 159–163. [Google Scholar] [CrossRef]
- Fiorillo, A.; Battistoni, A.; Ammendola, S.; Secli, V.; Rinaldo, S.; Cutruzzola, F.; Demitri, N.; Ilari, A. Structure and metal-binding properties of PA4063, a novel player in periplasmic zinc trafficking by Pseudomonas aeruginosa. Acta Crystallogr. Sect. D Struct. Biol. 2021, 77, 1401–1410. [Google Scholar]
- Fernández, M.; Rico-Jiménez, M.; Ortega, Á.; Daddaoua, A.; García, A.I.; Martín-Mora, D.; Mesa Torres, N.; Tajuelo, A.; Matilla, M.A.; Krell, T. Determination of ligand profiles for Pseudomonas aeruginosa solute binding proteins. Int. J. Mol. Sci. 2019, 20, 5156. [Google Scholar] [CrossRef]
- Ammendola, S.; Secli, V.; Pacello, F.; Mastropasqua, M.C.; Romão, M.A.; Gomes, C.M.; Battistoni, A. Zinc-binding metallophores protect Pseudomonas aeruginosa from calprotectin-mediated metal starvation. FEMS Microbiol. Lett. 2022, 369, fnac071. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Slimmed GO | Count of Unique Input Access | Molecular Function |
|---|---|---|
| GO:0003674 | 150 | Molecular Function |
| GO:0016787 | 138 | Hydrolase Activity |
| GO:0005215 | 129 | Transporter Activity |
| GO:0003676 | 113 | Nucleic Acid Binding |
| GO:0016740 | 109 | Transferase Activity |
| GO:0016491 | 96 | Oxidoreductase Activity |
| GO:0043167 | 70 | Ion Binding |
| GO:0008233 | 55 | Peptidase Activity |
| GO:0000166 | 41 | Nucleotide Binding |
| GO:0005488 | 36 | Binding |
| Slimmed GO | Count of Unique Input Access | Biological Function |
|---|---|---|
| GO:0008152 | 1967 | Metabolic Process |
| GO:0008150 | 1751 | Biological Process |
| GO:0044237 | 1724 | Cellular Metabolic Process |
| GO:0006807 | 1442 | Nitrogen Compound Metabolic Process |
| GO:0044238 | 1180 | Primary Metabolic Process |
| GO:0009987 | 946 | Cellular Process |
| GO:0046483 | 866 | Heterocycle Metabolic Process |
| GO:0006082 | 783 | Organic Acid Metabolic Process |
| GO:0006725 | 781 | Cellular Aromatic Compound Metabolic Process |
| GO:0043170 | 692 | Macromolecule Metabolic Process |
| Modeling Tool | Phyre2 | SwissModel | GALAXY | LOMETS |
|---|---|---|---|---|
| Residues Built | 1–79 | 1–79 | 1–79 | 1–79 |
| ProQ3 | 0.345 | 0.37 | 0.401 | 0.447 |
| ERRAT | 47.76 | 81.63 | 78.57 | 44.92 |
| PSICA Server | 0.49 | 0.47 | 0.53 | 0.43 |
| Most Favored | 85.5 | 71 | 83.9 | 66.1 |
| Additionally Allowed | 11.3 | 25.8 | 11.3 | 25.8 |
| Generously allowed | 3.2 | 3.2 | 1.3 | 6.5 |
| G-factor | −0.07 | −0.36 | −0.13 | −7.89 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tasleem, M.; Hussein, W.M.; El-Sayed, A.-A.A.A.; Alrehaily, A. An In Silico Bioremediation Study to Identify Essential Residues of Metallothionein Enhancing the Bioaccumulation of Heavy Metals in Pseudomonas aeruginosa. Microorganisms 2023, 11, 2262. https://doi.org/10.3390/microorganisms11092262
Tasleem M, Hussein WM, El-Sayed A-AAA, Alrehaily A. An In Silico Bioremediation Study to Identify Essential Residues of Metallothionein Enhancing the Bioaccumulation of Heavy Metals in Pseudomonas aeruginosa. Microorganisms. 2023; 11(9):2262. https://doi.org/10.3390/microorganisms11092262
Chicago/Turabian StyleTasleem, Munazzah, Wesam M. Hussein, Abdel-Aziz A. A. El-Sayed, and Abdulwahed Alrehaily. 2023. "An In Silico Bioremediation Study to Identify Essential Residues of Metallothionein Enhancing the Bioaccumulation of Heavy Metals in Pseudomonas aeruginosa" Microorganisms 11, no. 9: 2262. https://doi.org/10.3390/microorganisms11092262
APA StyleTasleem, M., Hussein, W. M., El-Sayed, A.-A. A. A., & Alrehaily, A. (2023). An In Silico Bioremediation Study to Identify Essential Residues of Metallothionein Enhancing the Bioaccumulation of Heavy Metals in Pseudomonas aeruginosa. Microorganisms, 11(9), 2262. https://doi.org/10.3390/microorganisms11092262