_Di_Marco.png)
Oxygen Exposure and Tolerance Shapes the Cell Wall-Associated Lipids of the Skin Commensal Cutibacterium acnes

 , , ,
, , ,  ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Strains and Cultivation
2.2. Determination of Protein Concentration
2.3. Quantification of Protein Carbonyls
2.4. Lipids Purification and Fractionation
2.5. MS Lipids Analysis
2.6. SFC-HRMS Analysis
2.7. Data Processing and Statistical Analysis
3. Results
3.1. The Proteome Carbonylation Is Affected by Presence of RoxP
3.2. Free Fatty Acid Changes in Bacteria Lacking the Antioxidant RoxP
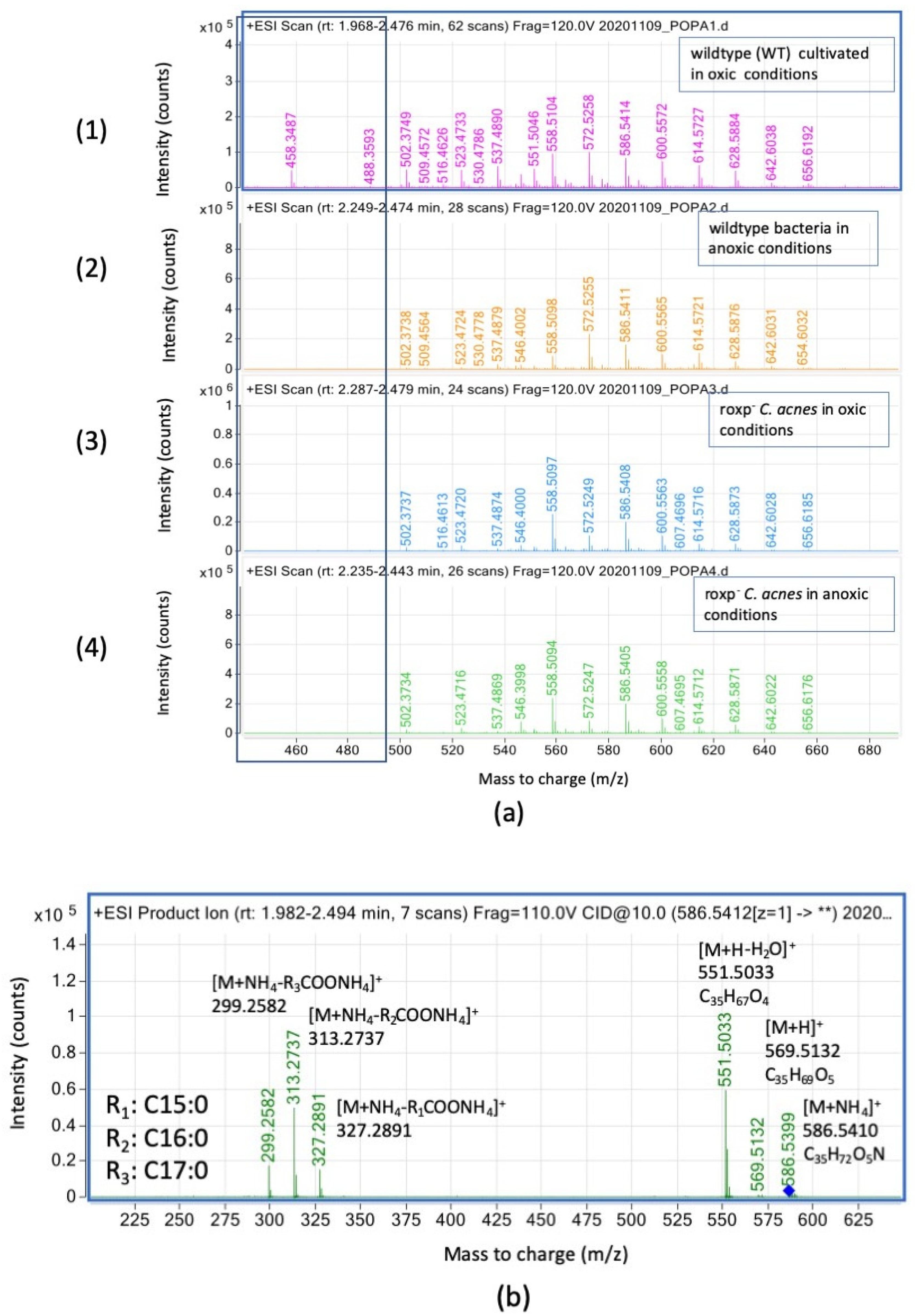
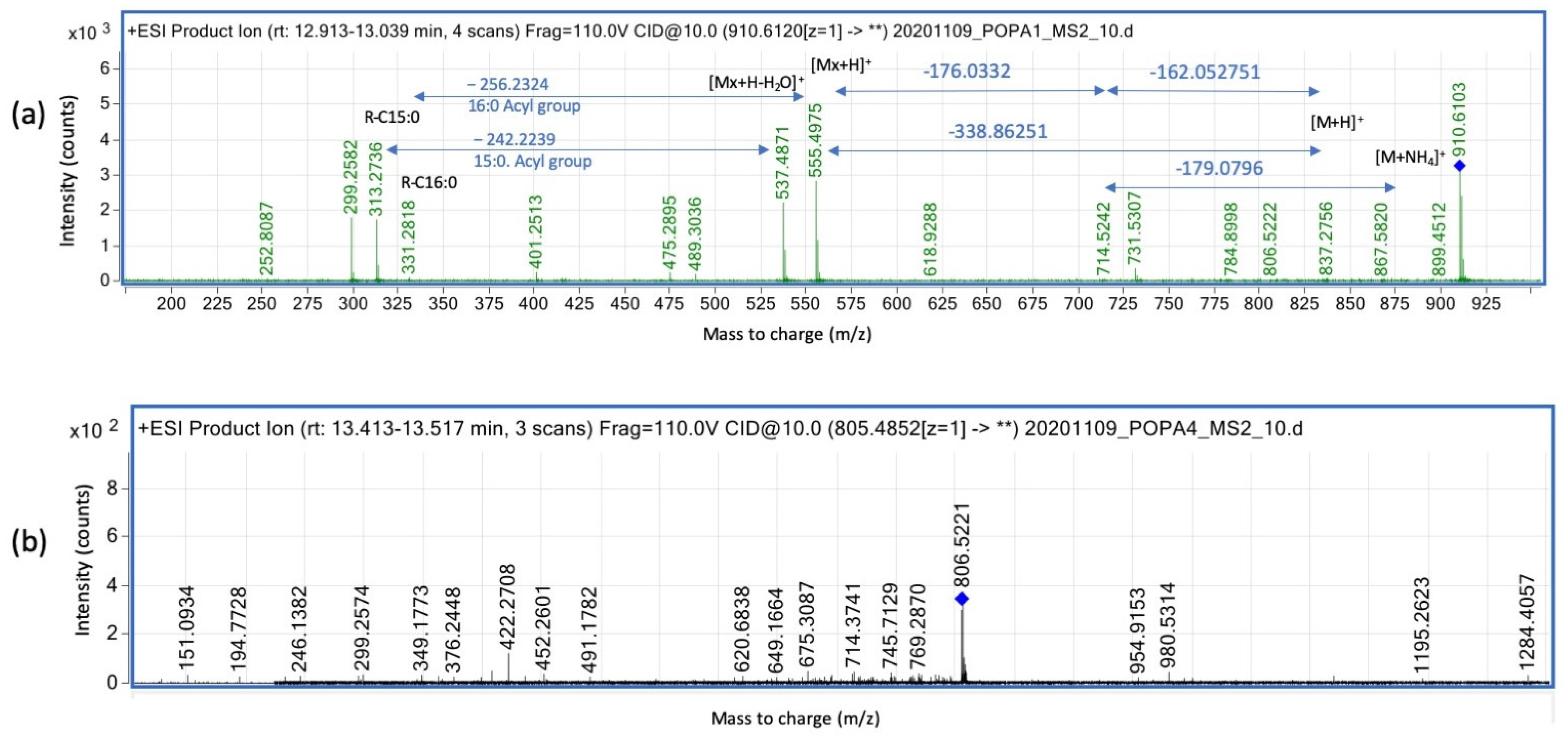
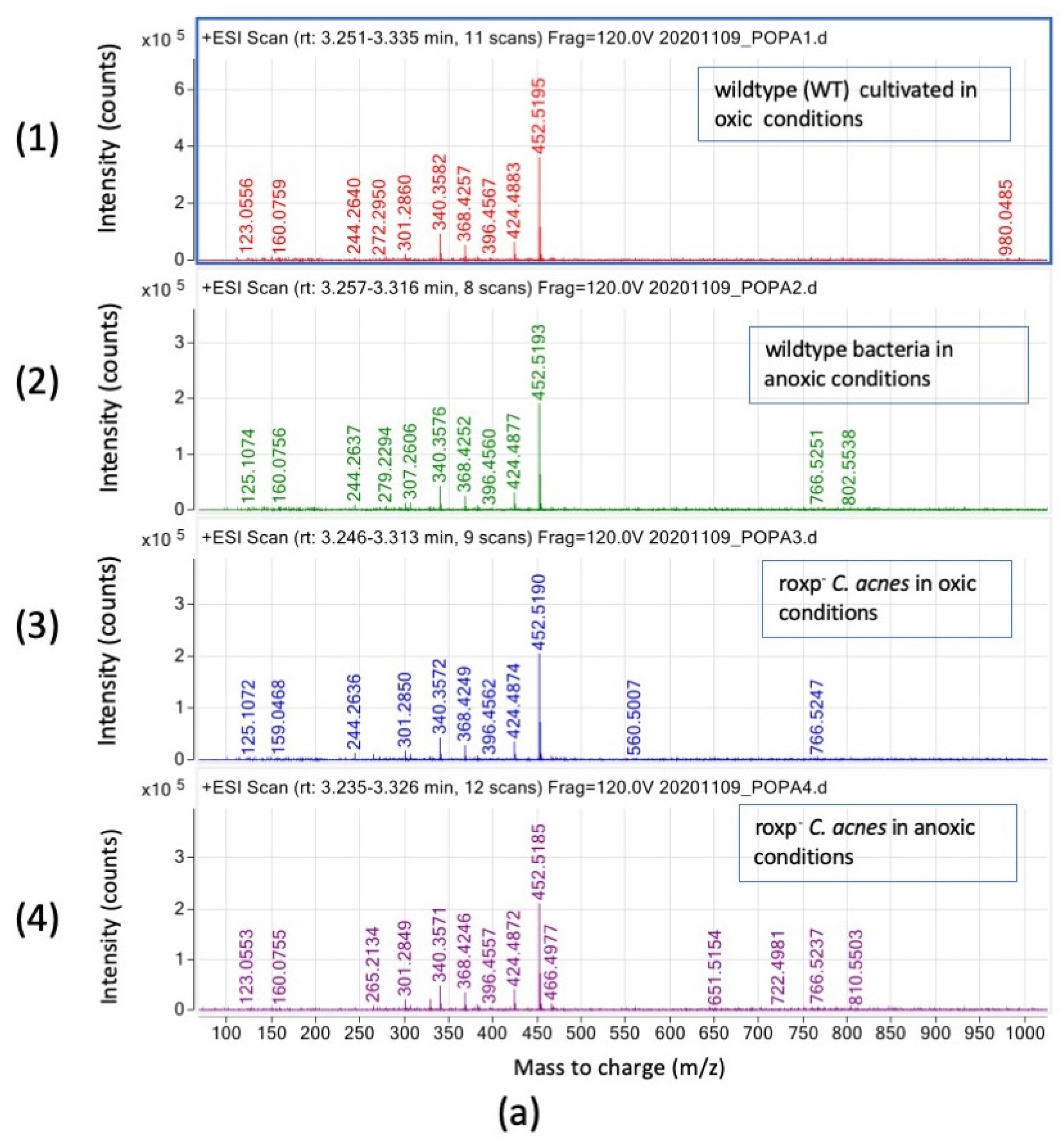
3.3. Complex Lipids Profile of C. acnes Cultivated in Oxic and Anoxic Conditions Vary Based on Presence of RoxP
3.3.1. Modified Diglyceride Relative Intensities (Counts) Are Positively Affected by Presence of the Antioxidant RoxP
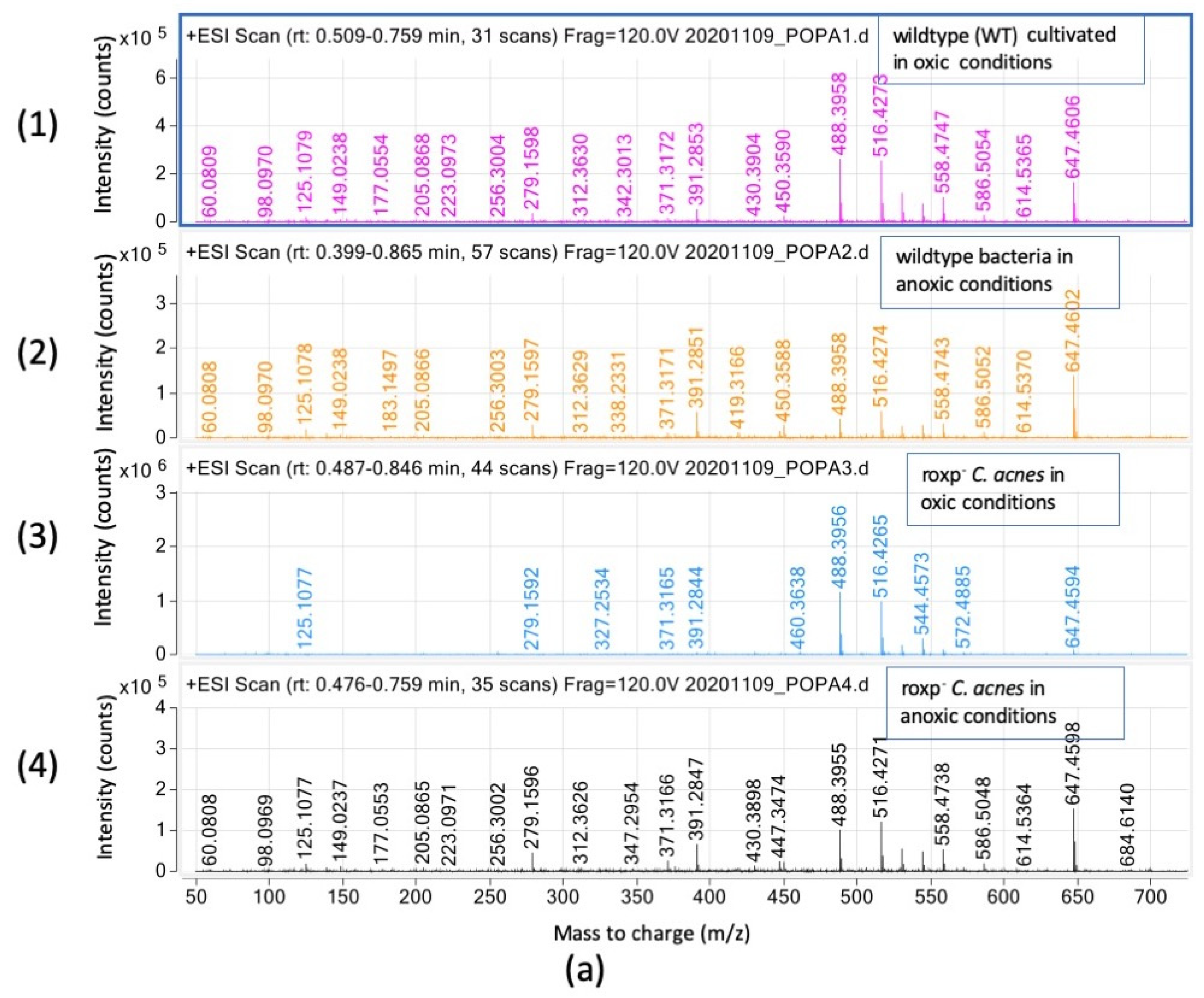
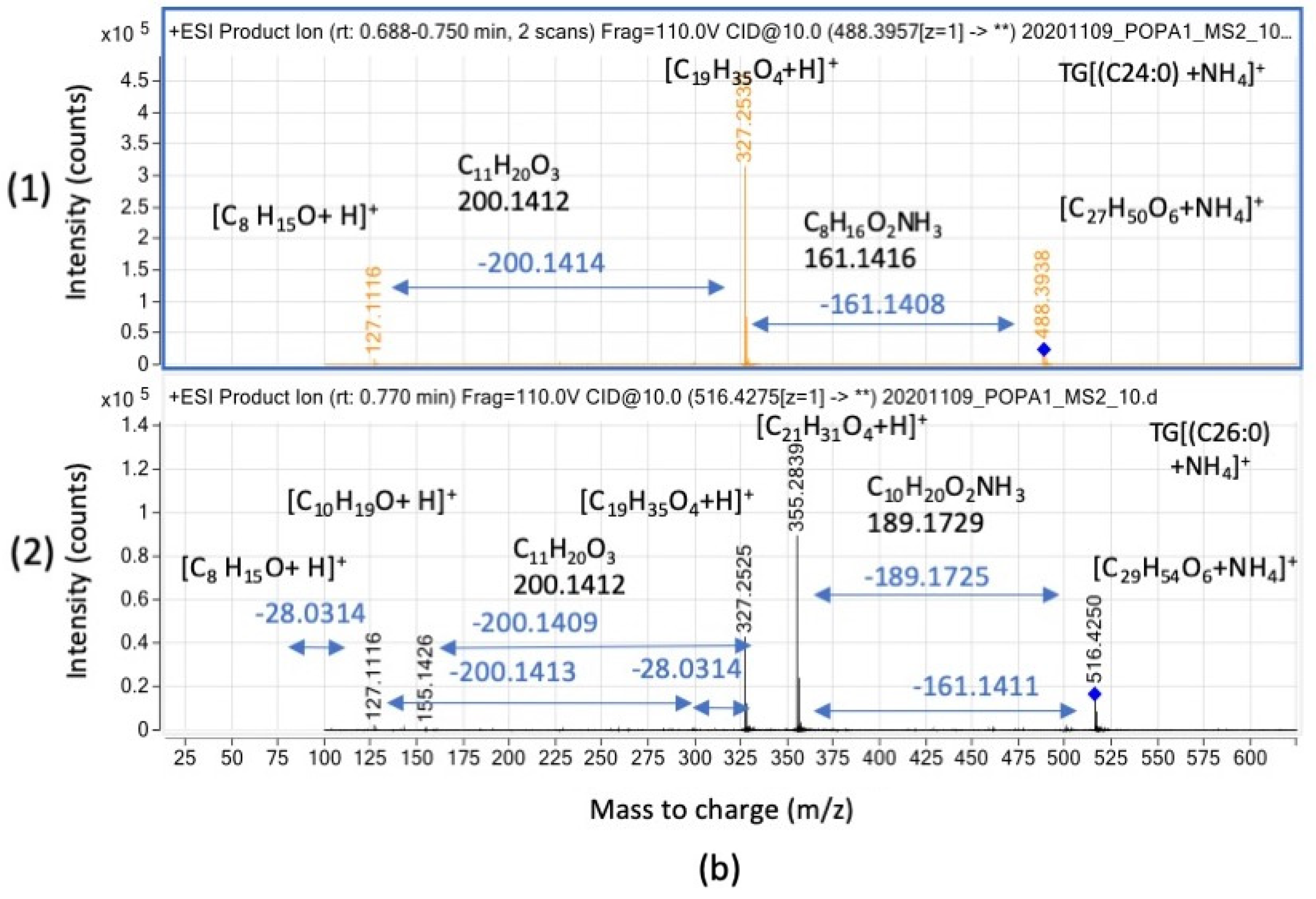
3.3.2. Triglyceride Relative Intensities (Counts) Are Negatively Affected in the Presence of Oxygen upon Lack of Functional Bacterial Antioxidant System
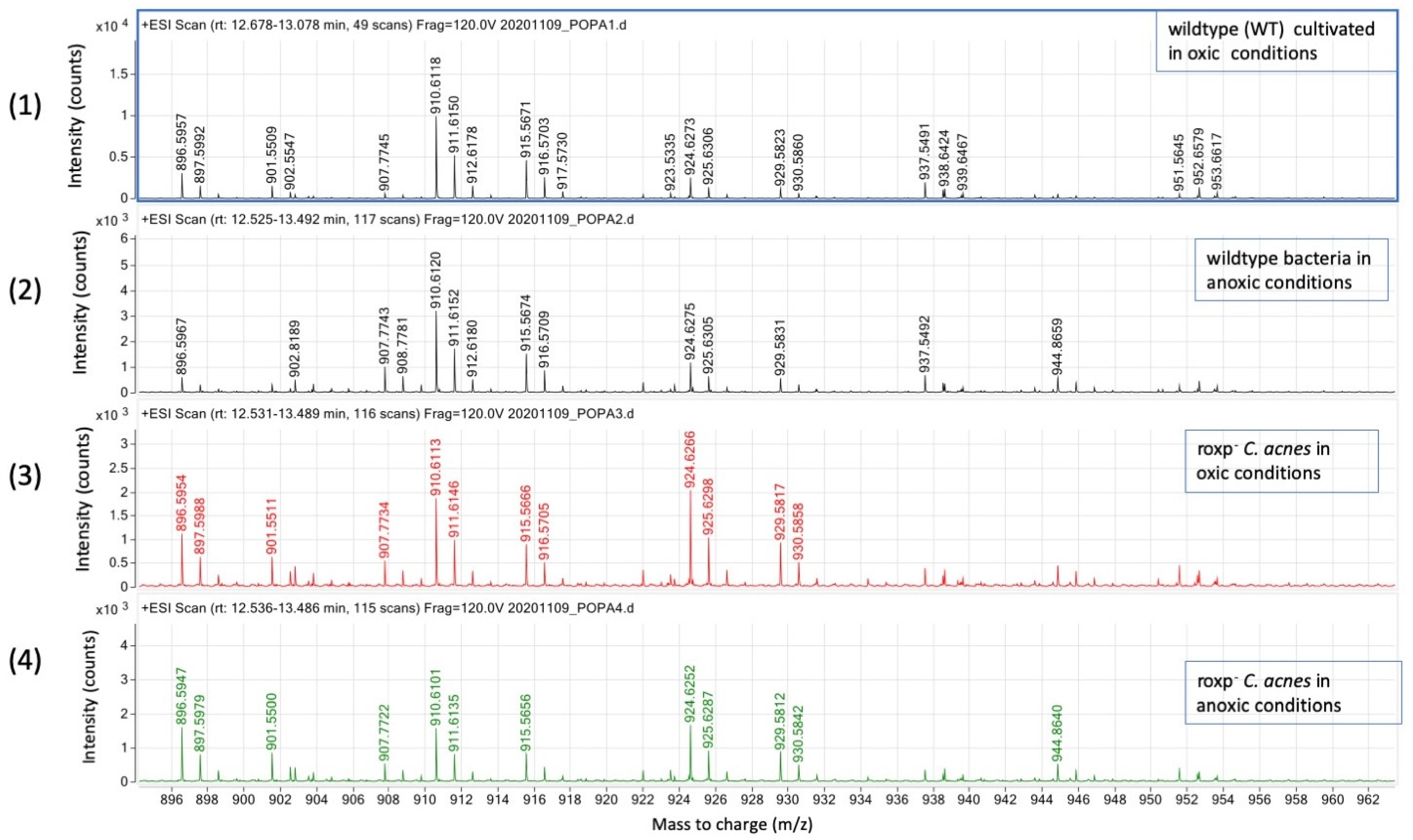
3.3.3. Presence of Antioxidants Is Critical for High Relative Intensities of DGDG and SQDG in C. acnes
3.3.4. Ceramide Species Are Mainly Unaffected by Oxic Stress in C. acnes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Coenye, T.; Spittaels, K.J.; Achermann, Y. The Role of Biofilm Formation in the Pathogenesis and Antimicrobial Susceptibility of Cutibacterium Acnes. Biofilm 2021, 4, 100063. [Google Scholar] [CrossRef] [PubMed]
- Holmberg, A.; Lood, R.; Mörgelin, M.; Söderquist, B.; Holst, E.; Collin, M.; Christensson, B.; Rasmussen, M. Biofilm Formation by Propionibacterium Acnes Is a Characteristic of Invasive Isolates. Clin. Microbiol. Infect. 2009, 15, 787–795. [Google Scholar] [CrossRef]
- Mayslich, C.; Grange, P.A.; Dupin, N. Cutibacterium Acnes as an Opportunistic Pathogen: An Update of Its Virulence-Associated Factors. Microorganisms 2021, 9, 303. [Google Scholar] [CrossRef]
- Lomholt, H.B.; Scholz, C.F.P.; Brüggemann, H.; Tettelin, H.; Kilian, M. A Comparative Study of Cutibacterium (Propionibacterium) Acnes Clones from Acne Patients and Healthy Controls. Anaerobe 2017, 47, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Allhorn, M.; Arve, S.; Brüggemann, H.; Lood, R. A Novel Enzyme with Antioxidant Capacity Produced by the Ubiquitous Skin Colonizer Propionibacterium Acnes. Sci. Rep. 2016, 6, 36412. [Google Scholar] [CrossRef] [PubMed]
- Andersson, T.; Ertürk Bergdahl, G.; Saleh, K.; Magnúsdóttir, H.; Stødkilde, K.; Andersen, C.B.F.; Lundqvist, K.; Jensen, A.; Brüggemann, H.; Lood, R. Common Skin Bacteria Protect Their Host from Oxidative Stress through Secreted Antioxidant RoxP. Sci. Rep. 2019, 9, 3596. [Google Scholar] [CrossRef] [PubMed]
- Bowe, W.P.; Logan, A.C. Clinical Implications of Lipid Peroxidation in Acne Vulgaris: Old Wine in New Bottles. Lipids Health Dis. 2010, 9, 141. [Google Scholar] [CrossRef]
- Capitanio, B.; Lora, V.; Ludovici, M.; Sinagra, J.L.; Ottaviani, M.; Mastrofrancesco, A.; Ardigò, M.; Camera, E. Modulation of Sebum Oxidation and Interleukin-1α Levels Associates with Clinical Improvement of Mild Comedonal Acne. J. Eur. Acad. Dermatol. Venereol. 2014, 28, 1792–1797. [Google Scholar] [CrossRef]
- Hawkins, C.L.; Morgan, P.E.; Davies, M.J. Quantification of Protein Modification by Oxidants. Free Radic. Biol. Med. 2009, 46, 965–988. [Google Scholar] [CrossRef]
- Folch, J.; Lees, M.; Sloane Stanley, G.H. A Simple Method for the Isolation and Purification of Total Lipides from Animal Tissues. J. Biol. Chem. 1957, 226, 497–509. [Google Scholar] [CrossRef]
- Bodennec, J.; Koul, O.; Aguado, I.; Brichon, G.; Zwingelstein, G.; Portoukalian, J. A Procedure for Fractionation of Sphingolipid Classes by Solid-Phase Extraction on Aminopropyl Cartridges. J. Lipid Res. 2000, 41, 1524–1531. [Google Scholar] [CrossRef]
- Popa, I.; Remoue, N.; Osta, B.; Pin, D.; Gatto, H.; Haftek, M.; Portoukalian, J. The Lipid Alterations in the Stratum Corneum of Dogs with Atopic Dermatitis Are Alleviated by Topical Application of a Sphingolipid-Containing Emulsion. Clin. Exp. Dermatol. 2012, 37, 665–671. [Google Scholar] [CrossRef]
- Chollet, C.; Boutet-Mercey, S.; Laboureur, L.; Rincon, C.; Méjean, M.; Jouhet, J.; Fenaille, F.; Colsch, B.; Touboul, D. Supercritical fluid chromatography coupled to mass spectrometry for lipidomics. J. Mass. Spectrom. 2019, 54, 791–801. [Google Scholar] [CrossRef]
- Ta, H.P.; Clarisse, C.; Maes, E.; Yamakawa, N.; Guérardel, Y.; Krzewinski, F.; Zarzycka, W.; Touboul, D.; Girardeau, A.; Fonseca, F.; et al. Membrane lipid composition of Carnobacterium maltaromaticum CNCM I-3298, a highly cryoresistant lactic bacterium. Chem. Phys. Lipids 2023, 255, 105326. [Google Scholar] [CrossRef]
- Jeon, J.; Park, S.C.; Her, J.; Lee, J.W.; Han, J.K.; Kim, Y.K.; Kim, K.P.; Ban, C. Comparative Lipidomic Profiling of the Human Commensal Bacterium Propionibacterium Acnes and Its Extracellular Vesicles. RSC Adv. 2018, 8, 15241–15247. [Google Scholar] [CrossRef]
- Pham, T.H.; Zaeem, M.; Fillier, T.A.; Nadeem, M.; Vidal, N.P.; Manful, C.; Cheema, S.; Cheema, M.; Thomas, R.H. Targeting Modified Lipids during Routine Lipidomics Analysis Using HILIC and C30 Reverse Phase Liquid Chromatography Coupled to Mass Spectrometry. Sci. Rep. 2019, 9, 5048. [Google Scholar] [CrossRef]
- Lopes, D.; Moreira, A.S.P.; Rey, F.; Da Costa, E.; Melo, T.; Maciel, E.; Rego, A.; Abreu, M.H.; Domingues, P.; Calado, R.; et al. Lipidomic Signature of the Green Macroalgae Ulva Rigida Farmed in a Sustainable Integrated Multi-Trophic Aquaculture. Appl. Phycol. 2019, 31, 1369–1381. [Google Scholar] [CrossRef]
- Staerck, C.; Gastebois, A.; Vandeputte, P.; Calenda, A.; Larcher, G.; Gillmann, L.; Papon, N.; Bouchara, J.P.; Fleury, M.J.J. Microbial antioxidant defense enzymes. Microb. Pathog. 2017, 110, 56–65. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira, M.A.; Tairum, C.A.; Soares Netto, L.E.; Pires de Oliveira, A.L.; Aleixo-Silva, R.L.; Montanhero Cabrera, V.I.; Breyer, C.A.; Cardoso dos Santos, M. Relevance of peroxiredoxins in pathogenic microorganisms. App Microb. Biotech. 2021, 105, 5701–5717. [Google Scholar] [CrossRef]
- Barry, H.; Gutteridge, J.M.C. Free Radicals in Biology and Medicine, 5th ed.; Oxford University Press: Oxford, UK, 2015. [Google Scholar] [CrossRef]
- Nakamura, K.; O’Neill, A.M.; Williams, M.R.; Cau, L.; Nakatsuji, T.; Horswill, A.R.; Gallo, R.L. Short Chain Fatty Acids Produced by Cutibacterium Acnes Inhibit Biofilm Formation by Staphylococcus Epidermidis. Sci. Rep. 2020, 10, 21237. [Google Scholar] [CrossRef] [PubMed]
- Obeid, L.M.; Hannun, Y.A. Ceramide: A Stress Signal and Mediator of Growth Suppression and Apoptosis. J. Cell Biochem. 1995, 58, 191–198. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Acyl FFA (%) | FFA (%) | OH-FFA (%) |
|---|---|---|---|
| 1-wt bacteria (oxic) | 21.70 | 76.40 | 2.00 |
| 2-wt bacteria (anoxic) | 45.10 | 54.70 | 0.20 |
| 3-roxp− bacteria (oxic) | 52.70 | 36.20 | 1.10 |
| 4-roxp− bacteria (anoxic) | 37.70 | 61.50 | 1.80 |
| DGDG (m/z) | 910.6112 | 915.5674 | 924.6276 | 929.5833 |
|---|---|---|---|---|
| wt bacteria (oxic) | 252,476 | 117,254 | 62,233 | 29,774 |
| wt bacteria (anoxic) | 191,071 | 88,784 | 67,414 | 31,552 |
| roxp− bacteria (oxic) | 108,911 | 51,809 | 116,441 | 53,098 |
| roxp− bacteria (anoxic) | 90,252 | 48,843 | 95,145 | 50,482 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Popa, I.; Touboul, D.; Andersson, T.; Fuentes-Lemus, E.; Santerre, C.; Davies, M.J.; Lood, R. Oxygen Exposure and Tolerance Shapes the Cell Wall-Associated Lipids of the Skin Commensal Cutibacterium acnes. Microorganisms 2023, 11, 2260. https://doi.org/10.3390/microorganisms11092260
Popa I, Touboul D, Andersson T, Fuentes-Lemus E, Santerre C, Davies MJ, Lood R. Oxygen Exposure and Tolerance Shapes the Cell Wall-Associated Lipids of the Skin Commensal Cutibacterium acnes. Microorganisms. 2023; 11(9):2260. https://doi.org/10.3390/microorganisms11092260
Chicago/Turabian StylePopa, Iuliana, David Touboul, Tilde Andersson, Eduardo Fuentes-Lemus, Cyrille Santerre, Michael J. Davies, and Rolf Lood. 2023. "Oxygen Exposure and Tolerance Shapes the Cell Wall-Associated Lipids of the Skin Commensal Cutibacterium acnes" Microorganisms 11, no. 9: 2260. https://doi.org/10.3390/microorganisms11092260
APA StylePopa, I., Touboul, D., Andersson, T., Fuentes-Lemus, E., Santerre, C., Davies, M. J., & Lood, R. (2023). Oxygen Exposure and Tolerance Shapes the Cell Wall-Associated Lipids of the Skin Commensal Cutibacterium acnes. Microorganisms, 11(9), 2260. https://doi.org/10.3390/microorganisms11092260