
Comparative Genomic Analysis of Multi-Drug Resistant Pseudomonas aeruginosa Sequence Type 235 Isolated from Sudan

 , , , ,
, , , ,  ,
,  ,
,  ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. Bacterial Identification
2.2. DNA Extraction
2.3. PCR
2.4. 16S rRNA Gene Sequencing
2.5. Antimicrobial Susceptibility Testing (AST)
2.6. Sequences Cleaning and Alignment
2.7. Whole-Genome Sequencing (WGS)
2.8. Genome Assembly, Annotation, and Typing
2.9. Comparative Genomic
2.10. Antibiotic Resistance Genes Detection
2.11. Analysis of Novel Mutations in the Antibiotic-Resistant Genes
2.12. Statistical Analysis
3. Results
3.1. Characteristics of the Collected Bacterial Isolates
3.2. Amplified 16S rRNA
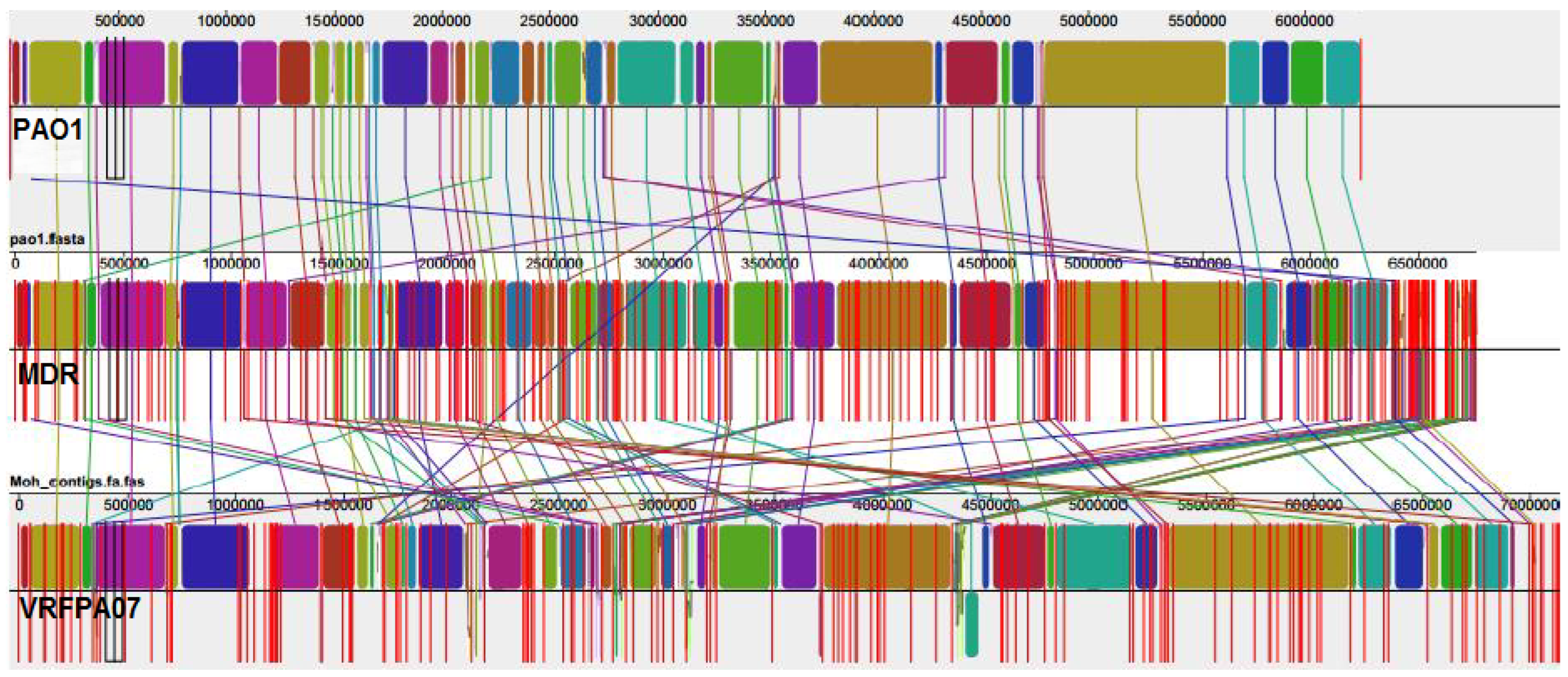
3.3. Comparative Genomic Results
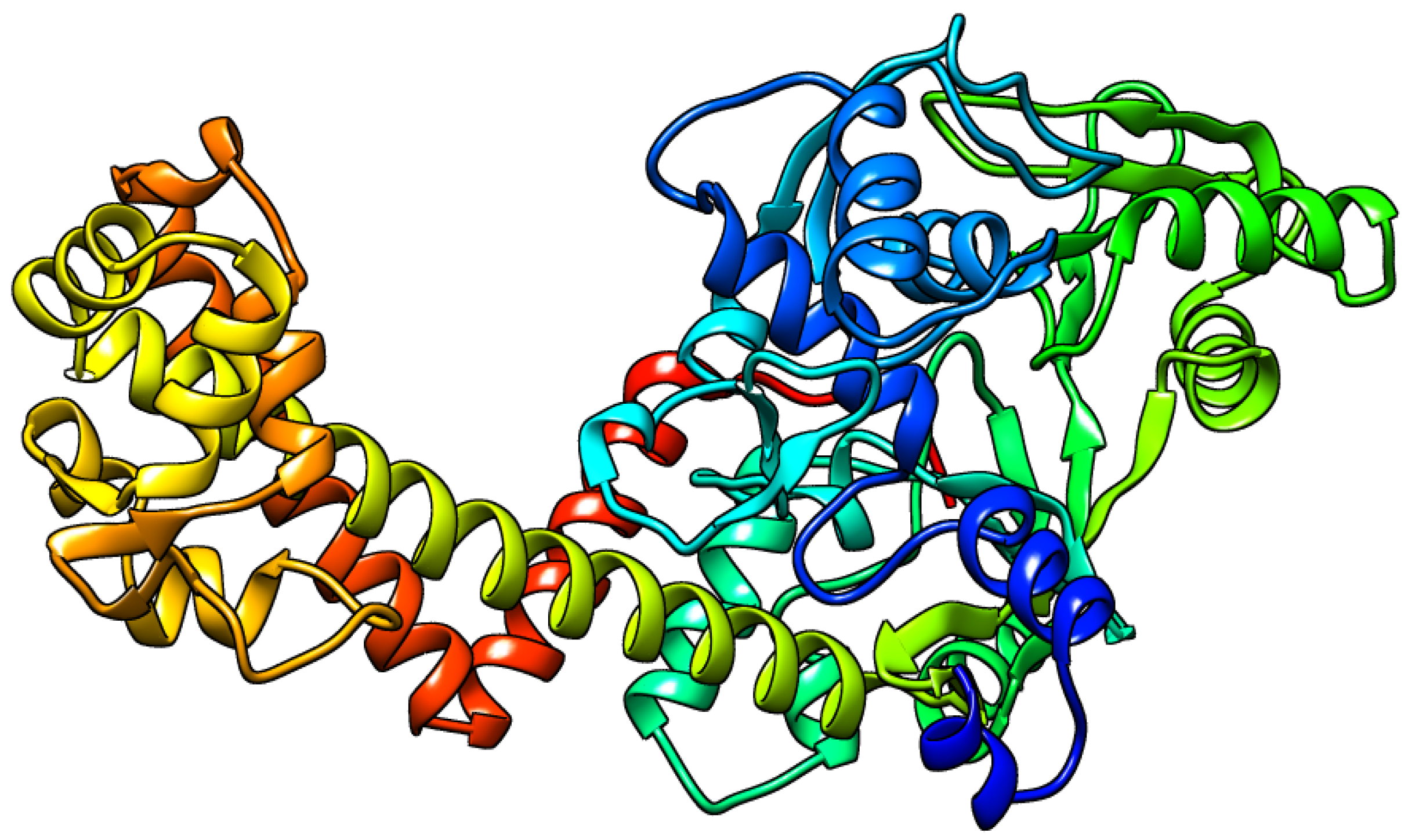
3.4. Detected Variants across Genomes
4. Discussions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Moradali, M.F.; Ghods, S.; Rehm, B.H.A. Pseudomonas aeruginosa Lifestyle: A Paradigm for Adaptation, Survival, and Persistence. Front. Cell. Infect. Microbiol. 2017, 7, 39. [Google Scholar] [CrossRef][Green Version]
- Nascimento, A.P.B.D.; Filho, F.M.; Pauer, H.; Antunes, L.C.M.; Sousa, H.; Senger, H.; Albano, R.M.; dos Santos, M.T.; Carvalho-Assef, A.P.D.; da Silva, F.A.B. Characterization of a SPM-1 metallo-beta-lactamase-producing Pseudomonas aeruginosa by comparative genomics and phenotypic analysis. Sci. Rep. 2020, 10, 13192. [Google Scholar] [CrossRef] [PubMed]
- Sekiguchi, J.; Asagi, T.; Miyoshi-Akiyama, T.; Kasai, A.; Mizuguchi, Y.; Araake, M.; Fujino, T.; Kikuchi, H.; Sasaki, S.; Watari, H.; et al. Outbreaks of multidrug-resistant Pseudomonas aeruginosa in community hospitals in Japan. J. Clin. Microbiol. 2007, 45, 979–989. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Paterson, D.L. The Epidemiological Profile of Infections with Multidrug-Resistant Pseudomonas aeruginosa and Acinetobacter Species. Clin. Infect. Dis. 2006, 43, S43–S48. [Google Scholar] [CrossRef][Green Version]
- Stover, C.K.; Pham, X.Q.; Erwin, A.L.; Mizoguchi, S.D.; Warrener, P.; Hickey, M.J.; Brinkman, F.S.L.; Hufnagle, W.O.; Kowalik, D.J.; Lagrou, M.; et al. Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature 2000, 406, 959–964. [Google Scholar] [CrossRef][Green Version]
- CDC. Antibiotic Resistance Threats in the United States, 2013; US Department of Health and Human Services Atlanta: Atlanta, GA, USA, 2013.
- Hussain, M.; Suliman, M.; Ahmed, A.; Altayb, H.; Elneima, E. Draft Genome Sequence of a Multidrug-Resistant Pseudomonas aeruginosa Strain Isolated from a Patient with a Urinary Tract Infection in Khartoum, Sudan. Genome Announc. 2017, 5, e00203-17. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Luzzaro, F.; Mantengoli, E.; Perilli, M.; Lombardi, G.; Orlandi, V.; Orsatti, A.; Amicosante, G.; Maria Rossolini, G.; Toniolo, A. Dynamics of a nosocomial outbreak of multi-drug-resistant Pseudomonas aeruginosa producing the PER-1 extended-spectrum beta-lactamase. J. Clin. Microbiol. 2001, 39, 1865–1870. [Google Scholar] [CrossRef][Green Version]
- Bert, F.; Branger, C.; Lambert-Zechovsky, N. Identification of PSE and OXA β-lactamase genes in Pseudomonas aeruginosa using PCR–restriction fragment length polymorphism. J. Antimicrob. Chemother. 2002, 50, 11–18. [Google Scholar] [CrossRef]
- Tada, T.; Miyoshi-Akiyama, T.; Shimada, K.; Shimojima, M.; Kirikae, T. IMP-43 and IMP-44 metallo-beta-lactamases with increased carbapenemase activities in multidrug-resistant Pseudomonas aeruginosa. Antimicrob Agents Chemother 2013, 57, 4427–4432. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Philippon, L.N.; Naas, T.; Bouthors, A.T.; Barakett, V.; Nordmann, P. OXA-18, a class D clavulanic acid-inhibited extended-spectrum beta-lactamase from Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 1997, 41, 2188–2195. [Google Scholar] [CrossRef][Green Version]
- Pagani, L.; Mantengoli, E.; Migliavacca, R.; Nucleo, E.; Pollini, S.; Spalla, M.; Daturi, R.; Romero, E.; Rossolini, G.M. Multifocal detection of multidrug-resistant Pseudomonas aeruginosa producing the PER-1 extended-spectrum β-lactamase in northern Italy. J. Clin. Microbiol. 2004, 42, 2523–2529. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Poole, K. Aminoglycoside Resistance in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2005, 49, 479–487. [Google Scholar] [CrossRef][Green Version]
- Shahid, M.; Malik, A. Resistance due to aminoglycoside modifying enzymes in Pseudomonas aeruginosa isolates from burns patients. Indian J. Med. Res. 2005, 122, 324–329. [Google Scholar] [PubMed]
- Yokoyama, K.; Doi, Y.; Yamane, K.; Kurokawa, H.; Shibata, N.; Shibayama, K.; Yagi, T.; Kato, H.; Arakawa, Y. Acquisition of 16S rRNA methylase gene in Pseudomonas aeruginosa. Lancet 2003, 362, 1888–1893. [Google Scholar] [CrossRef]
- Alibert-Franco, S.; Pradines, B.; Mahamoud, A.; Davin-Regli, A.; Pages, J.M. Efflux mechanism, an attractive target to combat mul-tidrug resistant Plasmodium falciparum and Pseudomonas aeruginosa. Curr. Med. Chem. 2009, 16, 301–317. [Google Scholar] [CrossRef] [PubMed]
- Lister, P.D.; Wolter, D.J.; Hanson, N.D. Antibacterial-Resistant Pseudomonas aeruginosa: Clinical Impact and Complex Regulation of Chromosomally Encoded Resistance Mechanisms. Clin. Microbiol. Rev. 2009, 22, 582–610. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhang, L.; Li, X.-Z.; Poole, K. Fluoroquinolone susceptibilities of efflux-mediated multidrug-resistant Pseudomonas aeruginosa, Stenotrophomonas maltophilia and Burkholderia cepacia. J. Antimicrob. Chemother. 2001, 48, 549–552. [Google Scholar] [CrossRef]
- Tegos, G.P.; Haynes, M.; Jacob Strouse, J.; Khan, M.M.T.; Bologa, C.G.; Oprea, T.I.; Larry, A.S. Microbial efflux pump inhibition: Tactics and strategies. Curr. Pharm. Des. 2011, 17, 1291–1302. [Google Scholar] [CrossRef][Green Version]
- Lambert, P.A. Bacterial resistance to antibiotics: Modified target sites. Adv. Drug Deliv. Rev. 2005, 57, 1471–1485. [Google Scholar] [CrossRef]
- Sun, S.; Selmer, M.; Andersson, D.I. Resistance to β-lactam antibiotics conferred by point mutations in penicillin-binding proteins PBP3, PBP4 and PBP6 in Salmonella enterica. PLoS ONE 2014, 9, e97202. [Google Scholar] [CrossRef][Green Version]
- Rigouts, L.; Coeck, N.; Gumusboga, M.; De Rijk, W.B.; Aung, K.J.M.; Hossain, M.A.; Fissette, K.; Rieder, H.L.; Meehan, C.J.; De Jong, B.C.; et al. Specific gyrA gene mutations predict poor treatment outcome in MDR-TB. J. Antimicrob. Chemother. 2016, 71, 314–323. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Breidenstein, E.B.M.; Khaira, B.K.; Wiegand, I.; Overhage, J.; Hancock, R.E.W. Complex Ciprofloxacin Resistome Revealed by Screening a Pseudomonas aeruginosa Mutant Library for Altered Susceptibility. Antimicrob. Agents Chemother. 2008, 52, 4486–4491. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sierra, J.M.; Martinez-Martinez, L.; Vázquez, F.; Giralt, E.; Vila, J. Relationship between Mutations in the gyrA Gene and Quinolone Resistance in Clinical Isolates of Corynebacterium striatum and Corynebacterium amycolatum. Antimicrob. Agents Chemother. 2005, 49, 1714–1719. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Glocker, E.; Kist, M. Rapid detection of point mutations in the gyrA gene of Helicobacter pylori conferring resistance to ciprof-loxacin by a fluorescence resonance energy transfer-based real-time PCR approach. J. Clin. Microbiol. 2004, 42, 2241–2246. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bébéar, C.M.; Grau, O.; Charron, A.; Renaudin, H.; Gruson, D. Cloning and Nucleotide Sequence of the DNA Gyrase (gyrA) Gene from Mycoplasma hominis and Characterization of Quinolone-Resistant Mutants Selected In Vitro with Trovafloxacin. Antimicrob. Agents Chemother. 2000, 44, 2719–2727. [Google Scholar] [CrossRef][Green Version]
- Srinivasan, R.; Karaoz, U.; Volegova, M.; MacKichan, J.; Kato-Maeda, M.; Miller, S.; Nadarajan, R.; Brodie, E.L.; Lynch, S.V. Use of 16S rRNA Gene for Identification of a Broad Range of Clinically Relevant Bacterial Pathogens. PLoS ONE 2015, 10, e0117617. [Google Scholar] [CrossRef]
- Jorgensen, J.H.; Turnidge, J.D. Susceptibility test methods: Dilution and disk diffusion methods. Manual of Clinical Microbiology. Am. Soc. Microbiol. 2015, 1253–1273. [Google Scholar] [CrossRef]
- Magiorakos, A.-P.; Srinivasan, A.; Carey, R.B.; Carmeli, Y.; Falagas, M.E.; Giske, C.G.; Harbarth, S.; Hindler, J.F.; Kahlmeter, G.; Olsson-Liljequist, B.; et al. Multidrug-resistant, extensively drug-resistant and pandrug-resistant bacteria: An international expert proposal for interim standard definitions for acquired resistance. Clin. Microbiol. Infect. 2012, 18, 268–281. [Google Scholar] [CrossRef][Green Version]
- Krumperman, P.H. Multiple antibiotic resistance indexing of Escherichia coli to identify high-risk sources of fecal contamination of foods. Appl. Environ. Microbiol. 1983, 46, 165–170. [Google Scholar] [CrossRef][Green Version]
- Geospiza. FinchTV 1.4.0; Geospiza, Inc. Seattle: Seattle, WA, USA, 2009. [Google Scholar]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Hall, T. BioEdit: An important software for molecular biology. GERF Bull Biosci 2011, 2, 60–61. [Google Scholar]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data; Babraham Bioinformatics, Babraham Institute: Cambridge, UK, 2010. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef][Green Version]
- Swindell, S.R.; Plasterer, T.N. SEQMAN: Contig assembly. Seq. Data Anal. Guideb. 1997, 70, 75–89. [Google Scholar] [CrossRef]
- Tatusova, T.; DiCuccio, M.; Badretdin, A.; Chetvernin, V.; Nawrocki, E.P.; Zaslavsky, L.; Lomsadze, A.; Pruitt, K.D.; Borodovsky, M.; Ostell, J. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 2016, 44, 6614–6624. [Google Scholar] [CrossRef]
- Larsen, M.V.; Cosentino, S.; Rasmussen, S.; Friis, C.; Hasman, H.; Marvig, R.L.; Jelsbak, L.; Sicheritz-Pontén, T.; Ussery, D.W.; Aarestrup, F.M.; et al. Multilocus sequence typing of total genome sequenced bacteria. J. Clin. Microbiol. 2012, 50, 1355–1361. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Murugan, N.; Malathi, J.; Umashankar, V.; Madhavan, H.N.R. Comparative Genomic Analysis of Multidrug-Resistant Pseudomonas aeruginosa Clinical Isolates VRFPA06 and VRFPA08 with VRFPA07. Genome Announc. 2014, 2, e00140-14. [Google Scholar] [CrossRef][Green Version]
- Darling, A.C.E.; Mau, B.; Blattner, F.R.; Perna, N.T. Mauve: Multiple Alignment of Conserved Genomic Sequence With Rearrangements. Genome Res. 2004, 14, 1394–1403. [Google Scholar] [CrossRef][Green Version]
- Darling, A.E.; Mau, B.; Perna, N.T. progressiveMauve: Multiple Genome Alignment with Gene Gain, Loss and Rearrangement. PLoS ONE 2010, 5, e11147. [Google Scholar] [CrossRef][Green Version]
- Yoon, S.H.; Park, Y.-K.; Kim, J.F. PAIDB v2.0: Exploration and analysis of pathogenicity and resistance islands. Nucleic Acids Res. 2015, 43, D624–D630. [Google Scholar] [CrossRef][Green Version]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST server: Rapid annotations using subsystems technology. BMC Genom. 2008, 9, 75. [Google Scholar] [CrossRef][Green Version]
- Carattoli, A.; Zankari, E.; Garcìa-Fernandez, A.; Larsen, M.; Lund, O.; Voldby Villa, L.; Møller Aarestrup, F.; Hasman, H. In Silico Detection and Typing of Plasmids. Antimicrob using PlasmidFinder and plasmid multilocus sequence typing. Agents Chemother. 2014, 58, 3895–3903. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Arndt, D.; Marcu, A.; Liang, Y.; Wishart, D.S. PHAST, PHASTER and PHASTEST: Tools for finding prophage in bacterial genomes. Briefings Bioinform. 2017, 20, 1560–1567. [Google Scholar] [CrossRef] [PubMed]
- Giardine, B.; Riemer, C.; Hardison, R.C.; Burhans, R.; Elnitski, L.; Shah, P.; Zhang, Y.; Blankenberg, D.; Albert, I.; Taylor, J.; et al. Galaxy: A platform for interactive large-scale genome analysis. Genome Res. 2005, 15, 1451–1455. [Google Scholar] [CrossRef][Green Version]
- Jia, B.; Raphenya, A.R.; Alcock, B.; Waglechner, N.; Guo, P.; Tsang, K.K.; Lago, B.A.; Dave, B.M.; Pereira, S.; Sharma, A.N.; et al. CARD 2017: Expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res. 2017, 45, D566–D573. [Google Scholar] [CrossRef]
- Capriotti, E.; Fariselli, P.; Casadio, R. I-Mutant2.0: Predicting stability changes upon mutation from the protein sequence or structure. Nucleic Acids Res. 2005, 33, W306–W310. [Google Scholar] [CrossRef][Green Version]
- Venselaar, H.; Beek, T.A.H.T.; Kuipers, R.K.P.; Hekkelman, M.L.; Vriend, G. Protein structure analysis of mutations causing inheritable diseases. An e-Science approach with life scientist friendly interfaces. BMC Bioinform. 2010, 11, 548. [Google Scholar] [CrossRef][Green Version]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J.E. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera? A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef][Green Version]
- Diggle, S.P.; Whiteley, M. Microbe Profile: Pseudomonas aeruginosa: Opportunistic pathogen and lab rat. Microbiology 2020, 166, 30–33. [Google Scholar] [CrossRef]
- Wellinghausen, N.; KÖthe, J.; Wirths, B.; Sigge, A.; Poppert, S.; Johnson, J.R.; Scheutz, F.; Ulleryd, P.; Kuskowski, M.A.; O’Bryan, T.T.; et al. Superiority of Molecular Techniques for Identification of Gram-Negative, Oxidase-Positive Rods, Including Morphologically Nontypical Pseudomonas aeruginosa, from Patients with Cystic Fibrosis. J. Clin. Microbiol. 2005, 43, 3895–3900. [Google Scholar] [CrossRef][Green Version]
- Trenholme, G.M.; Kaplan, R.L.; Karakusis, P.H.; Stine, T.; Fuhrer, J.; Landau, W.; Levin, S. Clinical impact of rapid identification and susceptibility testing of bacterial blood culture isolates. J. Clin. Microbiol. 1989, 27, 1342–1345. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Falagas, M.E.; Kasiakou, S.K. Toxicity of polymyxins: A systematic review of the evidence from old and recent studies. Crit. Care 2006, 10, R27. [Google Scholar] [CrossRef][Green Version]
- Ahmed, O.B.; Omar, A.O.; Asghar, A.H.; Elhassan, M.M.; Al-Munawwarah, A.-M.; Arabia, S. Prevalence of TEM, SHV and CTX-M genes in Escherichia coli and Klebsiella spp. Urinary Isolates from Sudan with confirmed ESBL phenotype. Life Sci. J. 2013, 10, 191–195. [Google Scholar]
- Araujo, B.F.; Ferreira, M.L.; Campos, P.A.; Royer, S.; Batistao, D.W.; Dantas, R.C.; Gonçalves, I.R.; Faria, A.L.S.; De Brito, C.S.; Yokosawa, J.; et al. Clinical and Molecular Epidemiology of Multi-drug-Resistant P. aeruginosa Carrying aac(6′)-Ib-cr, qnrS1 and blaSPM Genes in Brazil. PLoS ONE 2016, 11, e0155914. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tsukamoto, N.; Ohkoshi, Y.; Okubo, T.; Sato, T.; Kuwahara, O.; Fujii, N.; Tamura, Y.; Yokota, S.-I. High Prevalence of Cross-Resistance to Aminoglycosides in Fluoroquinolone-Resistant Escherichia coli Clinical Isolates. Chemotherapy 2013, 59, 379–384. [Google Scholar] [CrossRef]
- Yokota, S.-I.; Sato, T.; Okubo, T.; Ohkoshi, Y.; Okabayashi, T.; Kuwahara, O.; Tamura, Y.; Fujii, N. Prevalence of Fluoroquinolone-Resistant Escherichia coli O25:H4-ST131 (CTX-M-15-Nonproducing) Strains Isolated in Japan. Chemotherapy 2012, 58, 52–59. [Google Scholar] [CrossRef]
- Curran, B.; Jonas, D.; Grundmann, H.; Pitt, T.; Dowson, C.G. Development of a multilocus sequence typing scheme for the oppor-tunistic pathogen Pseudomonas aeruginosa. J. Clin. Microbiol. 2004, 42, 5644–5649. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bull, C.T.; Clarke, C.R.; Cai, R.; Vinatzer, B.A.; Jardini, T.M.; Koike, S.T. Multilocus sequence typing of Pseudomonas syringae sensu lato confirms previously described genomospecies and permits rapid identification of P. syringae pv. coriandricola and P. syringae pv. apii causing bacterial leaf spot on parsley. Phytopathology 2011, 101, 847–858. [Google Scholar] [CrossRef][Green Version]
- Silva, F.M.; Carmo, M.S.; Silbert, S.; Gales, A.C. SPM-1-producing Pseudomonas aeruginosa: Analysis of the ancestor relationship using multilocus sequence typing, pulsed-field gel electrophoresis, and automated ribotyping. Microb. Drug Resist. 2011, 17, 215–220. [Google Scholar] [CrossRef]
- Adams, M.D.; Goglin, K.; Molyneaux, N.; Hujer, K.M.; Lavender, H.; Jamison, J.J.; MacDonald, I.J.; Martin, K.M.; Russo, T.; Campagnari, A.A.; et al. Comparative Genome Sequence Analysis of Multidrug-Resistant Acinetobacter baumannii. J. Bacteriol. 2008, 190, 8053–8064. [Google Scholar] [CrossRef][Green Version]
- Wolfgang, M.C.; Kulasekara, B.R.; Liang, X.; Boyd, D.; Wu, K.; Yang, Q.; Garrett Miyada, C.; Lory, S. Conservation of genome content and virulence determinants among clinical and environmental isolates of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 2003, 100, 8484–8489. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Livermore, D.M.; Chen, H.Y. Quality of antimicrobial susceptibility testing in the UK: A Pseudomonas aeruginosa survey revisited. J. Antimicrob. Chemother. 1999, 43, 517–522. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Schindler, B.D.; Seo, S.M.; Jacinto, P.L.; Kumaraswami, M.; Birukou, I.; Brennan, R.G.; Kaatz, G.W. Functional consequences of substitution mutations in MepR, a repressor of the Staphylococcus aureus mepA multidrug efflux pump gene. J. Bacteriol. 2013, 195, 3651–3662. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hearn, E.M.; Gray, M.R.; Foght, J.M. Mutations in the central cavity and periplasmic domain affect efflux activity of the resistance-nodulation-division pump EmhB from Pseudomonas fluorescens cLP6a. J. Bacteriol. 2006, 188, 115–123. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Henrichfreise, B.; Wiegand, I.; Pfister, W.; Wiedemann, B. Resistance mechanisms of multiresistant Pseudomonas aeruginosa strains from Germany and correlation with hypermutation. Antimicrob. Agents Chemother. 2007, 51, 4062–4070. [Google Scholar] [CrossRef][Green Version]
- Van Nguyen, K.; Nguyen, T.V.; Nguyen, H.T.T.; Van Le, D.J.I.; Resistance, D. Mutations in the gyrA, parC, and mexR genes provide functional insights into the fluoroquinolone-resistant Pseudomonas aeruginosa isolated in Vietnam. Infect. Drug Resist. 2018, 11, 275–282. [Google Scholar] [CrossRef][Green Version]
- Poole, K.; Tetro, K.; Zhao, Q.; Neshat, S.; Heinrichs, D.E.; Bianco, N. Expression of the multidrug resistance operon mexA-mexB-oprM in Pseudomonas aeruginosa: mexR encodes a regulator of operon expression. Antimicrob. Agents Chemother. 1996, 40, 2021–2028. [Google Scholar] [CrossRef][Green Version]
- Middlemiss, J.K.; Poole, K. Differential impact of MexB mutations on substrate selectivity of the MexAB-OprM multidrug efflux pump of Pseudomonas aeruginosa. J. Bacteriol. 2004, 186, 1258–1269. [Google Scholar] [CrossRef][Green Version]
- Kim, J.H.; Cho, E.H.; Kim, K.S.; Kim, H.Y.; Kim, Y.M. Cloning and nucleotide sequence of the DNA gyrase gyrA gene from Serratia marcescens and characterization of mutations in gyrA of quinolone-resistant clinical isolates. Antimicrob. Agents Chemother. 1998, 42, 190–193. [Google Scholar] [CrossRef][Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibiotic | Sensitive % | Intermediate % | Resistant % |
|---|---|---|---|
| CIP 5 µg | (148) 74 | (10) 5.0 | (42) 21 |
| PB 300 IU | (189) 94.5 | (2.0) 1.0 | (9.0) 4.5 |
| PRL 100 µg | (44) 22 | (0.0) 0.00 | (156) 78 |
| MEM 10 µg | (184) 92 | (5.0) 2.5 | (11) 5.5 |
| CN 10 µg | (144) 72 | (6.0) 3.0 | (50) 25 |
| CAZ 30 µg | (159) 79.5 | (5.0) 2.5 | (36) 18 |
| Co-Resistance | Number/Percent |
|---|---|
| PRL-MEM | (11) 5.5% |
| PRL-CAZ | (35)17.5% |
| MEM-CAZ | (5.0) 2.5% |
| PRL-MEM-CAZ | (5.0) 2.5% |
| PRL-CIP | (40) 20% |
| PRL-CN | (47) 23.5% |
| PRL-PB | (8.0) 4.0% |
| CIP-CN | (37) 18.5% |
| Strain Features | MDR Isolate | Susceptible (VRFPA07) | Reference Genome (PAO1) |
|---|---|---|---|
| NCBI accession no. | MVDK00000000 | AZBO00000000 | NC_002516.2 |
| Genome coverage | 132 | 80 | --- |
| Genome size (bp) | 6,764,168 | 7,177,216 | 6,264,404 |
| Contigs (n) | 240 | 140 | --- |
| G + C content (%) | 64.6 | 65.90 | 66.6 |
| Genes (n) | 6557 | 6916 | 5697 |
| Pseudogenes (n) | 120 | 84 | 19 |
| Proteins (n) | 6373 | 6765 | 5572 |
| rRNAs (n) | 4 (5S,16S,23S) | 9 (5S, 16S, 23S) | 13 (5S, 16S, 23S) |
| tRNAs (n) | 56 | 57 | 63 |
| ncRNAs (n) | 4 | 1 | 30 |
| Gene Name | Number of Missense | Substituted Nucleotide | Substituted Amino Acid | Novelty/Effect |
|---|---|---|---|---|
| OXA-50 | 2 | A46G | Thr16Ala | Reported |
| A74G | Gln25Arg | Reported | ||
| mfd | 1 | G1171C | Ala391Pro | Reported |
| gyrA | 2 | A260G | asp87gly | Novel/deleterious |
| G83A | thr83Ile | Reported/deleterious | ||
| parC | 1 | C260T | Ser87Leu | Reported/deleterious |
| APH(3′)-IIb | 1 | C128A | Ala43Glu | Reported |
| Gene Name | Number of Missense | Substituted Nucleotide | Substituted Amino Acid |
|---|---|---|---|
| MexB | 2 | 2870GAG | Gly957Asp |
| 3120GTG | Ala1040Glu | ||
| MexC | 5 | 1147GAG | Pro383Ser |
| 988ACC | Ser330Ala | ||
| 929TCT | His310Arg | ||
| 227CTC | Arg76Gln | ||
| 92GAG | Ala31Val | ||
| MexD | 2 | 2944CTC | Ile982Val |
| 2533ACC | Ser845Ala | ||
| MexE | 3 | 5ATA | Glu2Val |
| 23CTC | Ser8Phe | ||
| 1103AGA | Gln368Arg | ||
| MexJ | 1 | 940CGG | Ala314Pro |
| MexH | 1 | 906CAA | Asp302Glu |
| MexI | 1 | 234CAA | Ala78Glu |
| MexM | 4 | 140TAA | Ile47Asn |
| 689TCC | Leu230Pro | ||
| 974ACC | Asp325Ala | ||
| 1139CTC | Ala380Val | ||
| MexN | 3 | 278CGC | Thr93Ser |
| 2351AGG | Ser784Phe | ||
| 3067AGG | Thr1023Ala | ||
| MexP | 2 | 1097CAA | Arg366Leu |
| 890GTT | Ala297Glu | ||
| MexQ | 4 | 1967CTT | Arg656Lys |
| 1514ACTT | Gly505Asp | ||
| 1150CTT | Val38Ile | ||
| 880CAA | Ile294Val | ||
| MexV | 3 | 673GTG | Ala225Ser |
| 686CGG | Ala229Gly | ||
| 962AGG | Gln321Arg | ||
| MexW | 2 | 779GTG | Arg260Gln |
| 1532AGG | Gln511Arg | ||
| OprJ | 2 | 800TCT | Gly267ARg |
| 205TCT | Met69Val | ||
| OpmD | 2 | 335GAA | Ser112Asn |
| 805GAG | Gly269Ser | ||
| OprN | 1 | 37TCC | Ser13Pro |
| OpmE | 3 | 1072CTC | Ala358Thr |
| 1060AGG | Trp354Arg | ||
| 523ATT | Ser175Thr | ||
| AmrA | 4 | 1072AGG | Trp358Arg |
| 991ACC | Leu331Val | ||
| 985TGG | Lys329Gln | ||
| 88CTC | Ala30Thr | ||
| AmrB | 1 | 1627TCC | Thr543Ala |
| TriA | 5 | 872ACC | Glu291Ala |
| 911TAT | Vla304Asp | ||
| 942CTC | Asp314Gln | ||
| C956TC | Arg319Val | ||
| 76GAA | Gly26Ser | ||
| TriC | 1 | 1019GAA | Arg340Gln |
| Position | WT | NEW | pH | Temp | SVMs Prediction Effect | DDG Value Prediction |
|---|---|---|---|---|---|---|
| 87 | D | G | 7.0 | 25 | Decrease | –0.75 Kcal/mol |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hussain, M.A.; Mohamed, M.S.; Altayb, H.N.; Mohamed, A.O.; Ashour, A.; Osman, W.; Sherif, A.E.; Ghazawi, K.F.; Miski, S.F.; Ibrahim, S.R.M.; et al. Comparative Genomic Analysis of Multi-Drug Resistant Pseudomonas aeruginosa Sequence Type 235 Isolated from Sudan. Microorganisms 2023, 11, 1432. https://doi.org/10.3390/microorganisms11061432
Hussain MA, Mohamed MS, Altayb HN, Mohamed AO, Ashour A, Osman W, Sherif AE, Ghazawi KF, Miski SF, Ibrahim SRM, et al. Comparative Genomic Analysis of Multi-Drug Resistant Pseudomonas aeruginosa Sequence Type 235 Isolated from Sudan. Microorganisms. 2023; 11(6):1432. https://doi.org/10.3390/microorganisms11061432
Chicago/Turabian StyleHussain, Mohamed A., Malik Suliman Mohamed, Hisham N. Altayb, Ahmed Osman Mohamed, Ahmed Ashour, Wadah Osman, Asmaa E. Sherif, Kholoud F. Ghazawi, Samar F. Miski, Sabrin R. M. Ibrahim, and et al. 2023. "Comparative Genomic Analysis of Multi-Drug Resistant Pseudomonas aeruginosa Sequence Type 235 Isolated from Sudan" Microorganisms 11, no. 6: 1432. https://doi.org/10.3390/microorganisms11061432
APA StyleHussain, M. A., Mohamed, M. S., Altayb, H. N., Mohamed, A. O., Ashour, A., Osman, W., Sherif, A. E., Ghazawi, K. F., Miski, S. F., Ibrahim, S. R. M., Mohamed, G. A., Sindi, I. A., Alshamrani, A. A., & Elgaml, A. (2023). Comparative Genomic Analysis of Multi-Drug Resistant Pseudomonas aeruginosa Sequence Type 235 Isolated from Sudan. Microorganisms, 11(6), 1432. https://doi.org/10.3390/microorganisms11061432