
COVID Variants, Villain and Victory: A Bioinformatics Perspective


Abstract
1. Introduction
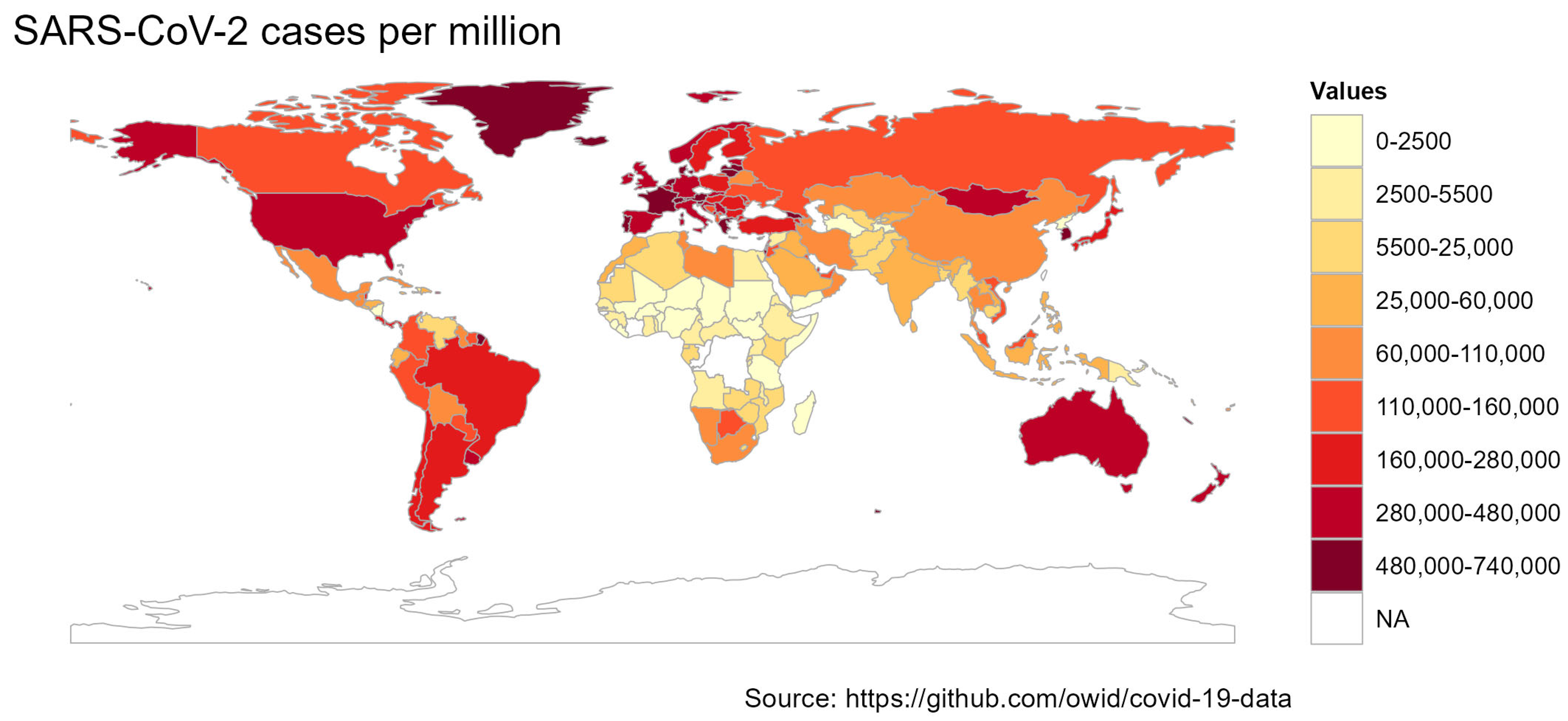
2. Tracking of SARS-CoV-2
3. Vaccine Status and Development
4. Discussion and Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Centers for Disease Control Control and Prevention. SARS-CoV-2 Variant Classifications and Definitions. 2023. Available online: https://www.cdc.gov/coronavirus/2019-ncov/variants/variant-classifications.html (accessed on 25 March 2023).
- WHO. Updated Working Definitions and Primary Actions for SARS-CoV-2 Variants. 2023. Available online: https://www.who.int/publications/m/item/updated-working-definitions-and-primary-actions-for--sars-cov-2-variants (accessed on 14 June 2023).
- World Health Organization. Statement on the Update of WHO’s Working Definitions and Tracking System for SARS-CoV-2 Variants of Concern and Variants of Interest. 2023. Available online: https://www.who.int/news/item/16-03-2023-statement-on-the-update-of-who-s-working-definitions-and-tracking-system-for-sars-cov-2-variants-of-concern-and-variants-of-interest (accessed on 14 June 2023).
- Mathieu, E.; Ritchie, H.; Rodés-Guirao, L.; Appel, C.; Gavrilov, D.; Giattino, C.; Hasell, J.; Macdonald, B.; Dattani, S.; Beltekian, D.; et al. Coronavirus Pandemic (COVID-19). 2020. Available online: https://ourworldindata.org/covid-cases/ (accessed on 16 June 2023).
- Davis, H.E.; Assaf, G.S.; McCorkell, L.; Wei, H.; Low, R.J.; Re’Em, Y.; Redfield, S.; Austin, J.P.; Akrami, A. Characterizing long COVID in an international cohort: 7 months of symptoms and their impact. Eclinicalmedicine 2021, 38, 101019. [Google Scholar] [CrossRef]
- Rambaut, A.; Loman, N.; Pybus, O.; Barclay, W.; Barrett, J.; Carabelli, A.; Connor, T.; Peacock, T.; Robertson, D.L.; Volz, E. Preliminary Genomic Characterisation of an Emergent SARS-CoV-2 Lineage in the UK Defined by a Novel Set of Spike Mutations. 2020. Available online: https://virological.org/t/preliminary-genomic-characterisation-of-an-emergent-sars-cov-2-lineage-in-the-uk-defined-by-a-novel-set-of-spike-mutations/563 (accessed on 3 July 2023).
- Volz, E.; Mishra, S.; Chand, M.; Barrett, J.C.; Johnson, R.; Geidelberg, L.; Ferguson, N.M. Assessing transmissibility of SARS-CoV-2 lineage B.1.1.7 in England. Nature 2021, 593, 266–269. [Google Scholar] [CrossRef] [PubMed]
- Weisblum, Y.; Schmidt, F.; Zhang, F.; DaSilva, J.; Poston, D.; Lorenzi, J.C.; Bieniasz, P.D. Escape from neutralizing antibodies by SARS-CoV-2 spike protein variants. eLife 2020, 9, e61312. [Google Scholar] [CrossRef] [PubMed]
- Mlcochova, P.; Kemp, S.A.; Dhar, M.S.; Papa, G.; Meng, B.; Ferreira, I.A.T.M.; Datir, R.; Collier, D.A.; Albecka, A.; Singh, S.; et al. SARS-CoV-2 B.1.617.2 Delta variant replication and immune evasion. Nature 2021, 599, 114–119. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Ma, X. Acute respiratory failure in COVID-19: Is it “typical” ARDS? Crit. Care 2020, 24, 198. [Google Scholar] [CrossRef] [PubMed]
- Khandia, R.; Singhal, S.; Alqahtani, T.; Kamal, M.A.; El-Shall, N.A.; Nainu, F.; Desingu, P.A.; Dhama, K. Emergence of SARS-CoV-2 Omicron (B.1.1.529) variant, salient features, high global health concerns and strategies to counter it amid ongoing COVID-19 pandemic. Environ. Res. 2022, 209, 112816. [Google Scholar] [CrossRef]
- Hyams, C.; Challen, R.; Marlow, R.; Nguyen, J.; Begier, E.; Southern, J.; Szasz-Benczur, Z. Severity of Omicron (B.1.1.529) and Delta (B.1.617.2) SARS-CoV-2 infection among hospitalised adults: A prospective cohort study in Bristol, United Kingdom. Lancet Reg. Health Eur. 2023, 25, 100556. [Google Scholar] [CrossRef]
- Carabelli, A.M.; Peacock, T.P.; Thorne, L.G.; Harvey, W.T.; Hughes, J. SARS-CoV-2 variant biology: Immune escape, transmission and fitness. Nat. Rev. Microbiol. 2023, 21, 162–177. [Google Scholar] [CrossRef]
- Willett, B.J.; Grove, J.; MacLean, O.A.; Wilkie, C.; De Lorenzo, G.; Furnon, W.; Thomson, E.C. SARS-CoV-2 Omicron is an immune escape variant with an altered cell entry pathway. Nat. Microbiol. 2022, 7, 1161–1179. [Google Scholar] [CrossRef]
- Meng, B.; Abdullahi, A.; Ferreira, I.A.; Goonawardane, N.; Saito, A.; Kimura, I.; Gupta, R.K. Altered TMPRSS2 usage by SARS-CoV-2 Omicron impacts infectivity and fusogenicity. Nature 2022, 603, 706–714. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, R.; Yamasoba, D.; Kimura, I.; Wang, L.; Kishimoto, M.; Ito, J.; Sato, K. Attenuated fusogenicity and pathogenicity of SARS-CoV-2 Omicron variant. Nature 2022, 603, 700–705. [Google Scholar] [CrossRef]
- Bentley, E.G.; Kirby, A.; Sharma, P.; Kipar, A.; Mega, D.F.; Bramwell, C.; Stewart, J.P. SARS-CoV-2 Omicron-B.1.1.529 Variant leads to less severe disease than Pango B and Delta variants strains in a mouse model of severe COVID-19. BioRxiv, 2021; Preprint. [Google Scholar] [CrossRef]
- Bowe, B.; Xie, Y.; Al-Aly, Z. Acute and postacute sequelae associated with SARS-CoV-2 reinfection. Nat. Med. 2022, 28, 2398–2405. [Google Scholar] [CrossRef] [PubMed]
- Kedor, C.; Freitag, H.; Meyer-Arndt, L.; Wittke, K.; Hanitsch, L.G.; Zoller, T.; Scheibenbogen, C. A prospective observational study of post-COVID-19 chronic fatigue syndrome following the first pandemic wave in Germany and biomarkers associated with symptom severity. Nat. Commun. 2022, 13, 5104. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Liu, B.; Zhang, S.; Huang, N.; Zhao, T.; Lu, Q.B.; Cui, F. Differences in incidence and fatality of COVID-19 by SARS-CoV-2 Omicron variant versus Delta variant in relation to vaccine coverage: A world-wide review. J. Med. Virol. 2023, 95, e28118. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Shaman, J.L. Author response: COVID-19 pandemic dynamics in South Africa and epidemiological characteristics of three variants of concern (Beta, Delta, and Omicron). eLife 2022, 11, e78933. [Google Scholar] [CrossRef]
- Ray, M.; Sable, M.N.; Sarkar, S.; Hallur, V. Essential interpretations of bioinformatics in COVID-19 pandemic. Meta Gene 2020, 27, 100844. [Google Scholar] [CrossRef]
- Guzzi, P.H.; Petrizzelli, F.; Mazza, T. Disease spreading modeling and analysis: A survey. Brief. Bioinform. 2022, 23, bbac230. [Google Scholar] [CrossRef]
- Kumar Das, J.; Tradigo, G.; Veltri, P.; Guzzi, P.H.; Roy, S. Data science in unveiling COVID-19 pathogenesis and diagnosis: Evolutionary origin to drug repurposing. Brief. Bioinform. 2021, 22, 855–872. [Google Scholar] [CrossRef]
- Ma, J.; Deng, Y.; Zhang, M.; Yu, J. The role of multi-omics in the diagnosis of COVID-19 and the prediction of new therapeutic targets. Virulence 2022, 13, 1101–1110. [Google Scholar] [CrossRef]
- Siminea, N.; Popescu, V.; Martin, J.A.S.; Florea, D.; Gavril, G.; Gheorghe, A.-M.; Iţcuş, C.; Kanhaiya, K.; Pacioglu, O.; Popa, L.I.; et al. Network analytics for drug repurposing in COVID-19. Brief. Bioinform. 2021, 23, bbab490. [Google Scholar] [CrossRef]
- Hu, T.; Li, J.; Zhou, H.; Li, C.; Holmes, E.C.; Shi, W. Bioinformatics resources for SARS-CoV-2 discovery and surveillance. Brief. Bioinform. 2021, 22, 631–641. [Google Scholar] [CrossRef]
- Mehta, P.; Swaminathan, A.; Yadav, A.; Chattopadhyay, P.; Shamim, U.; Pandey, R. Integrative genomics important to understand host–pathogen interactions. Brief. Funct. Genom. 2022, elac021. [Google Scholar] [CrossRef]
- Wood, D.E.; Lu, J.; Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-Source, Platform-Independent, Community-Supported Software for Describing and Comparing Microbial Communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [PubMed]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, Interactive, Scalable and Extensible Microbiome Data Science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Hao, S.; Andersen-Nissen, E.; Mauck, W.M., 3rd; Zheng, S.; Butler, A.; Lee, M.J.; Wilk, A.J.; Darby, C.; Zager, M.; et al. Integrated analysis of multimodal single-cell data. Cell 2021, 184, 3573–3587.e29. [Google Scholar] [CrossRef] [PubMed]
- Aran, D.; Looney, A.P.; Liu, L.; Wu, E.; Fong, V.; Hsu, A.; Chak, S.; Naikawadi, R.P.; Wolters, P.J.; Abate, A.R.; et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat. Immunol. 2019, 20, 163–172. [Google Scholar] [CrossRef]
- Fu, R.; Gillen, A.E.; Sheridan, R.M.; Tian, C.; Daya, M.; Hao, Y.; Riemondy, K.A. clustifyr: An R package for automated single-cell RNA sequencing cluster classification. F1000Research 2020, 9, 223. [Google Scholar] [CrossRef]
- Chen, J.; Wang, R.; Wang, M.; Wei, G.W. Mutations Strengthened SARS-CoV-2 Infectivity. J. Mol. Biol. 2020, 432, 5212–5226. [Google Scholar] [CrossRef]
- Bate, N.; Savva, C.G.; Moody, P.C.; Brown, E.A.; Evans, S.E.; Ball, J.K.; Brindle, N.P. In vitro evolution predicts emerging SARS-CoV-2 mutations with high affinity for ACE2 and cross-species binding. PLoS Pathog. 2022, 18, e1010733. [Google Scholar] [CrossRef]
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-time tracking of pathogen evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef] [PubMed]
- Markov, P.V.; Ghafari, M.; Beer, M.; Lythgoe, K.; Simmonds, P.; Stilianakis, N.I.; Katzourakis, A. The evolution of SARS-CoV-2. Nat. Rev. Microbiol. 2023, 21, 361–379. [Google Scholar] [CrossRef]
- Hufsky, F.; Lamkiewicz, K.; Almeida, A.; Aouacheria, A.; Arighi, C.; Bateman, A.; Marz, M. Computational strategies to combat COVID-19: Useful tools to accelerate SARS-CoV-2 and coronavirus research. Brief. Bioinform. 2021, 22, 642–663. [Google Scholar] [CrossRef] [PubMed]
- Corman, V.M.; Landt, O.; Kaiser, M.; Molenkamp, R.; Meijer, A.; Chu, D.K.W.; Bleicker, T.; Brünink, S.; Schneider, J.; Schmidt, M.L.; et al. Detection of 2019 novel coronavirus (2019-nCoV) by real-time RT-PCR. Eurosurveillance 2020, 25, 2000045. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Monaghan, M.T.; Reinert, K. PriSeT: Efficient de novo primer discovery. In Proceedings of the 12th ACM Conference on Bioinformatics, Computational Biology, and Health Informatics, Gaineswille, FL, USA, 1–4 August 2021; pp. 1–12. [Google Scholar]
- Sumon, T.A.; Hussain, A.; Hasan, T.; Hasan, M.; Jang, W.J.; Bhuiya, E.H.; Chowdhury, A.A.M.; Sharifuzzaman, S.M.; Brown, C.L.; Kwon, H.-J.; et al. A Revisit to the Research Updates of Drugs, Vaccines, and Bioinformatics Approaches in Combating COVID-19 Pandemic. Front. Mol. Biosci. 2021, 7, 585899. [Google Scholar] [CrossRef]
- Kumar, V.M.; Pandi-Perumal, S.R.; Trakht, I.; Thyagarajan, S.P. Strategy for COVID-19 vaccination in India: The country with the second highest population and number of cases. NPJ Vaccines 2021, 6, 60. [Google Scholar] [CrossRef] [PubMed]
- Posada-Céspedes, S.; Seifert, D.; Topolsky, I.; Jablonski, K.P.; Metzner, K.J.; Beerenwinkel, N. V-pipe: A computational pipeline for assessing viral genetic diversity from high-throughput data. Bioinformatics 2021, 37, 1673–1680. [Google Scholar] [CrossRef]
- Fritz, A.; Bremges, A.; Deng, Z.L.; Lesker, T.R.; Götting, J.; Ganzenmueller, T.; McHardy, A.C. Haploflow: Strain-resolved de novo assembly of viral genomes. Genome Biol. 2021, 22, 212. [Google Scholar] [CrossRef]
- Gohl, D.M.; Garbe, J.; Grady, P.; Daniel, J.; Watson, R.H.; Auch, B.; Beckman, K.B. A rapid, cost-effective tailed amplicon method for sequencing SARS-CoV-2. BMC Genom. 2020, 21, 863. [Google Scholar] [CrossRef]
- Rosenthal, S.H.; Gerasimova, A.; Ruiz-Vega, R.; Livingston, K.; Kagan, R.M.; Liu, Y.; Lacbawan, F. Development and validation of a high throughput SARS-CoV-2 whole genome sequencing workflow in a clinical laboratory. Sci. Rep. 2022, 12, 2054. [Google Scholar] [CrossRef]
- Schäffer, A.A.; Hatcher, E.L.; Yankie, L.; Shonkwiler, L.; Brister, J.R.; Mizrachi, I.K.; Nawrocki, E.P. VADR: Validation and annotation of virus sequence submissions to GenBank. BMC Bioinform. 2020, 21, 211. [Google Scholar] [CrossRef]
- Libin, P.J.K.; Deforche, K.; Abecasis, A.B.; Theys, K. VIRULIGN: Fast codon-correct alignment and annotation of viral genomes. Bioinformatics 2018, 35, 1763–1765. [Google Scholar] [CrossRef]
- Kalvari, I.; Argasinska, J.; Quinones-Olvera, N.; Nawrocki, E.P.; Rivas, E.; Eddy, S.R.; Bateman, A.; Finn, R.D.; I Petrov, A. Rfam 13.0: Shifting to a genome-centric resource for non-coding RNA families. Nucleic Acids Res. 2018, 46, D335–D342. [Google Scholar] [CrossRef] [PubMed]
- Bouckaert, R.; Heled, J.; Kühnert, D.; Vaughan, T.; Wu, C.-H.; Xie, D.; Suchard, M.A.; Rambaut, A.; Drummond, A.J. BEAST 2: A Software Platform for Bayesian Evolutionary Analysis. PLoS Comput. Biol. 2014, 10, e1003537. [Google Scholar] [CrossRef]
- Hoops, S.; Sahle, S.; Gauges, R.; Lee, C.; Pahle, J.; Simus, N.; Kummer, U. COPASI—A COmplex PAthway SImulator. Bioinformatics 2006, 22, 3067–3074. [Google Scholar] [CrossRef] [PubMed]
- Adam, D. Special report: The simulations driving the world’s response to COVID-19. Nature 2020, 580, 316–318. [Google Scholar] [CrossRef] [PubMed]
- Singer, J.; Gifford, R.; Cotten, M.; Robertson, D. CoV-GLUE: A Web Application for Tracking SARS-CoV-2 Genomic Variation. Preprints.org 2020, 2020060225. [Google Scholar] [CrossRef]
- Sathyaseelan, C.; Magateshvaren Saras, M.A.; Prasad Patro, L.P.; Uttamrao, P.P.; Rathinavelan, T. CoVe-Tracker: An Interactive SARS-CoV-2 Pan Proteome Evolution Tracker. J. Proteome Res. 2023, 22, 1984–1996. [Google Scholar] [CrossRef]
- Cacciabue, M.; Aguilera, P.; Gismondi, M.I.; Taboga, O. Covidex: An ultrafast and accurate tool for SARS-CoV-2 subtyping. Infect Genet. Evol. 2022, 99, 105261. [Google Scholar] [CrossRef]
- O’Toole, Á.; Scher, E.; Underwood, A.; Jackson, B.; Hill, V.; McCrone, J.T.; Colquhoun, R.; Ruis, C.; Abu-Dahab, K.; Taylor, B.; et al. Assignment of Epidemiological Lineages in an Emerging Pandemic Using the Pangolin Tool. Virus Evol. 2021, 7, veab064. [Google Scholar] [CrossRef]
- Flower, D.R. Bioinformatics for Vaccinology; John Wiley & Sons: Hoboken, NJ, USA, 2008; ISBN 9780470027110. [Google Scholar]
- WHO. COVID-19 Vaccines with WHO Emergency Use Listing. Available online: https://extranet.who.int/pqweb/vaccines/vaccinescovid-19-vaccine-eul-issued (accessed on 15 June 2023).
- Wang, D.; Mai, J.; Zhou, W.; Yu, W.; Zhan, Y.; Wang, N.; Yang, Y. Immunoinformatic Analysis of T- and B-Cell Epitopes for SARS-CoV-2 Vaccine Design. Vaccines 2020, 8, 355. [Google Scholar] [CrossRef] [PubMed]
- Ullah, A.; Sarkar, B.; Islam, S.S. Exploiting the reverse vaccinology approach to design novel subunit vaccines against Ebola virus. Immunobiology 2020, 225, 151949. [Google Scholar] [CrossRef] [PubMed]
- Doytchinova, I.A.; Flower, D.R. VaxiJen: A server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinform. 2007, 8, 4. [Google Scholar] [CrossRef]
- Bukhari SN, H.; Jain, A.; Haq, E.; Mehbodniya, A.; Webber, J. Machine Learning Techniques for the Prediction of B-Cell and T-Cell Epitopes as Potential Vaccine Targets with a Specific Focus on SARS-CoV-2 Pathogen: A Review. Pathogens 2022, 11, 146. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Zhao, L.; Wei, F.; Li, J. DeepNetBim: Deep learning model for predicting HLA-epitope interactions based on network analysis by harnessing binding and immunogenicity information. BMC Bioinform. 2021, 22, 231. [Google Scholar] [CrossRef]
- Kames, J.; Holcomb, D.D.; Kimchi, O.; DiCuccio, M.; Hamasaki-Katagiri, N.; Wang, T.; Kimchi-Sarfaty, C. Sequence analysis of SARS-CoV-2 genome reveals features important for vaccine design. Sci. Rep. 2020, 10, 15643. [Google Scholar] [CrossRef]
- Taherzadeh, G.; Yang, Y.; Zhang, T.; Liew, A.W.; Zhou, Y. Sequence-based prediction of protein–peptide binding sites using support vector machine. J. Comput. Chem. 2016, 37, 1223–1229. [Google Scholar] [CrossRef]
- Müller, A.T.; Gabernet, G.; Hiss, J.A.; Schneider, G. modlAMP: Python for antimicrobial peptides. Bioinformatics 2017, 33, 2753–2755. [Google Scholar] [CrossRef]
- De Vries, S.J.; Rey, J.; Schindler, C.E.M.; Zacharias, M.; Tuffery, P. The pepATTRACT web server for blind, large-scale peptide–protein docking. Nucleic Acids Res. 2017, 45, W361–W364. [Google Scholar] [CrossRef]
- Lamiable, A.; Thévenet, P.; Rey, J.; Vavrusa, M.; Derreumaux, P.; Tufféry, P. PEP-FOLD3: Faster de novo structure prediction for linear peptides in solution and in complex. Nucleic Acids Res. 2016, 44, W449–W454. [Google Scholar] [CrossRef]
- Vita, R.; Mahajan, S.; Overton, J.A.; Dhanda, S.K.; Martini, S.; Cantrell, J.R.; Wheeler, D.K.; Sette, A.; Peters, B. The Immune Epitope Database (IEDB): 2018 update. Nucleic Acids Res. 2019, 47, D339–D343. [Google Scholar] [CrossRef] [PubMed]
- Guan, P.; Hattotuwagama, C.K.; Doytchinova, I.A.; Flower, D.R. MHCPred 2.0: An updated quantitative T-cell epitope prediction server. Appl. Bioinform. 2006, 5, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Yao, B.; Zhang, L.; Liang, S.; Zhang, C. SVMTriP: A Method to Predict Antigenic Epitopes Using Support Vector Machine to Integrate Tri-Peptide Similarity and Propensity. PLoS ONE 2012, 7, e45152. [Google Scholar] [CrossRef]
- Dimitrov, I.; Bangov, I.; Flower, D.R.; Doytchinova, I. AllerTOP v.2—A server for in silico prediction of allergens. J. Mol. Model. 2014, 20, 2278. [Google Scholar] [CrossRef]
- Harper, L.; Kalfa, N.; Beckers, G.; Kaefer, M.; Nieuwhof-Leppink, A.; Fossum, M.; Herbst, K.; Bagli, D. The impact of COVID-19 on research. J. Pediatr. Urol. 2020, 16, 715–716. [Google Scholar] [CrossRef]
- Venkatesh, V. Impacts of COVID-19: A research agenda to support people in their fight. Int. J. Inf. Manag. 2020, 55, 102197. [Google Scholar] [CrossRef] [PubMed]
- WHO. Statement on the Fifteenth Meeting of the IHR (2005) Emergency Committee on the COVID-19 Pandemic. Available online: https://www.who.int/news/item/05-05-2023-statement-on-the-fifteenth-meeting-of-the-international-health-regulations-(2005)-emergency-committee-regarding-the-coronavirus-disease-(covid-19)-pandemic (accessed on 6 July 2023).




{kind=link}
{kind=link}
{kind=link}
| Areas Supporting Candidate Vaccine Development | Description | Examples |
|---|---|---|
| Genome Sequencing | Rapid processing and analysis of sequenced SARS-CoV-2 genomes aid in the rapid identification of mutations and thus aid in developing potential therapeutic targets and surveillance. | Trimmomatic, BWA, SAMTools, Seurat, GATK, SPAdes, Pangolin, IGV, Deseq2/EdgeR |
| Molecular Modeling | Various computational tools for molecular docking and molecular dynamics simulations facilitate the design and optimize therapeutic candidates. | PyMOL, Chimera, GROMACS, CHARMM, AMBER |
| Epitope Prediction | Numerous bioinformatics tools are being used to predict potential epitopes (antigenic determinants) that could be used to induce an immune response in the host and accelerate research/development. | NetMHC, IEDB, BepiPred, DiscoTope, Ellipro, ABCpred |
| Data Mining | Large-scale data mining and synthesizing of large-scale databases and information have speeded up the process of understanding virulence factors of new mutations, predicting their behavior, and creating appropriate drugs/vaccines to mitigate severe illness and curb spread, and inform public health policy. | Weka, Orange, KNIME, Cytoscape, TANGARA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shukla, N.; Srivastava, N.; Gupta, R.; Srivastava, P.; Narayan, J. COVID Variants, Villain and Victory: A Bioinformatics Perspective. Microorganisms 2023, 11, 2039. https://doi.org/10.3390/microorganisms11082039
Shukla N, Srivastava N, Gupta R, Srivastava P, Narayan J. COVID Variants, Villain and Victory: A Bioinformatics Perspective. Microorganisms. 2023; 11(8):2039. https://doi.org/10.3390/microorganisms11082039
Chicago/Turabian StyleShukla, Nityendra, Neha Srivastava, Rohit Gupta, Prachi Srivastava, and Jitendra Narayan. 2023. "COVID Variants, Villain and Victory: A Bioinformatics Perspective" Microorganisms 11, no. 8: 2039. https://doi.org/10.3390/microorganisms11082039
APA StyleShukla, N., Srivastava, N., Gupta, R., Srivastava, P., & Narayan, J. (2023). COVID Variants, Villain and Victory: A Bioinformatics Perspective. Microorganisms, 11(8), 2039. https://doi.org/10.3390/microorganisms11082039