
Molecular Mechanisms of Persistence in Protozoan Parasites
 and
and
Abstract
:1. Introduction
2. Mechanisms of Persistence in Plasmodium spp.
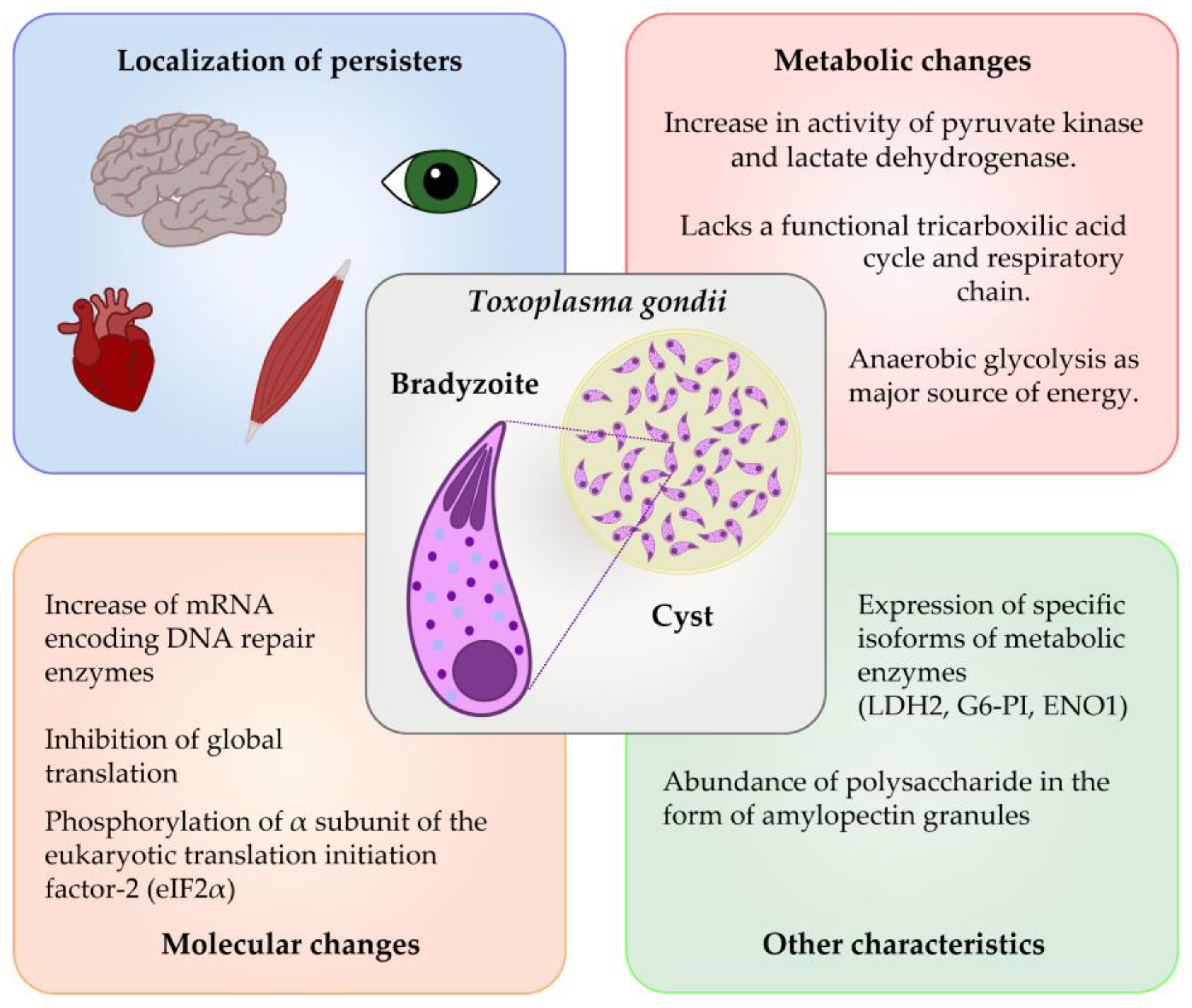
3. Mechanisms of Persistence in Toxoplasma spp.
4. Mechanisms of Persistence in Trypanosoma spp.
5. Mechanisms of Persistence in Leishmania spp.
6. Concluding Remarks
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| ATF | Adipose Tissue Forms |
| DSB | Double strand break |
| INH | Isonicotinic Acid Hydrazide |
| PYR | Pyrimethamine |
| SDZ | Sulfadiazine |
| GFP | Green Fluorescent Protein |
| TA | Toxin-Antitoxin |
| DNA | Deoxyribonucleic Acid |
| MRD | Minimal Residual Disease |
| NTD | Neglected Tropical Disease |
| CRT | Chloroquine Resistance Transporter |
| MDR1 | Multidrug Resistance 1 |
| VSA | Variant Surface Antigens |
| DHA | Dihydroartemisinin |
| RNA | Ribonucleic Acid |
| DDX6 | DEAD-Box Helicase 6 |
| Puf1 and Puf2 | named after two founding members Pumilio in Drosophila and fem-3-binding factor (FBF) in Caenorhabditis elegans |
| eIF2α | Eukaryotic Initiation Factor 2 alpha kinases |
| mRNA | Messenger Ribonucleic Acid |
| TCA cycle | tricarboxylic acid cycle |
| ATP | Adenosine triphosphate |
| ENO2/ENO1 | α-Enolase |
| LDH1/LDH2 | Lactate Dehydrogenase |
| G6-PI | glucose-6-phosphate isomerase |
| NO | Nitric Oxide |
| UPRT | uracil phosphoribosyl transferase |
| CD4 | cluster of differentiation 4 |
| IFN-γ | Interferon Gamma |
| BZN | benznidazole |
| NFX | nifurtimox |
| GPI | Glycosylphosphatidylinositol |
| CL | Cutaneous Leishmaniasis |
| VL | Visceral Leishmaniasis |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| MHC | Major Histocompatibility Complex |
| miRNA | MicroRNA |
| kDNA | Kinetoplast DNA |
| RBP | RNA-binding proteins |
References
- Reygaert, W.C. An overview of the antimicrobial resistance mechanisms of bacteria. AIMS Microbiol. 2018, 4, 482–501. [Google Scholar] [CrossRef] [PubMed]
- Hazlehurst, L.; Hacker, M. Chapter 15—Drug Resistance. In Pharmacology; Hacker, M., Messer, W., Bachmann, K., Eds.; Academic Press: San Diego, CA, USA, 2009; pp. 371–385. [Google Scholar]
- Wood, T.K.; Knabel, S.J.; Kwan, B.W. Bacterial persister cell formation and dormancy. Appl. Environ. Microbiol. 2013, 79, 7116–7121. [Google Scholar] [CrossRef] [PubMed]
- Lewis, K. Persister cells, dormancy and infectious disease. Nat. Rev. Microbiol. 2007, 5, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Vogwill, T.; Comfort, A.C.; Furio, V.; MacLean, R.C. Persistence and resistance as complementary bacterial adaptations to antibiotics. J. Evol. Biol. 2016, 29, 1223–1233. [Google Scholar] [CrossRef]
- Prestinaci, F.; Pezzotti, P.; Pantosti, A. Antimicrobial resistance: A global multifaceted phenomenon. Pathog. Glob. Health 2015, 109, 309–318. [Google Scholar] [CrossRef]
- Fisher, R.A.; Gollan, B.; Helaine, S. Persistent bacterial infections and persister cells. Nat. Rev. Microbiol. 2017, 15, 453–464. [Google Scholar] [CrossRef]
- Balaban, N.Q.; Merrin, J.; Chait, R.; Kowalik, L.; Leibler, S. Bacterial Persistence as a Phenotypic Switch. Science 2004, 305, 1622–1625. [Google Scholar] [CrossRef]
- Brown, S.P.; Grenfell, B.T. An unlikely partnership: Parasites, concomitant immunity and host defence. Proc. Biol. Sci. 2001, 268, 2543–2549. [Google Scholar] [CrossRef] [PubMed]
- Wainwright, J.; Hobbs, G.; Nakouti, I. Persister cells: Formation, resuscitation and combative therapies. Arch. Microbiol. 2021, 203, 5899–5906. [Google Scholar] [CrossRef]
- Shah, D.; Zhang, Z.; Khodursky, A.B.; Kaldalu, N.; Kurg, K.; Lewis, K. Persisters: A distinct physiological state of E. coli. BMC Microbiol. 2006, 6, 53. [Google Scholar] [CrossRef]
- Wang, X.; Wood, T.K. Toxin-antitoxin systems influence biofilm and persister cell formation and the general stress response. Appl. Environ. Microbiol. 2011, 77, 5577–5583. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.-H.; Ryu, C.-M.; Kim, J.-S. Bacterial persistence: Fundamentals and clinical importance. J. Microbiol. 2019, 57, 829–835. [Google Scholar] [CrossRef] [PubMed]
- Jayaraman, R. Bacterial persistence: Some new insights into an old phenomenon. J. Biosci. 2008, 33, 795–805. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; Vagner, S.; Robert, C. Persistent Cancer Cells: The Deadly Survivors. Cell 2020, 183, 860–874. [Google Scholar] [CrossRef] [PubMed]
- Andrews, K.T.; Fisher, G.; Skinner-Adams, T.S. Drug repurposing and human parasitic protozoan diseases. Int. J. Parasitol. Drugs Drug Resist. 2014, 4, 95–111. [Google Scholar] [CrossRef]
- WHO. Leishmaniasis. Available online: https://www.who.int/health-topics/leishmaniasis#tab=tab_3 (accessed on 23 March 2023).
- World Health Organization. Malaria. Available online: https://www.who.int/news-room/fact-sheets/detail/malaria (accessed on 3 May 2023).
- Fèvre, E.M.; Wissmann, B.V.; Welburn, S.C.; Lutumba, P. The burden of human African trypanosomiasis. PLoS Negl. Trop. Dis. 2008, 2, e333. [Google Scholar] [CrossRef]
- Mandell, M.A.; Beverley, S.M. Continual renewal and replication of persistent Leishmania major parasites in concomitantly immune hosts. Proc. Natl. Acad. Sci. USA 2017, 114, E801–E810. [Google Scholar] [CrossRef]
- Sanchez-Valdez, F.J.; Padilla, A.; Wang, W.; Orr, D.; Tarleton, R.L. Spontaneous dormancy protects Trypanosoma cruzi during extended drug exposure. eLife 2018, 7, e34039. [Google Scholar] [CrossRef]
- Barrett, M.P.; Kyle, D.E.; Sibley, L.D.; Radke, J.B.; Tarleton, R.L. Protozoan persister-like cells and drug treatment failure. Nat. Rev. Microbiol. 2019, 17, 607–620. [Google Scholar] [CrossRef]
- Fairlamb, A.H.; Gow, N.A.R.; Matthews, K.R.; Waters, A.P. Drug resistance in eukaryotic microorganisms. Nat. Microbiol. 2016, 1, 16092. [Google Scholar] [CrossRef]
- CDC. Malaria. Available online: https://www.cdc.gov/malaria/about/ (accessed on 14 March 2023).
- Howes, R.E.; Battle, K.E.; Mendis, K.N.; Smith, D.L.; Cibulskis, R.E.; Baird, J.K.; Hay, S.I. Global Epidemiology of Plasmodium vivax. Am. J. Trop. Med. Hyg. 2016, 95, 15–34. [Google Scholar] [CrossRef] [PubMed]
- Sato, S. Plasmodium-a brief introduction to the parasites causing human malaria and their basic biology. J. Physiol. Anthr. 2021, 40, 1. [Google Scholar] [CrossRef]
- Sattabongkot, J.; Suansomjit, C.; Nguitragool, W.; Sirichaisinthop, J.; Warit, S.; Tiensuwan, M.; Buates, S. Prevalence of asymptomatic Plasmodium infections with sub-microscopic parasite densities in the northwestern border of Thailand: A potential threat to malaria elimination. Malar. J. 2018, 17, 329. [Google Scholar] [CrossRef] [PubMed]
- Hyde, J.E. Drug-resistant malaria—An insight. FEBS J. 2007, 274, 4688–4698. [Google Scholar] [CrossRef] [PubMed]
- Cowman, A.F.; Morry, M.J.; Biggs, B.A.; Cross, G.A.; Foote, S.J. Amino acid changes linked to pyrimethamine resistance in the dihydrofolate reductase-thymidylate synthase gene of Plasmodium falciparum. Proc. Natl. Acad. Sci. USA 1988, 85, 9109–9113. [Google Scholar] [CrossRef]
- Valderramos, S.G.; Fidock, D.A. Transporters involved in resistance to antimalarial drugs. Trends Pharmacol. Sci. 2006, 27, 594–601. [Google Scholar] [CrossRef] [PubMed]
- Price, R.N.; Uhlemann, A.C.; Brockman, A.; McGready, R.; Ashley, E.; Phaipun, L.; Patel, R.; Laing, K.; Looareesuwan, S.; White, N.J.; et al. Mefloquine resistance in Plasmodium falciparum and increased pfmdr1 gene copy number. Lancet 2004, 364, 438–447. [Google Scholar] [CrossRef]
- Wicht, K.J.; Mok, S.; Fidock, D.A. Molecular Mechanisms of Drug Resistance in Plasmodium falciparum Malaria. Annu. Rev. Microbiol. 2020, 74, 431–454. [Google Scholar] [CrossRef]
- Schieck, E.; Poole, E.J.; Rippert, A.; Peshu, J.; Sasi, P.; Borrmann, S.; Bull, P.C. Plasmodium falciparum variant erythrocyte surface antigens: A pilot study of antibody acquisition in recurrent natural infections. Malar. J. 2017, 16, 450. [Google Scholar] [CrossRef]
- Schneider, V.M.; Visone, J.E.; Harris, C.T.; Florini, F.; Hadjimichael, E.; Zhang, X.; Gross, M.R.; Rhee, K.Y.; Ben Mamoun, C.; Kafsack, B.F.C.; et al. The human malaria parasite Plasmodium falciparum can sense environmental changes and respond by antigenic switching. Proc. Natl. Acad. Sci. USA 2023, 120, e2302152120. [Google Scholar] [CrossRef]
- Markus, M.B. Do hypnozoites cause relapse in malaria? Trends Parasitol. 2015, 31, 239–245. [Google Scholar] [CrossRef] [PubMed]
- LaCrue, A.N.; Scheel, M.; Kennedy, K.; Kumar, N.; Kyle, D.E. Effects of artesunate on parasite recrudescence and dormancy in the rodent malaria model Plasmodium vinckei. PLoS ONE 2011, 6, e26689. [Google Scholar] [CrossRef]
- Henrici, R.C.; van Schalkwyk, D.A.; Sutherland, C.J. Transient temperature fluctuations severely decrease P. falciparum susceptibility to artemisinin in vitro. Int. J. Parasitol. Drugs Drug Resist. 2019, 9, 23–26. [Google Scholar] [CrossRef] [PubMed]
- Menard, S.; Ben Haddou, T.; Ramadani, A.P.; Ariey, F.; Iriart, X.; Beghain, J.; Bouchier, C.; Witkowski, B.; Berry, A.; Mercereau-Puijalon, O.; et al. Induction of Multidrug Tolerance in Plasmodium falciparum by Extended Artemisinin Pressure. Emerg. Infect. Dis. 2015, 21, 1733–1741. [Google Scholar] [CrossRef] [PubMed]
- Nakazawa, S.; Maoka, T.; Uemura, H.; Ito, Y.; Kanbara, H. Malaria parasites giving rise to recrudescence in vitro. Antimicrob. Agents Chemother. 2002, 46, 958–965. [Google Scholar] [CrossRef]
- Teuscher, F.; Gatton, M.L.; Chen, N.; Peters, J.; Kyle, D.E.; Cheng, Q. Artemisinin-induced dormancy in Plasmodium falciparum: Duration, recovery rates, and implications in treatment failure. J. Infect. Dis. 2010, 202, 1362–1368. [Google Scholar] [CrossRef]
- Thapar, M.M.; Gil, J.P.; Bjorkman, A. In vitro recrudescence of Plasmodium falciparum parasites suppressed to dormant state by atovaquone alone and in combination with proguanil. Trans. R Soc. Trop. Med. Hyg. 2005, 99, 62–70. [Google Scholar] [CrossRef]
- Cheng, Q.; Kyle, D.E.; Gatton, M.L. Artemisinin resistance in Plasmodium falciparum: A process linked to dormancy? Int. J. Parasitol. Drugs Drug Resist. 2012, 2, 249–255. [Google Scholar] [CrossRef]
- Saggu, G.S. Apicoplast Journey and Its Essentiality as a Compartment for Malaria Parasite Survival. Front. Cell Infect. Microbiol. 2022, 12, 881825. [Google Scholar] [CrossRef]
- Chen, N.; LaCrue, A.N.; Teuscher, F.; Waters, N.C.; Gatton, M.L.; Kyle, D.E.; Cheng, Q. Fatty acid synthesis and pyruvate metabolism pathways remain active in dihydroartemisinin-induced dormant ring stages of Plasmodium falciparum. Antimicrob. Agents Chemother. 2014, 58, 4773–4781. [Google Scholar] [CrossRef]
- Peatey, C.L.; Chavchich, M.; Chen, N.; Gresty, K.J.; Gray, K.A.; Gatton, M.L.; Waters, N.C.; Cheng, Q. Mitochondrial Membrane Potential in a Small Subset of Artemisinin-Induced Dormant Plasmodium falciparum Parasites In Vitro. J. Infect. Dis. 2015, 212, 426–434. [Google Scholar] [CrossRef] [PubMed]
- Connelly, S.V.; Manzella-Lapeira, J.; Levine, Z.C.; Brzostowski, J.; Krymskaya, L.; Rahman, R.S.; Ellis, A.C.; Amin, S.N.; Sa, J.M.; Wellems, T.E. Restructured Mitochondrial-Nuclear Interaction in Plasmodium falciparum Dormancy and Persister Survival after Artemisinin Exposure. mBio 2021, 12, e0075321. [Google Scholar] [CrossRef]
- Witkowski, B.; Lelievre, J.; Barragan, M.J.; Laurent, V.; Su, X.Z.; Berry, A.; Benoit-Vical, F. Increased tolerance to artemisinin in Plasmodium falciparum is mediated by a quiescence mechanism. Antimicrob. Agents Chemother. 2010, 54, 1872–1877. [Google Scholar] [CrossRef] [PubMed]
- Vivax Sporozoite Consortium. Transcriptome and histone epigenome of Plasmodium vivax salivary-gland sporozoites point to tight regulatory control and mechanisms for liver-stage differentiation in relapsing malaria. Int. J. Parasitol. 2019, 49, 501–513. [Google Scholar] [CrossRef]
- Ruberto, A.A.; Maher, S.P.; Vantaux, A.; Joyner, C.J.; Bourke, C.; Balan, B.; Jex, A.; Mueller, I.; Witkowski, B.; Kyle, D.E. Single-cell RNA profiling of Plasmodium vivax-infected hepatocytes reveals parasite- and host- specific transcriptomic signatures and therapeutic targets. Front. Cell Infect. Microbiol. 2022, 12, 986314. [Google Scholar] [CrossRef] [PubMed]
- Ngwa, C.J.; Scheuermayer, M.; Mair, G.R.; Kern, S.; Brugl, T.; Wirth, C.C.; Aminake, M.N.; Wiesner, J.; Fischer, R.; Vilcinskas, A.; et al. Changes in the transcriptome of the malaria parasite Plasmodium falciparum during the initial phase of transmission from the human to the mosquito. BMC Genom. 2013, 14, 256. [Google Scholar] [CrossRef]
- Simon, N.; Kuehn, A.; Williamson, K.C.; Pradel, G. Adhesion protein complexes of malaria gametocytes assemble following parasite transmission to the mosquito. Parasitol. Int. 2016, 65, 27–30. [Google Scholar] [CrossRef]
- Barnes, K.I.; Little, F.; Mabuza, A.; Mngomezulu, N.; Govere, J.; Durrheim, D.; Roper, C.; Watkins, B.; White, N.J. Increased gametocytemia after treatment: An early parasitological indicator of emerging sulfadoxine-pyrimethamine resistance in falciparum malaria. J. Infect. Dis. 2008, 197, 1605–1613. [Google Scholar] [CrossRef]
- Peatey, C.L.; Skinner-Adams, T.S.; Dixon, M.W.; McCarthy, J.S.; Gardiner, D.L.; Trenholme, K.R. Effect of antimalarial drugs on Plasmodium falciparum gametocytes. J. Infect. Dis. 2009, 200, 1518–1521. [Google Scholar] [CrossRef]
- Thommen, B.T.; Passecker, A.; Buser, T.; Hitz, E.; Voss, T.S.; Brancucci, N.M.B. Revisiting the Effect of Pharmaceuticals on Transmission Stage Formation in the Malaria Parasite Plasmodium falciparum. Front. Cell Infect. Microbiol. 2022, 12, 802341. [Google Scholar] [CrossRef]
- Portugaliza, H.P.; Miyazaki, S.; Geurten, F.J.; Pell, C.; Rosanas-Urgell, A.; Janse, C.J.; Cortes, A. Artemisinin exposure at the ring or trophozoite stage impacts Plasmodium falciparum sexual conversion differently. eLlife 2020, 9, e60058. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. WHO Guidelines for Malaria; WHO: Geneva, Switzerland, 2023. [Google Scholar]
- Lamour, S.D.; Straschil, U.; Saric, J.; Delves, M.J. Changes in metabolic phenotypes of Plasmodium falciparum in vitro cultures during gametocyte development. Malar. J. 2014, 13, 468. [Google Scholar] [CrossRef] [PubMed]
- Mair, G.R.; Braks, J.A.; Garver, L.S.; Wiegant, J.C.; Hall, N.; Dirks, R.W.; Khan, S.M.; Dimopoulos, G.; Janse, C.J.; Waters, A.P. Regulation of sexual development of Plasmodium by translational repression. Science 2006, 313, 667–669. [Google Scholar] [CrossRef] [PubMed]
- Gomes-Santos, C.S.; Braks, J.; Prudencio, M.; Carret, C.; Gomes, A.R.; Pain, A.; Feltwell, T.; Khan, S.; Waters, A.; Janse, C.; et al. Transition of Plasmodium sporozoites into liver stage-like forms is regulated by the RNA binding protein Pumilio. PLoS Pathog. 2011, 7, e1002046. [Google Scholar] [CrossRef]
- Hall, N.; Karras, M.; Raine, J.D.; Carlton, J.M.; Kooij, T.W.; Berriman, M.; Florens, L.; Janssen, C.S.; Pain, A.; Christophides, G.K.; et al. A comprehensive survey of the Plasmodium life cycle by genomic, transcriptomic, and proteomic analyses. Science 2005, 307, 82–86. [Google Scholar] [CrossRef]
- Cui, L.; Fan, Q.; Li, J. The malaria parasite Plasmodium falciparum encodes members of the Puf RNA-binding protein family with conserved RNA binding activity. Nucleic Acids Res. 2002, 30, 4607–4617. [Google Scholar] [CrossRef]
- Müller, K.; Matuschewski, K.; Silvie, O. The Puf-Family RNA-Binding Protein Puf2 Controls Sporozoite Conversion to Liver Stages in the Malaria Parasite. PLoS ONE 2011, 6, e19860. [Google Scholar] [CrossRef]
- Zhang, M.; Fennell, C.; Ranford-Cartwright, L.; Sakthivel, R.; Gueirard, P.; Meister, S.; Caspi, A.; Doerig, C.; Nussenzweig, R.S.; Tuteja, R.; et al. The Plasmodium eukaryotic initiation factor-2alpha kinase IK2 controls the latency of sporozoites in the mosquito salivary glands. J. Exp. Med. 2010, 207, 1465–1474. [Google Scholar] [CrossRef]
- Zhang, M.; Mishra, S.; Sakthivel, R.; Rojas, M.; Ranjan, R.; Sullivan, W.J., Jr.; Fontoura, B.M.; Menard, R.; Dever, T.E.; Nussenzweig, V. PK4, a eukaryotic initiation factor 2alpha(eIF2alpha) kinase, is essential for the development of the erythrocytic cycle of Plasmodium. Proc. Natl. Acad. Sci. USA 2012, 109, 3956–3961. [Google Scholar] [CrossRef]
- Zhang, M.; Gallego-Delgado, J.; Fernandez-Arias, C.; Waters, N.C.; Rodriguez, A.; Tsuji, M.; Wek, R.C.; Nussenzweig, V.; Sullivan, W.J., Jr. Inhibiting the Plasmodium eIF2alpha Kinase PK4 Prevents Artemisinin-Induced Latency. Cell Host Microbe 2017, 22, 766–776 e764. [Google Scholar] [CrossRef]
- Cowman, A.F.; Crabb, B.S. Invasion of red blood cells by malaria parasites. Cell 2006, 124, 755–766. [Google Scholar] [CrossRef]
- Imwong, M.; Snounou, G.; Pukrittayakamee, S.; Tanomsing, N.; Kim, J.R.; Nandy, A.; Guthmann, J.P.; Nosten, F.; Carlton, J.; Looareesuwan, S.; et al. Relapses of Plasmodium vivax infection usually result from activation of heterologous hypnozoites. J. Infect. Dis. 2007, 195, 927–933. [Google Scholar] [CrossRef]
- Markus, M.B. The hypnozoite concept, with particular reference to malaria. Parasitol. Res. 2011, 108, 247–252. [Google Scholar] [CrossRef]
- Hutchison, W.M.; Dunachie, J.F.; Siim, J.C.; Work, K. Life cycle of Toxoplasma gondii. Br. Med. J. 1969, 4, 806. [Google Scholar] [CrossRef]
- Tenter, A.M.; Heckeroth, A.R.; Weiss, L.M. Toxoplasma gondii: From animals to humans. Int. J. Parasitol. 2000, 30, 1217–1258. [Google Scholar] [CrossRef] [PubMed]
- Luft, B.J.; Remington, J.S. AIDS commentary. Toxoplasmic encephalitis. J. Infect. Dis. 1988, 157, 1–6. [Google Scholar] [CrossRef] [PubMed]
- McLeod, R.; Mack, D.; Brown, C. Toxoplasma gondii-new advances in cellular and molecular biology. Exp. Parasitol. 1991, 72, 109–121. [Google Scholar] [CrossRef]
- Wong, S.Y.; Remington, J.S. Biology of Toxoplasma gondii. AIDS 1993, 7, 299–316. [Google Scholar] [CrossRef]
- Lainson, R. Observations on the development and nature of pseudocysts and cysts of Toxoplasma gondii. Trans. R. Soc. Trop. Med. Hyg. 1958, 52, 396–407. [Google Scholar] [CrossRef]
- Jeffers, V.; Tampaki, Z.; Kim, K.; Sullivan, W.J., Jr. A latent ability to persist: Differentiation in Toxoplasma gondii. Cell Mol. Life Sci. 2018, 75, 2355–2373. [Google Scholar] [CrossRef] [PubMed]
- Luft, B.J.; Brooks, R.G.; Conley, F.K.; McCabe, R.E.; Remington, J.S. Toxoplasmic Encephalitis in Patients with Acquired Immune Deficiency Syndrome. JAMA 1984, 252, 913–917. [Google Scholar] [CrossRef] [PubMed]
- Amikura, T.; Kikuchi, T.; Kato, J.; Koda, Y.; Sakurai, M.; Yamazaki, R.; Mikita, K.; Saburi, M.; Nakazato, T.; Mori, T. Toxoplasmosis after allogeneic hematopoietic stem cell transplantation: Impact of serostatus-based management. Transpl. Infect. Dis. 2021, 23, e13506. [Google Scholar] [CrossRef] [PubMed]
- Rougier, S.; Montoya, J.G.; Peyron, F. Lifelong Persistence of Toxoplasma Cysts: A Questionable Dogma? Trends Parasitol. 2017, 33, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, W.J., Jr.; Jeffers, V. Mechanisms of Toxoplasma gondii persistence and latency. FEMS Microbiol. Rev. 2012, 36, 717–733. [Google Scholar] [CrossRef]
- Montoya, J.G.; Liesenfeld, O. Toxoplasmosis. Lancet 2004, 363, 1965–1976. [Google Scholar] [CrossRef]
- Dzierszinski, F.; Nishi, M.; Ouko, L.; Roos, D.S. Dynamics of Toxoplasma gondii differentiation. Eukaryot. Cell 2004, 3, 992–1003. [Google Scholar] [CrossRef]
- Radke, J.R.; Behnke, M.S.; Mackey, A.J.; Radke, J.B.; Roos, D.S.; White, M.W. The transcriptome of Toxoplasma gondii. BMC Biol. 2005, 3, 26. [Google Scholar] [CrossRef]
- Denton, H.; Roberts, C.W.; Alexander, J.; Thong, K.W.; Coombs, G.H. Enzymes of energy metabolism in the bradyzoites and tachyzoites of Toxoplasma gondii. FEMS Microbiol. Lett. 1996, 137, 103–108. [Google Scholar] [CrossRef]
- Yang, S.; Parmley, S.F. Toxoplasma gondii expresses two distinct lactate dehydrogenase homologous genes during its life cycle in intermediate hosts. Gene 1997, 184, 1–12. [Google Scholar] [CrossRef]
- Ferguson, D.J.; Parmley, S.F.; Tomavo, S. Evidence for nuclear localisation of two stage-specific isoenzymes of enolase in Toxoplasma gondii correlates with active parasite replication. Int. J. Parasitol. 2002, 32, 1399–1410. [Google Scholar] [CrossRef]
- Manger, I.D.; Hehl, A.; Parmley, S.; Sibley, L.D.; Marra, M.; Hillier, L.; Waterston, R.; Boothroyd, J.C. Expressed sequence tag analysis of the bradyzoite stage of Toxoplasma gondii: Identification of developmentally regulated genes. Infect. Immun. 1998, 66, 1632–1637. [Google Scholar] [CrossRef] [PubMed]
- Yahiaoui, B.; Dzierszinski, F.; Bernigaud, A.; Slomianny, C.; Camus, D.; Tomavo, S. Isolation and characterization of a subtractive library enriched for developmentally regulated transcripts expressed during encystation of Toxoplasma gondii. Mol. Biochem. Parasitol. 1999, 99, 223–235. [Google Scholar] [CrossRef] [PubMed]
- Soete, M.; Camus, D.; Dubremetz, J.F. Experimental induction of bradyzoite-specific antigen expression and cyst formation by the RH strain of Toxoplasma gondii in vitro. Exp. Parasitol. 1994, 78, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Bohne, W.; Heesemann, J.; Gross, U. Reduced replication of Toxoplasma gondii is necessary for induction of bradyzoite-specific antigens: A possible role for nitric oxide in triggering stage conversion. Infect. Immun. 1994, 62, 1761–1767. [Google Scholar] [CrossRef] [PubMed]
- Fox, B.A.; Gigley, J.P.; Bzik, D.J. Toxoplasma gondii lacks the enzymes required for de novo arginine biosynthesis and arginine starvation triggers cyst formation. Int. J. Parasitol. 2004, 34, 323–331. [Google Scholar] [CrossRef]
- Fox, B.A.; Ristuccia, J.G.; Gigley, J.P.; Bzik, D.J. Efficient gene replacements in Toxoplasma gondii strains deficient for nonhomologous end joining. Eukaryot. Cell 2009, 8, 520–529. [Google Scholar] [CrossRef]
- Fox, B.A.; Falla, A.; Rommereim, L.M.; Tomita, T.; Gigley, J.P.; Mercier, C.; Cesbron-Delauw, M.F.; Weiss, L.M.; Bzik, D.J. Type II Toxoplasma gondii KU80 knockout strains enable functional analysis of genes required for cyst development and latent infection. Eukaryot. Cell 2011, 10, 1193–1206. [Google Scholar] [CrossRef]
- Pereira-Chioccola, V.L.; Vidal, J.E.; Su, C. Toxoplasma gondii infection and cerebral toxoplasmosis in HIV-infected patients. Future Microbiol. 2009, 4, 1363–1379. [Google Scholar] [CrossRef]
- Suzuki, Y.; Orellana, M.A.; Schreiber, R.D.; Remington, J.S. Interferon-gamma: The major mediator of resistance against Toxoplasma gondii. Science 1988, 240, 516–518. [Google Scholar] [CrossRef]
- Wek, R.C.; Jiang, H.-Y.; Anthony, T.G. Coping with stress: eIF2 kinases and translational control. Biochem. Soc. Trans. 2006, 34, 7–11. [Google Scholar] [CrossRef]
- Sutherland, C.S.; Yukich, J.; Goeree, R.; Tediosi, F. A literature review of economic evaluations for a neglected tropical disease: Human African trypanosomiasis (“sleeping sickness”). PLoS Negl. Trop. Dis. 2015, 9, e0003397. [Google Scholar] [CrossRef] [PubMed]
- Funk, S.; Nishiura, H.; Heesterbeek, H.; Edmunds, W.J.; Checchi, F. Identifying transmission cycles at the human-animal interface: The role of animal reservoirs in maintaining gambiense human african trypanosomiasis. PLoS Comput. Biol. 2013, 9, e1002855. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, J.; Steiner, I.; Kennedy, P.G.E. Generation of neuroinflammation in human African trypanosomiasis. Neurol. Neuroimmunol. Neuroinflamm 2019, 6, e610. [Google Scholar] [CrossRef]
- Simarro, P.P.; Cecchi, G.; Paone, M.; Franco, J.R.; Diarra, A.; Ruiz, J.A.; Fevre, E.M.; Courtin, F.; Mattioli, R.C.; Jannin, J.G. The Atlas of human African trypanosomiasis: A contribution to global mapping of neglected tropical diseases. Int. J. Health Geogr. 2010, 9, 57. [Google Scholar] [CrossRef]
- Alvarez-Rodriguez, A.; Jin, B.K.; Radwanska, M.; Magez, S. Recent progress in diagnosis and treatment of Human African Trypanosomiasis has made the elimination of this disease a realistic target by 2030. Front. Med. 2022, 9, 1037094. [Google Scholar] [CrossRef] [PubMed]
- Perez-Molina, J.A.; Molina, I. Chagas disease. Lancet 2018, 391, 82–94. [Google Scholar] [CrossRef] [PubMed]
- Hemmige, V.; Tanowitz, H.; Sethi, A. Trypanosoma cruzi infection: A review with emphasis on cutaneous manifestations. Int. J. Dermatol. 2012, 51, 501–508. [Google Scholar] [CrossRef]
- Magalhaes, L.M.D.; Gollob, K.J.; Zingales, B.; Dutra, W.O. Pathogen diversity, immunity, and the fate of infections: Lessons learned from Trypanosoma cruzi human-host interactions. Lancet Microbe 2022, 3, e711–e722. [Google Scholar] [CrossRef]
- Echeverria, L.E.; Morillo, C.A. American Trypanosomiasis (Chagas Disease). Infect. Dis. Clin. North Am. 2019, 33, 119–134. [Google Scholar] [CrossRef]
- Garcia, M.N.; Woc-Colburn, L.; Aguilar, D.; Hotez, P.J.; Murray, K.O. Historical Perspectives on the Epidemiology of Human Chagas Disease in Texas and Recommendations for Enhanced Understanding of Clinical Chagas Disease in the Southern United States. PLoS Negl. Trop. Dis. 2015, 9, e0003981. [Google Scholar] [CrossRef]
- Alvar, J.; Alves, F.; Bucheton, B.; Burrows, L.; Büscher, P.; Carrillo, E.; Felger, I.; Hübner, M.P.; Moreno, J.; Pinazo, M.J.; et al. Implications of asymptomatic infection for the natural history of selected parasitic tropical diseases. Semin. Immunopathol. 2020, 42, 231–246. [Google Scholar] [CrossRef] [PubMed]
- Buscher, P.; Cecchi, G.; Jamonneau, V.; Priotto, G. Human African trypanosomiasis. Lancet 2017, 390, 2397–2409. [Google Scholar] [CrossRef] [PubMed]
- Kratz, J.M.; Garcia Bournissen, F.; Forsyth, C.J.; Sosa-Estani, S. Clinical and pharmacological profile of benznidazole for treatment of Chagas disease. Expert. Rev. Clin. Pharmacol. 2018, 11, 943–957. [Google Scholar] [CrossRef] [PubMed]
- Dumoulin, P.C.; Burleigh, B.A. Stress-Induced Proliferation and Cell Cycle Plasticity of Intracellular Trypanosoma cruzi Amastigotes. mBio 2018, 9, e00673-18. [Google Scholar] [CrossRef]
- Resende, B.C.; Oliveira, A.C.S.; Guanabens, A.C.P.; Repoles, B.M.; Santana, V.; Hiraiwa, P.M.; Pena, S.D.J.; Franco, G.R.; Macedo, A.M.; Tahara, E.B.; et al. The Influence of Recombinational Processes to Induce Dormancy in Trypanosoma cruzi. Front. Cell Infect. Microbiol. 2020, 10, 5. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, M.C.; Andrews, N.W. Host cell invasion by Trypanosoma cruzi: A unique strategy that promotes persistence. FEMS Microbiol. Rev. 2012, 36, 734–747. [Google Scholar] [CrossRef]
- Burleigh, B.A.; Andrews, N.W. The mechanisms of Trypanosoma cruzi invasion of mammalian cells. Annu. Rev. Microbiol. 1995, 49, 175–200. [Google Scholar] [CrossRef]
- Li, Y.; Shah-Simpson, S.; Okrah, K.; Belew, A.T.; Choi, J.; Caradonna, K.L.; Padmanabhan, P.; Ndegwa, D.M.; Temanni, M.R.; Corrada Bravo, H.; et al. Transcriptome Remodeling in Trypanosoma cruzi and Human Cells during Intracellular Infection. PLoS Pathog. 2016, 12, e1005511. [Google Scholar] [CrossRef]
- Desale, H.; Herrera, C.; Dumonteil, E. Trypanosoma cruzi amastigote transcriptome analysis reveals heterogenous populations with replicating and dormant parasites. Res. Sq. 2023, preprint. [Google Scholar] [CrossRef]
- Trindade, S.; De Niz, M.; Costa-Sequeira, M.; Bizarra-Rebelo, T.; Bento, F.; Dejung, M.; Narciso, M.V.; Lopez-Escobar, L.; Ferreira, J.; Butter, F.; et al. Slow growing behavior in African trypanosomes during adipose tissue colonization. Nat. Commun. 2022, 13, 7548. [Google Scholar] [CrossRef]
- Dumoulin, P.C.; Burleigh, B.A. Metabolic flexibility in Trypanosoma cruzi amastigotes: Implications for persistence and drug sensitivity. Curr. Opin. Microbiol. 2021, 63, 244–249. [Google Scholar] [CrossRef]
- Mougneau, E.; Bihl, F.; Glaichenhaus, N. Cell biology and immunology of Leishmania. Immunol. Rev. 2011, 240, 286–296. [Google Scholar] [CrossRef]
- Peacock, C.S.; Seeger, K.; Harris, D.; Murphy, L.; Ruiz, J.C.; Quail, M.A.; Peters, N.; Adlem, E.; Tivey, A.; Aslett, M.; et al. Comparative genomic analysis of three Leishmania species that cause diverse human disease. Nat. Genet. 2007, 39, 839–847. [Google Scholar] [CrossRef]
- CDC. Parasites—Leishmaniasis. Available online: https://www.cdc.gov/parasites/leishmaniasis/epi.html (accessed on 26 March 2023).
- World Health Organization. Manual de Procedimientos para Vigilancia y Control de las Leishmaniasis en las Américas; World Health Organization: Geneva, Switzerland, 2019; p. 166. [Google Scholar]
- Boelaert, M.; Sundar, S. 47—Leishmaniasis. In Manson’s Tropical Infectious Diseases, 23rd ed.; Farrar, J., Hotez, P.J., Junghanss, T., Kang, G., Lalloo, D., White, N.J., Eds.; W.B. Saunders: London, UK, 2014; pp. 631–651.e634. [Google Scholar]
- Gutierrez Guarnizo, S.A.; Tikhonova, E.B.; Karamyshev, A.L.; Muskus, C.E.; Karamysheva, Z.N. Translational reprogramming as a driver of antimony-drug resistance in Leishmania. Nat. Commun. 2023, 14, 2605. [Google Scholar] [CrossRef]
- Gutierrez Guarnizo, S.A.; Tikhonova, E.B.; Zabet-Moghaddam, M.; Zhang, K.; Muskus, C.; Karamyshev, A.L.; Karamysheva, Z.N. Drug-Induced Lipid Remodeling in Leishmania Parasites. Microorganisms 2021, 9, 790. [Google Scholar] [CrossRef]
- Gutierrez Guarnizo, S.A.; Karamysheva, Z.N.; Galeano, E.; Muskus, C.E. Metabolite Biomarkers of Leishmania Antimony Resistance. Cells 2021, 10, 1063. [Google Scholar] [CrossRef] [PubMed]
- Mitropoulos, P.; Konidas, P.; Durkin-Konidas, M. New World cutaneous leishmaniasis: Updated review of current and future diagnosis and treatment. J. Am. Acad. Dermatol. 2010, 63, 309–322. [Google Scholar] [CrossRef]
- Karamian, M.; Bojd, M.S.; Salehabadi, A.; Hemmati, M.; Barati, D.A. Effectiveness of meglumine antimoniate against L. tropica in a recently emerged focus of cutaneous leishmaniasis in Birjand, eastern Islamic Republic of Iran. East. Mediterr. Health J. 2015, 21, 280–286. [Google Scholar] [CrossRef] [PubMed]
- Van Zandbergen, G.; Bollinger, A.; Wenzel, A.; Kamhawi, S.; Voll, R.; Klinger, M.; Müller, A.; Hölscher, C.; Herrmann, M.; Sacks, D. Leishmania disease development depends on the presence of apoptotic promastigotes in the virulent inoculum. Proc. Natl. Acad. Sci. USA 2006, 103, 13837–13842. [Google Scholar] [CrossRef] [PubMed]
- Kaye, P.; Scott, P. Leishmaniasis: Complexity at the host–pathogen interface. Nat. Rev. Microbiol. 2011, 9, 604–615. [Google Scholar] [CrossRef] [PubMed]
- Saunders, E.C.; Ng, W.W.; Kloehn, J.; Chambers, J.M.; Ng, M.; McConville, M.J. Induction of a stringent metabolic response in intracellular stages of Leishmania mexicana leads to increased dependence on mitochondrial metabolism. PLoS Pathog. 2014, 10, e1003888. [Google Scholar] [CrossRef] [PubMed]
- Jara, M.; Berg, M.; Caljon, G.; de Muylder, G.; Cuypers, B.; Castillo, D.; Maes, I.; Orozco, M.D.C.; Vanaerschot, M.; Dujardin, J.C.; et al. Macromolecular biosynthetic parameters and metabolic profile in different life stages of Leishmania braziliensis: Amastigotes as a functionally less active stage. PLoS ONE 2017, 12, e0180532. [Google Scholar] [CrossRef]
- Chaparro, V.; Graber, T.E.; Alain, T.; Jaramillo, M. Transcriptional profiling of macrophages reveals distinct parasite stage-driven signatures during early infection by Leishmania donovani. Sci. Rep. 2022, 12, 6369. [Google Scholar] [CrossRef]
- Buates, S.; Matlashewski, G. General suppression of macrophage gene expression during Leishmania donovani infection. J. Immunol. 2001, 166, 3416–3422. [Google Scholar] [CrossRef]
- Chaparro, V.; Leroux, L.P.; Masvidal, L.; Lorent, J.; Graber, T.E.; Zimmermann, A.; Arango Duque, G.; Descoteaux, A.; Alain, T.; Larsson, O.; et al. Translational profiling of macrophages infected with Leishmania donovani identifies mTOR- and eIF4A-sensitive immune-related transcripts. PLoS Pathog. 2020, 16, e1008291. [Google Scholar] [CrossRef]
- Kong, F.; Saldarriaga, O.A.; Spratt, H.; Osorio, E.Y.; Travi, B.L.; Luxon, B.A.; Melby, P.C. Transcriptional Profiling in Experimental Visceral Leishmaniasis Reveals a Broad Splenic Inflammatory Environment that Conditions Macrophages toward a Disease-Promoting Phenotype. PLOS Pathog. 2017, 13, e1006165. [Google Scholar] [CrossRef]
- Shadab, M.; Das, S.; Banerjee, A.; Sinha, R.; Asad, M.; Kamran, M.; Maji, M.; Jha, B.; Deepthi, M.; Kumar, M.; et al. RNA-Seq Revealed Expression of Many Novel Genes Associated with Leishmania donovani Persistence and Clearance in the Host Macrophage. Front. Cell Infect. Microbiol. 2019, 9, 17. [Google Scholar] [CrossRef] [PubMed]
- Piccirillo, C.A.; Bjur, E.; Topisirovic, I.; Sonenberg, N.; Larsson, O. Translational control of immune responses: From transcripts to translatomes. Nat. Immunol. 2014, 15, 503–511. [Google Scholar] [CrossRef]
- William, M.; Leroux, L.P.; Chaparro, V.; Graber, T.E.; Alain, T.; Jaramillo, M. Translational repression of Ccl5 and Cxcl10 by 4E-BP1 and 4E-BP2 restrains the ability of mouse macrophages to induce migration of activated T cells. Eur. J. Immunol. 2019, 49, 1200–1212. [Google Scholar] [CrossRef] [PubMed]
- Jara, M.; Barrett, M.; Maes, I.; Regnault, C.; Imamura, H.; Domagalska, M.A.; Dujardin, J.C. Transcriptional Shift and Metabolic Adaptations during Leishmania Quiescence Using Stationary Phase and Drug Pressure as Models. Microorganisms 2022, 10, 97. [Google Scholar] [CrossRef] [PubMed]
- Matte, C.; Casgrain, P.A.; Seguin, O.; Moradin, N.; Hong, W.J.; Descoteaux, A. Leishmania major Promastigotes Evade LC3-Associated Phagocytosis through the Action of GP63. PLoS Pathog. 2016, 12, e1005690. [Google Scholar] [CrossRef] [PubMed]
- Jaramillo, M.; Gomez, M.A.; Larsson, O.; Shio, M.T.; Topisirovic, I.; Contreras, I.; Luxenburg, R.; Rosenfeld, A.; Colina, R.; McMaster, R.W.; et al. Leishmania repression of host translation through mTOR cleavage is required for parasite survival and infection. Cell Host Microbe 2011, 9, 331–341. [Google Scholar] [CrossRef]
- Guay-Vincent, M.M.; Matte, C.; Berthiaume, A.M.; Olivier, M.; Jaramillo, M.; Descoteaux, A. Revisiting Leishmania GP63 host cell targets reveals a limited spectrum of substrates. PLoS Pathog. 2022, 18, e1010640. [Google Scholar] [CrossRef] [PubMed]
- Matheoud, D.; Moradin, N.; Bellemare-Pelletier, A.; Shio, M.T.; Hong, W.J.; Olivier, M.; Gagnon, E.; Desjardins, M.; Descoteaux, A. Leishmania evades host immunity by inhibiting antigen cross-presentation through direct cleavage of the SNARE VAMP8. Cell Host Microbe 2013, 14, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Marr, A.K.; MacIsaac, J.L.; Jiang, R.; Airo, A.M.; Kobor, M.S.; McMaster, W.R. Leishmania donovani infection causes distinct epigenetic DNA methylation changes in host macrophages. PLoS Pathog. 2014, 10, e1004419. [Google Scholar] [CrossRef]
- Singh, A.K.; Pandey, R.K.; Siqueira-Neto, J.L.; Kwon, Y.J.; Freitas-Junior, L.H.; Shaha, C.; Madhubala, R. Proteomic-based approach to gain insight into reprogramming of THP-1 cells exposed to Leishmania donovani over an early temporal window. Infect. Immun. 2015, 83, 1853–1868. [Google Scholar] [CrossRef]
- Karamysheva, Z.N.; Gutierrez Guarnizo, S.A.; Karamyshev, A.L. Regulation of Translation in the Protozoan Parasite Leishmania. Int. J. Mol. Sci. 2020, 21, 2981. [Google Scholar] [CrossRef]
- Szentmary, N.; Daas, L.; Shi, L.; Laurik, K.L.; Lepper, S.; Milioti, G.; Seitz, B. Acanthamoeba keratitis—Clinical signs, differential diagnosis and treatment. J. Curr. Ophthalmol. 2019, 31, 16–23. [Google Scholar] [CrossRef]
- Siddiqui, R.; Khan, N.A. Balamuthia mandrillaris: Morphology, biology, and virulence. Trop. Parasitol. 2015, 5, 15–22. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Traits | Description | References |
|---|---|---|
| Location of the dormant cells | Host’s red blood cells and hepatocytes (P. vivax, P. ovale) | [48,66,67,68] |
| Transcriptome changes | Stage-specific gene expression, variant surface antigens (VSAs) expression (including the var gene family, which encodes the erythrocyte membrane protein 1 PfEMP1), utilizing non-coding RNAs, inducing stress-responsive pathways. Overexpression of genes encoding specific cellular RBPs and proteases like Vivapains | [33,34,49] |
| Translatome changes | DOZI binding to the ribonucleoprotein complex, with translation repression. Phosphorylation of eIF2α and formation of stress granules. Translational repression | [41,58,59,63,64,65] |
| Metabolomic changes | Metabolic activity decrease, nutrient uptake decrease, and active apicoplasts and mitochondria. Restructuring of mitochondria–nucleus interaction. | [43,46,57] |
| Traits | Description | References |
|---|---|---|
| Location of the dormant cells | Blood, lymph, and subcutaneous tissues (particularly the cardiac muscle) and CNS | [111,112] |
| Transcriptomic changes | Downregulation of transcripts of major polymorphic surface proteins, reduced expression of genes coding proteins involved in flagellar assembly and motility, shortening of the single T. cruzi flagellum; increased abundance of δ-amastin; increased transcript levels of GPI-inositol deacylase, membrane-bound/secreted phospholipase A1, and surface-localized phosphatidylinositol-phospholipase C (PI-PLC). Upregulation of proteins in charge of DSB repairing | [110,113,114] |
| Proteome changes | Differential expression of plasma membrane proteins, protein kinases, and phosphatases. Reduced protein synthesis in adipose tissue (T. brucei) | [113,115] |
| Metabolome changes | Signaling pathway retooling with differentially expressed protein kinases and phosphatases. | [116] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tarannum, A.; Rodríguez-Almonacid, C.C.; Salazar-Bravo, J.; Karamysheva, Z.N. Molecular Mechanisms of Persistence in Protozoan Parasites. Microorganisms 2023, 11, 2248. https://doi.org/10.3390/microorganisms11092248
Tarannum A, Rodríguez-Almonacid CC, Salazar-Bravo J, Karamysheva ZN. Molecular Mechanisms of Persistence in Protozoan Parasites. Microorganisms. 2023; 11(9):2248. https://doi.org/10.3390/microorganisms11092248
Chicago/Turabian StyleTarannum, Asfiha, Cristian Camilo Rodríguez-Almonacid, Jorge Salazar-Bravo, and Zemfira N. Karamysheva. 2023. "Molecular Mechanisms of Persistence in Protozoan Parasites" Microorganisms 11, no. 9: 2248. https://doi.org/10.3390/microorganisms11092248
APA StyleTarannum, A., Rodríguez-Almonacid, C. C., Salazar-Bravo, J., & Karamysheva, Z. N. (2023). Molecular Mechanisms of Persistence in Protozoan Parasites. Microorganisms, 11(9), 2248. https://doi.org/10.3390/microorganisms11092248