
Propidium Monoazide (PMAxx)-Recombinase Polymerase Amplification Exo (RPA Exo) Assay for Rapid Detection of Burkholderia cepacia Complex in Chlorhexidine Gluconate (CHX) and Benzalkonium Chloride (BZK) Solutions
Abstract
1. Introduction
2. Materials and Methods
2.1. Primer and Probe Design
2.2. 10 μM PMAxx-RPA Exo Assay
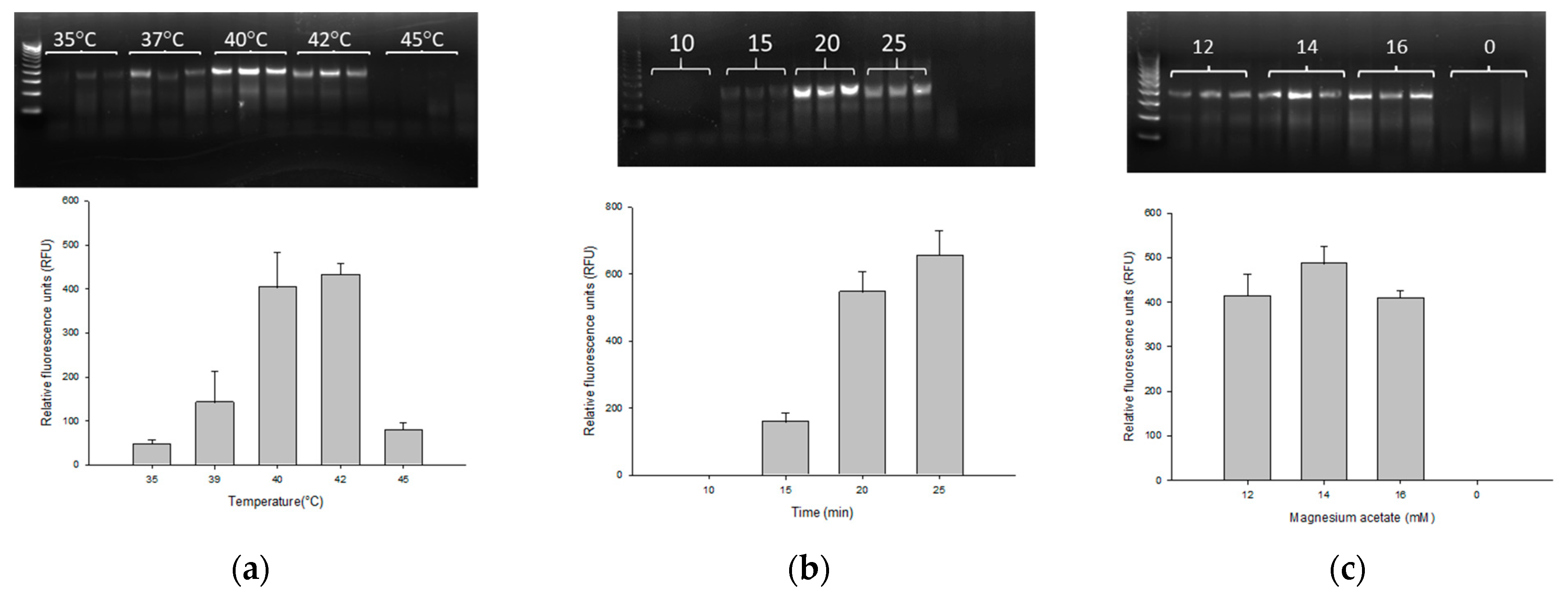
2.3. Optimization of Temperature, Reaction Time and Concentration of Magnesium Acetate for PMAxx-RPA Exo Assay
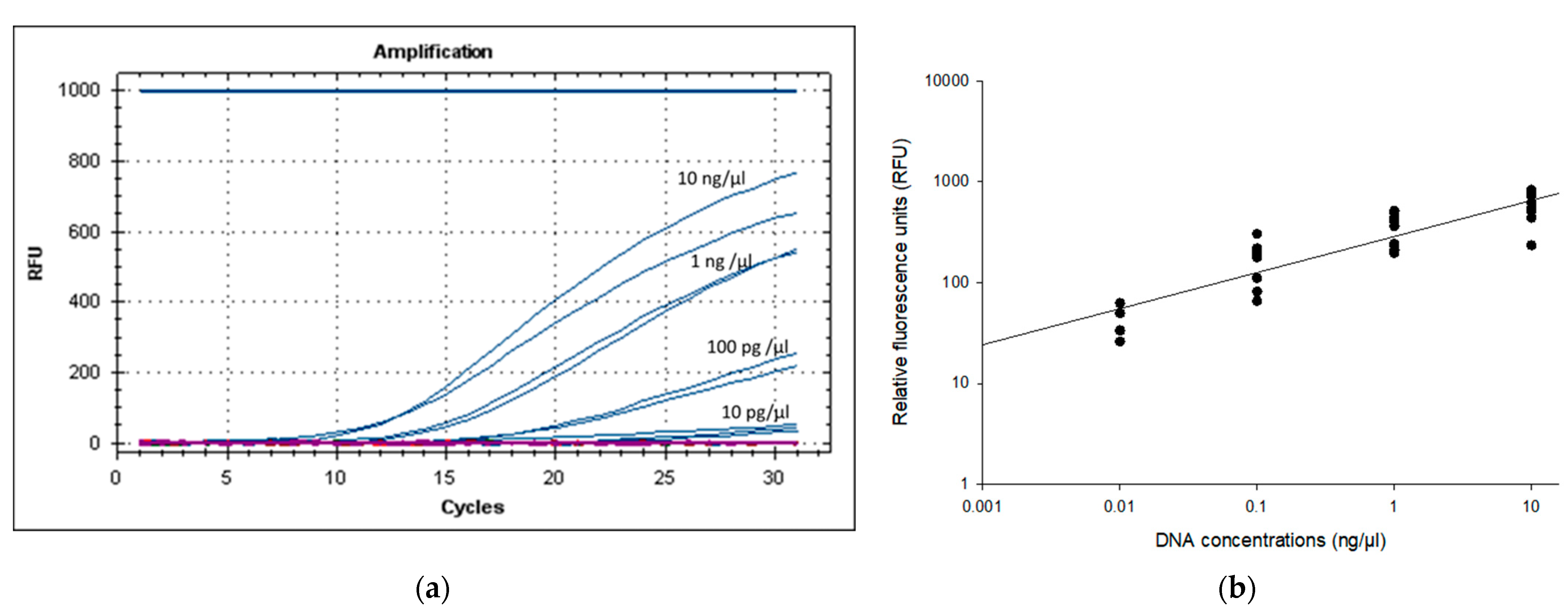
2.4. True-Negative Rate (Specificity) and Limit of Detection (LOD)
2.5. Effect of PMAxx-RPA Exo Assay in Presence of Antiseptics and Cell Lysates
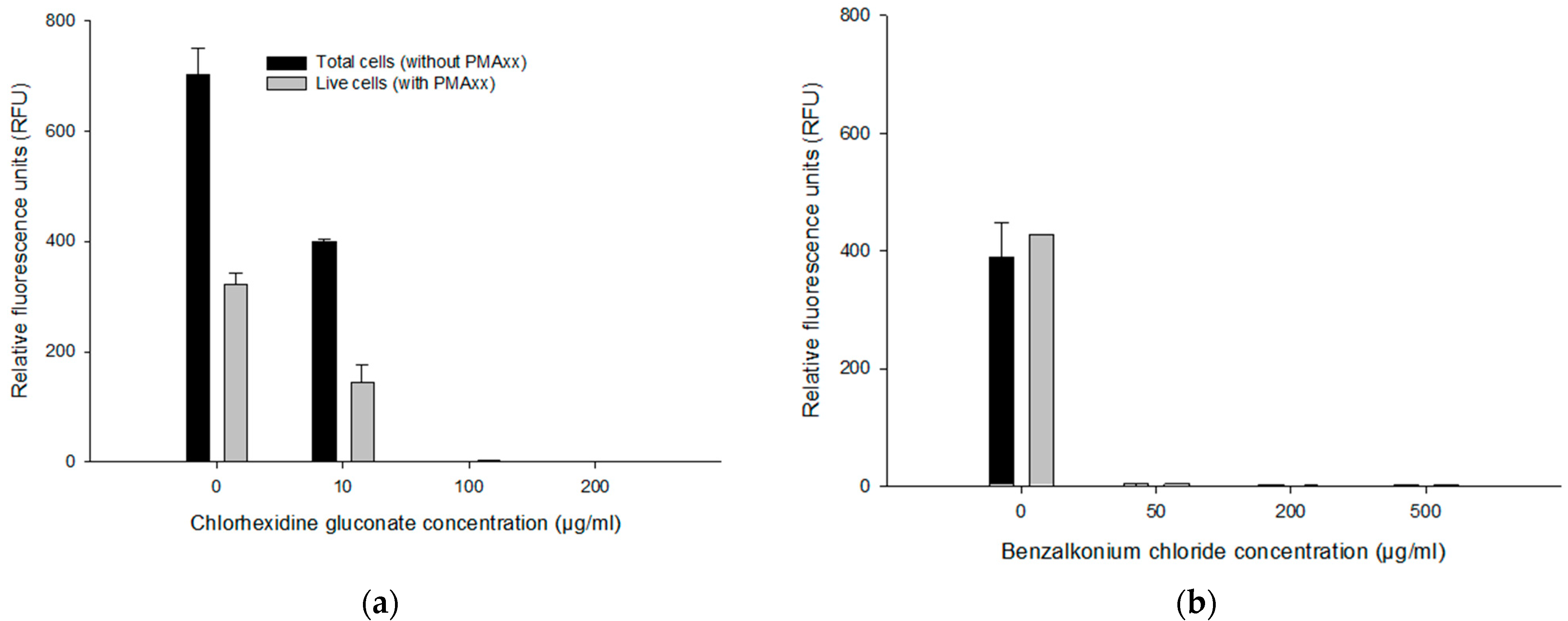
2.5.1. Evaluation of Various Concentrations of CHX and BZK
2.5.2. Comparing DNA Extraction Methods Using the Boiling Method and a Commercial Kit
2.6. Detection of Live/Dead B. multivorans HI2229 in CHX and BZK Solutions
3. Results
3.1. Optimization of the PMAxx-RPA Exo Assay
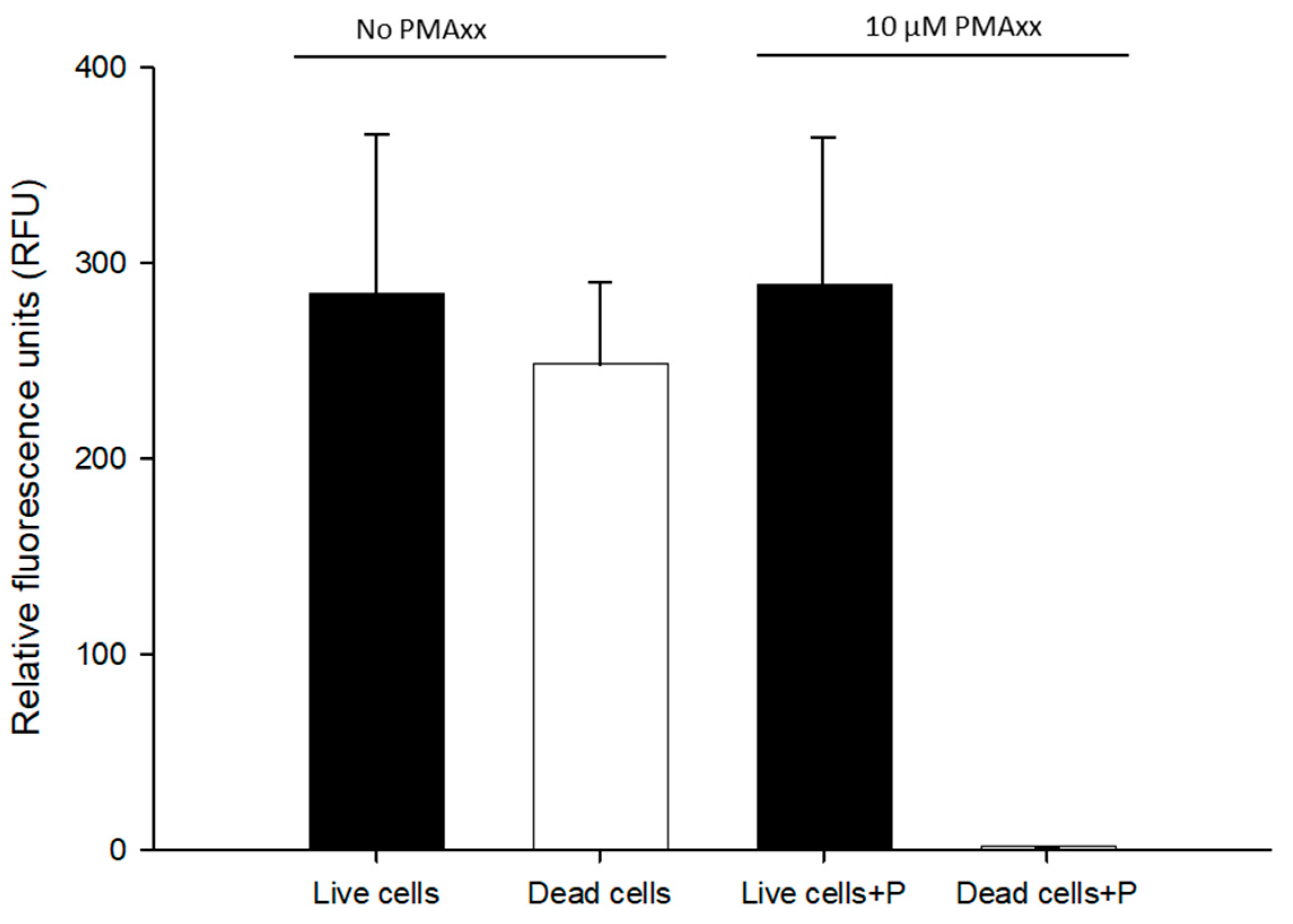
3.2. Evaluation of Live/Dead Cells with PMAxx Treatment
3.3. Specificity and LOD of PMAxx-RPA Exo Assay
3.4. Assessment of PMAxx-RPA Exo Conditions
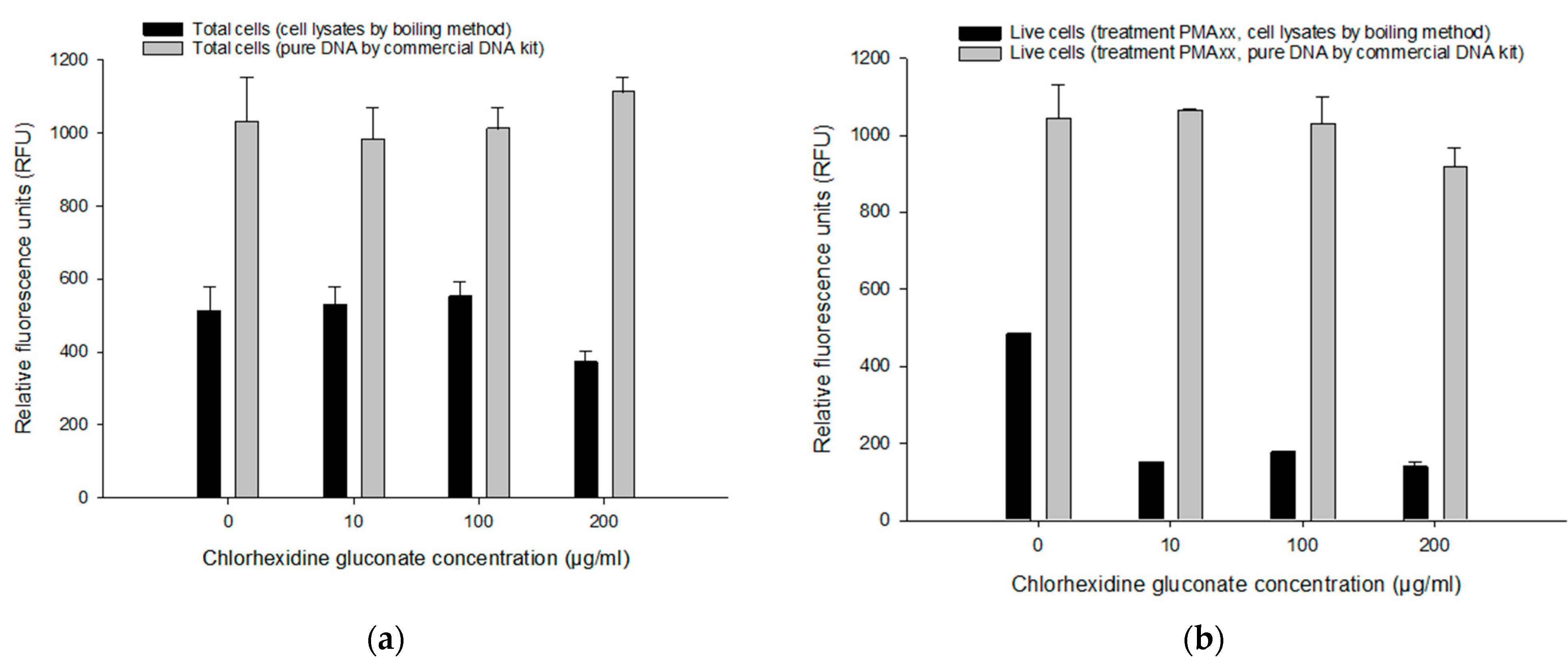
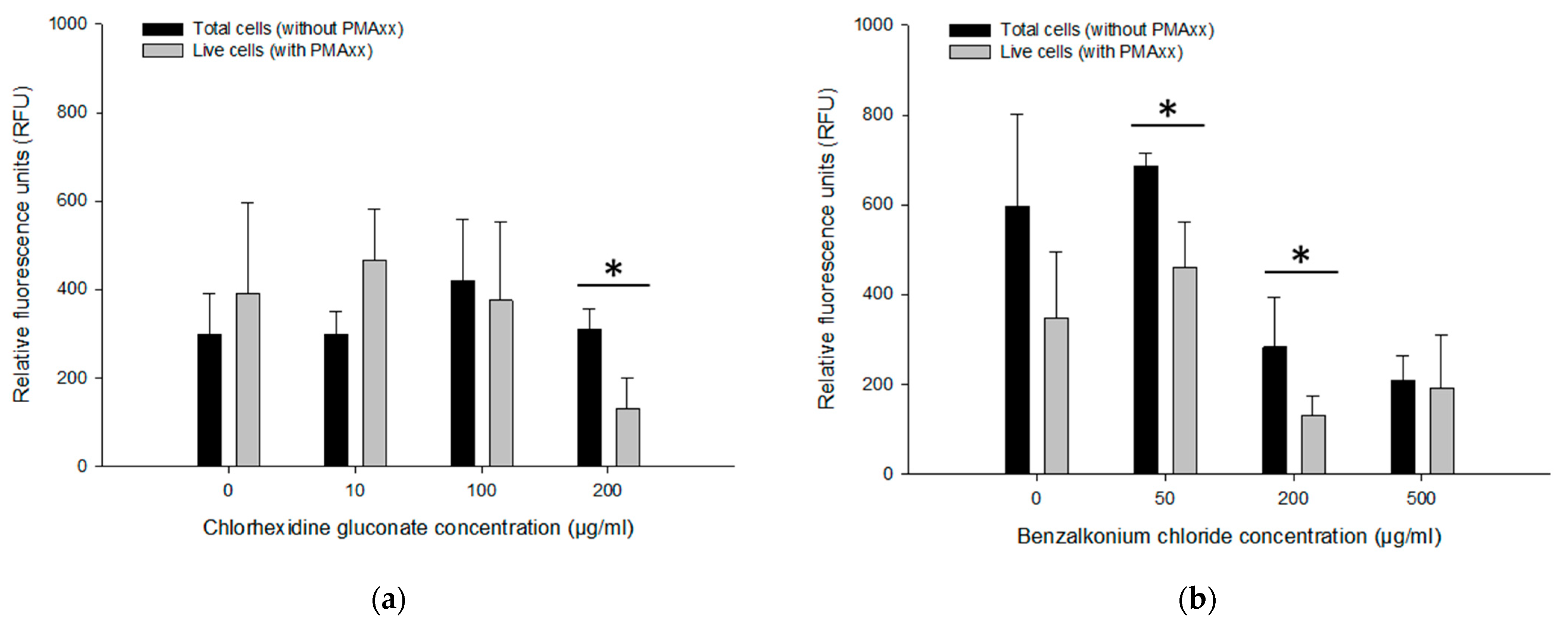
3.4.1. Effect of CHX and BZK on the PMAxx-RPA Exo Assay
3.4.2. Comparing Pure DNA and Cell Lysates on the PMAxx-RPA Exo Assay
3.5. Assessing Live/Dead Cells in CHX and BZK Solutions
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Depoorter, E.; De Canck, E.; Peeters, C.; Wieme, A.D.; Cnockaert, M.; Zlosnik, J.E.A.; LiPuma, J.J.; Coenye, T.; Vandamme, P. Burkholderia cepacia complex taxon K: Where to split? Front. Microbiol. 2020, 11, 1594. [Google Scholar] [CrossRef]
- Tavares, M.; Kozak, M.; Balola, A.; Sa-Correia, I. Burkholderia cepacia complex bacteria: A feared contamination risk in water-based pharmaceutical products. Clin. Microbiol. Rev. 2020, 33, e00139-19. [Google Scholar] [CrossRef] [PubMed]
- Scoffone, V.C.; Trespidi, G.; Barbieri, G.; Irudal, S.; Israyilova, A.; Buroni, S. Methodological tools to study species of the genus Burkholderia. Appl. Microbiol. Biotechnol. 2021, 105, 9019–9034. [Google Scholar] [CrossRef]
- Angrup, A.; Kanaujia, R.; Biswal, M.; Ray, P. Systematic review of ultrasound gel associated Burkholderia cepacia complex outbreaks: Clinical presentation, sources and control of outbreak. Am. J. Infect. Control 2022, 50, 1253–1257. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, L. Analysis of FDA Enforcement Reports (2012–2019) to Determine the Microbial Diversity in Contaminated Non-Sterile and Sterile Drugs. Am. Pharm. Rev. 2019, 4, 1–21. Available online: https://www.americanpharmaceuticalreview.com/Featured-Articles/518912-Analysis-of-FDA-Enforcement-Reports-2012-2019-to-Determine-the-Microbial-Diversity-in-Contaminated-Non-Sterile-and-Sterile-Drugs/ (accessed on 3 April 2023).
- USP. <60> Microbiological Examination of Nonsterile Products—Tests for Burkholderia cepacia Complex. 2018. Available online: http://www.usppf.com/pf/pub/data/v445/CHA_IPR_445_c60.xml (accessed on 17 January 2023).
- Ahn, Y.; Gibson, B.; Williams, A.; Alusta, P.; Buzatu, D.A.; Lee, Y.J.; LiPuma, J.J.; Hussong, D.; Marasa, B.; Cerniglia, C.E. A comparison of culture-based, real-time PCR, droplet digital PCR and flow cytometric methods for the detection of Burkholderia cepacia complex in nuclease-free water and antiseptics. J. Ind. Microbiol. Biotechnol. 2020, 47, 475–484. [Google Scholar] [CrossRef]
- Jimenez, L.; Jashari, T.; Vasquez, J.; Zapata, S.; Bochis, J.; Kulko, M.; Ellman, V.; Gardner, M.; Choe, T. Real-Time PCR detection of Burkholderia cepacia in pharmaceutical products contaminated with low levels of bacterial contamination. PDA J. Pharm. Sci. Technol. 2018, 72, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Lobato, I.M.; O’Sullivan, C.K. Recombinase polymerase amplification: Basics, applications and recent advances. Trends Analyt. Chem. 2018, 98, 19–35. [Google Scholar] [CrossRef]
- Li, J.; Macdonald, J.; von Stetten, F. Review: A comprehensive summary of a decade development of the recombinase polymerase amplification. Analyst 2018, 144, 31–67. [Google Scholar] [CrossRef]
- Piepenburg, O.; Williams, C.H.; Stemple, D.L.; Armes, N.A. DNA detection using recombination proteins. PLoS Biol. 2006, 4, e204. [Google Scholar] [CrossRef]
- Daddy Gaoh, S.; Kweon, O.; Lee, Y.-J.; LiPuma, J.J.; Hussong, D.; Marasa, B.; Ahn, Y. Loop-mediated isothermal amplification (LAMP) assay for detecting Burkholderia cepacia complex in non-sterile pharmaceutical products. Pathogens 2021, 10, 1071. [Google Scholar] [CrossRef] [PubMed]
- Daher, R.K.; Stewart, G.; Boissinot, M.; Bergeron, M.G. Recombinase polymerase amplification for diagnostic applications. Clin. Chem. 2016, 62, 947–958. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Zheng, X.; Kan, B.; Li, W.; Zhang, W.; Jiang, T.; Lu, J.; Qin, A. Rapid detection of Burkholderia pseudomallei with a lateral flow recombinase polymerase amplification assay. PLoS ONE 2019, 14, e0213416. [Google Scholar] [CrossRef]
- Li, J.; Zhong, Q.; Shang, M.Y.; Li, M.; Jiang, Y.S.; Zou, J.J.; Ma, S.S.; Huang, Q.; Lu, W.P. Preliminary evaluation of rapid visual identification of Burkholderia pseudomallei using a newly developed lateral flow strip-based recombinase polymerase amplification (LF-RPA) system. Front. Cell Infect. Microbiol. 2021, 11, 804737. [Google Scholar] [CrossRef] [PubMed]
- Saxena, A.; Pal, V.; Tripathi, N.K.; Goel, A.K. A recombinase polymerase amplification lateral flow assay for rapid detection of Burkholderia pseudomallei, the causative agent of melioidosis. Braz. J. Microbiol. 2022, 53, 185–193. [Google Scholar] [CrossRef]
- Saxena, A.; Pal, V.; Tripathi, N.K.; Goel, A.K. Development of a rapid and sensitive recombinase polymerase amplification-lateral flow assay for detection of Burkholderia mallei. Transbound. Emerg. Dis. 2019, 66, 1016–1022. [Google Scholar] [CrossRef]
- Fu, H.; Gan, L.; Tian, Z.; Han, J.; Du, B.; Xue, G.; Feng, Y.; Zhao, H.; Cui, J.; Yan, C.; et al. Rapid detection of Burkholderia cepacia complex carrying the 16S rRNA gene in clinical specimens by recombinase-aided amplification. Front. Cell. Infect. Microbiol. 2022, 12, 984140. [Google Scholar] [CrossRef]
- Wong, M.Y.; Tseng, Y.H.; Huang, T.Y.; Lin, B.S.; Tung, C.W.; Chu, C.S.; Huang, Y.K. Comparison of microbiological characteristics and genetic diversity between Burkholderia cepacia complex isolates from vascular access and other clinical infections. Microorganisms 2021, 9, 51. [Google Scholar] [CrossRef]
- Daddy Gaoh, S.; Kweon, O.; Lee, Y.J.; Hussong, D.; Marasa, B.; Ahn, Y. A Propidium monoazide (PMAxx)-droplet digital PCR (ddPCR) for the detection of viable Burkholderia cepacia complex in nuclease-Free water and antiseptics. Microorganisms 2022, 10, 943. [Google Scholar] [CrossRef]
- Daddy Gaoh, S.; Williams, A.; Le, D.; Kweon, O.; Alusta, P.; Buzatu, D.A.; Ahn, Y. Specific detection and enumeration of Burkholderia cepacia complex by flow cytometry using a fluorescence-labeled oligonucleotide probe. Microorganisms 2022, 10, 1170. [Google Scholar] [CrossRef]
- Higgins, M.; Ravenhall, M.; Ward, D.; Phelan, J.; Ibrahim, A.; Forrest, M.S.; Clark, T.G.; Campino, S. PrimedRPA: Primer design for recombinase polymerase amplification assays. Bioinformatics 2019, 35, 682–684. [Google Scholar] [CrossRef] [PubMed]
- Georgoutsou-Spyridonos, M.; Filippidou, M.; Kaprou, G.D.; Mastellos, D.C.; Chatzandroulis, S.; Tserepi, A. Isothermal recombinase polymerase amplification (RPA) of E. coli gDNA in commercially fabricated PCB-based microfluidic platforms. Micromachines 2021, 12, 1387. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.M.; Ahn, Y.; LiPuma, J.J.; Hussong, D.; Cerniglia, C.E. Survival and susceptibility of Burkholderia cepacia complex in chlorhexidine gluconate and benzalkonium chloride. J. Ind. Microbiol. Biotechnol. 2015, 42, 905–913. [Google Scholar] [CrossRef] [PubMed]
- Furlan, J.P.R.; Pitondo-Silva, A.; Braz, V.S.; Gallo, I.F.L.; Stehling, E.G. Evaluation of different molecular and phenotypic methods for identification of environmental Burkholderia cepacia complex. World J. Microb. Biot. 2019, 35, 39. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Zhou, J.L.; Zhou, J.; Hu, M.D.; Zhang, Q.; Kong, N.; Ren, H.G.; Liang, L.; Yue, J.J. Genome-based classification of Burkholderia cepacia complex provides new insight into its taxonomic status. Biol. Direct 2020, 15, 6. [Google Scholar] [CrossRef]
- Martinucci, M.; Roscetto, E.; Iula, V.D.; Votsi, A.; Catania, M.R.; De Gregorio, E. Accurate identification of members of the Burkholderia cepacia complex in cystic fibrosis sputum. Lett. Appl. Microbiol. 2016, 62, 221–229. [Google Scholar] [CrossRef]
- Jordan, L.; Kurtz, R. Optical Design of CFX96™ Real-Time PCR Detection System Eliminates the Requirement of a Passive Reference Dye. 2010. Available online: https://www.bio-rad.com/en-us/applications-technologies/normalization-real-time-pcr-fluorescence-data-with-rox-passive-reference-dye?ID=MW472W15#Normalization_of_Fluorescence_Data_Using_ROX (accessed on 17 January 2023).
- Li, J.; Zhou, D.; Xie, G.; Deng, M.; Feng, X.; Xu, H. PMAxx combined with recombinase aided amplification technique for specific and rapid detection of Salmonella in milk. Food Anal. Methods 2022, 15, 1769–1777. [Google Scholar] [CrossRef]
- Kersting, S.; Rausch, V.; Bier, F.F.; von Nickisch-Rosenegk, M. Rapid detection of Plasmodium falciparum with isothermal recombinase polymerase amplification and lateral flow analysis. Malar. J. 2014, 13, 99. [Google Scholar] [CrossRef]
- Liu, H.B.; Zang, Y.X.; Du, X.J.; Li, P.; Wang, S. Development of an isothermal amplification-based assay for the rapid visual detection of Salmonella bacteria. J. Dairy Sci. 2017, 100, 7016–7025. [Google Scholar] [CrossRef]
- Merk, S.; Meyer, H.; Greiser-Wilke, I.; Sprague, L.D.; Neubauer, H. Detection of Burkholderia cepacia DNA from artificially infected EDTA-blood and lung tissue comparing different DNA isolation methods. J. Vet. Med. B 2006, 53, 281–285. [Google Scholar] [CrossRef]
- Ahn, Y.; Kim, J.M.; Kweon, O.; Kim, S.J.; Jones, R.C.; Woodling, K.; Gamboa da Costa, G.; LiPuma, J.J.; Hussong, D.; Marasa, B.S.; et al. Intrinsic resistance of Burkholderia cepacia complex to benzalkonium chloride. MBio 2016, 7, e01716-16. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.J.; Bentham, R.H.; Ross, K.E. Limitations of using propidium monoazide with qPCR to discriminate between live and dead Legionella in biofilm samples. Microbiol. Insights 2014, 7, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Exterkate, R.A.M.; Zaura, E.; Brandt, B.W.; Buijs, M.J.; Koopman, J.; Crielaard, W.; ten Cate, J.M. The effect of propidium monoazide treatment on the measured bacterial composition of clinical samples after the use of a mouthwash. Clin. Oral Investig. 2015, 19, 813–822. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer/Probe | Sequence (5′–3′) | Primer Length (nt) |
|---|---|---|
| gbpT-F gbpT-R | ACGCTGTCGTCGACGATCATCAGCCTCGTGCT ACCATCGACAGCGCCATCATGATCGTCTGGTT | 32 32 |
| Probe | TCGGCCGCGTGCCGGGGATCCTGTCGACGG[FAM-dT]G[THF]-[BHQ1-dT]CTTCGCGATGCCGCC | 49 |
| Group | Species | Strain | Results |
|---|---|---|---|
| BCC | Burkholderia cepacia | PC783 | + |
| AU24442 | + | ||
| Burkholderia stabilis | AU23340 | + | |
| Burkholderia ambifaria | HI2468 | + | |
| Burkholderia anthina | HI2738 | + | |
| Burkholderia metallica | AU0553 | + | |
| AU16697 | + | ||
| Burkholderia contaminans | HI3429 | + | |
| AU24637 | + | ||
| Burkholderia diffusa | AU1075 | + | |
| Burkholderia arboris | ES0263a | + | |
| AU22095 | + | ||
| Burkholderia lata | HI4002 | + | |
| Burkholderia multivorans | HI2229 | + | |
| AU24571 | + | ||
| Burkholderia vietnamiensis | HI2212 | + | |
| AU24694 | + | ||
| Burkholderia cenocepacia | AU1054 | + | |
| AU0222 | + | ||
| AU19236 | + | ||
| HI2976 | + | ||
| HI2485 | + | ||
| J2315 | + | ||
| Non-BCC | Burkholderia glumae | AU6208 | + |
| AU12450 | + | ||
| Burkholderia gladioli | AU26454 | + | |
| AU29541 | + | ||
| AU30473 | + | ||
| AU16341 | + | ||
| Burkholderia concitans | AU12121 | – | |
| Burkholderia oklahomensis | ES0634 | + | |
| Burkholderia plantarii | AU9801 | + | |
| AU37486 | + | ||
| Burkholderia thailandensis | AU13555 | + | |
| AU36262 | + | ||
| Burkholderia tropica | AU15822 | – | |
| AU19944 | – | ||
| Burkholderia fungorum | AU18377 | – | |
| AU35949 | – | ||
| Non-Burkholderia | Caballeronia zhejiangensis | AU10475 | – |
| AU12096 | – | ||
| Enterococcus faecalis | ATCC29212 | – | |
| Enterococcus durans | ATCC6056 | – | |
| Proteus mirabilis | ATCC7002 | – | |
| Enterococcus faecium | ATCC35667 | – | |
| ATCC49624 | – | ||
| Bacillus subtilis | ATCC6051 | – | |
| Citrobacter freundii | ATCC8090 | – | |
| Pseudomonas aeruginosa | PAO1 | – | |
| ATCC27853 | – | ||
| Yersinia enterocolitica subsp. entrocolitica | ATCC27729 | – | |
| Shigella sonnei | ATCC9290 | – | |
| Lactobacillus salivarius subsp. salivarius | ATCC11741 | – | |
| Enterobacter aerogenes | ATCC13048 | – | |
| Klebsiella pneumoniae | ATCC13883 | – | |
| Candida albicans (Robin) Berkhout | ATCC10231 | – | |
| Salmonella enterica | isolates | – | |
| Paenibacillus lautus | isolates | – | |
| Brevibacillus laterosporus | isolates | – |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Daddy Gaoh, S.; Kweon, O.; Ahn, Y. Propidium Monoazide (PMAxx)-Recombinase Polymerase Amplification Exo (RPA Exo) Assay for Rapid Detection of Burkholderia cepacia Complex in Chlorhexidine Gluconate (CHX) and Benzalkonium Chloride (BZK) Solutions. Microorganisms 2023, 11, 1401. https://doi.org/10.3390/microorganisms11061401
Daddy Gaoh S, Kweon O, Ahn Y. Propidium Monoazide (PMAxx)-Recombinase Polymerase Amplification Exo (RPA Exo) Assay for Rapid Detection of Burkholderia cepacia Complex in Chlorhexidine Gluconate (CHX) and Benzalkonium Chloride (BZK) Solutions. Microorganisms. 2023; 11(6):1401. https://doi.org/10.3390/microorganisms11061401
Chicago/Turabian StyleDaddy Gaoh, Soumana, Ohgew Kweon, and Youngbeom Ahn. 2023. "Propidium Monoazide (PMAxx)-Recombinase Polymerase Amplification Exo (RPA Exo) Assay for Rapid Detection of Burkholderia cepacia Complex in Chlorhexidine Gluconate (CHX) and Benzalkonium Chloride (BZK) Solutions" Microorganisms 11, no. 6: 1401. https://doi.org/10.3390/microorganisms11061401
APA StyleDaddy Gaoh, S., Kweon, O., & Ahn, Y. (2023). Propidium Monoazide (PMAxx)-Recombinase Polymerase Amplification Exo (RPA Exo) Assay for Rapid Detection of Burkholderia cepacia Complex in Chlorhexidine Gluconate (CHX) and Benzalkonium Chloride (BZK) Solutions. Microorganisms, 11(6), 1401. https://doi.org/10.3390/microorganisms11061401