
Rumen Microbiota Predicts Feed Efficiency of Primiparous Nordic Red Dairy Cows


Abstract
1. Introduction
2. Materials and Methods
2.1. Animals, Experimental Design, and Diet
2.2. Sampling and Data Recording
2.3. DNA Extraction, Sequencing
2.4. Sequencing Data Processing
2.5. Statistical Analyses
3. Results
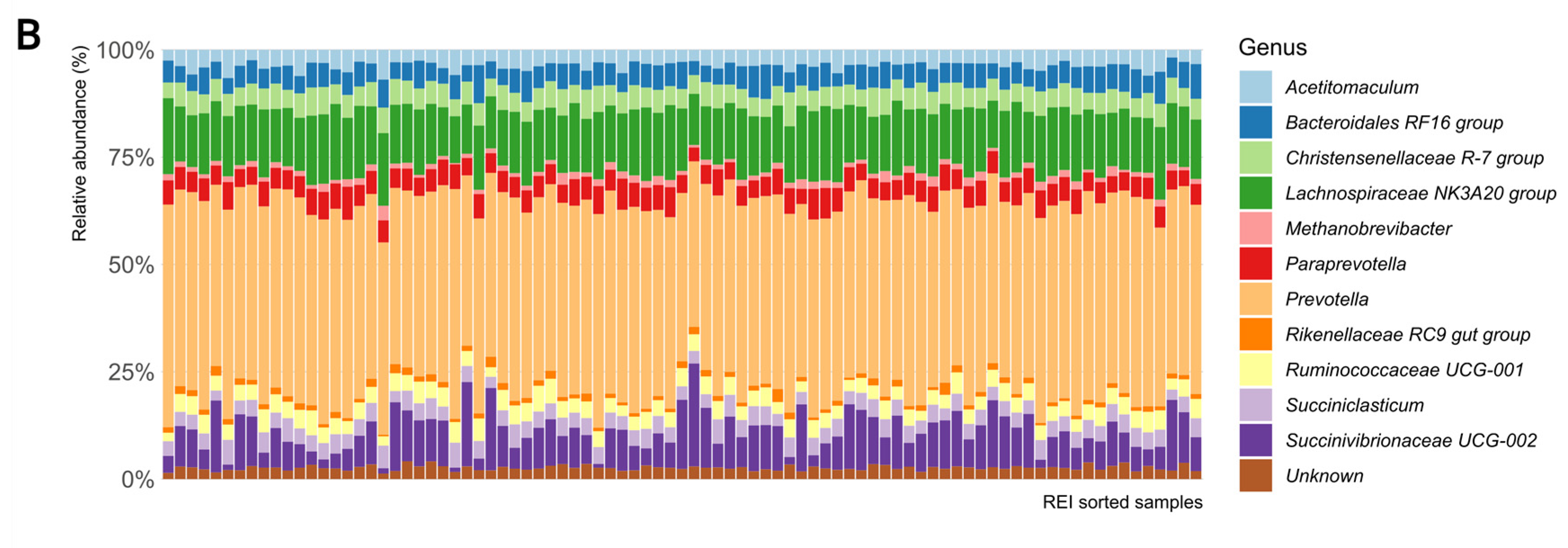
3.1. REI Association with VFA, Alpha, and Beta Diversities
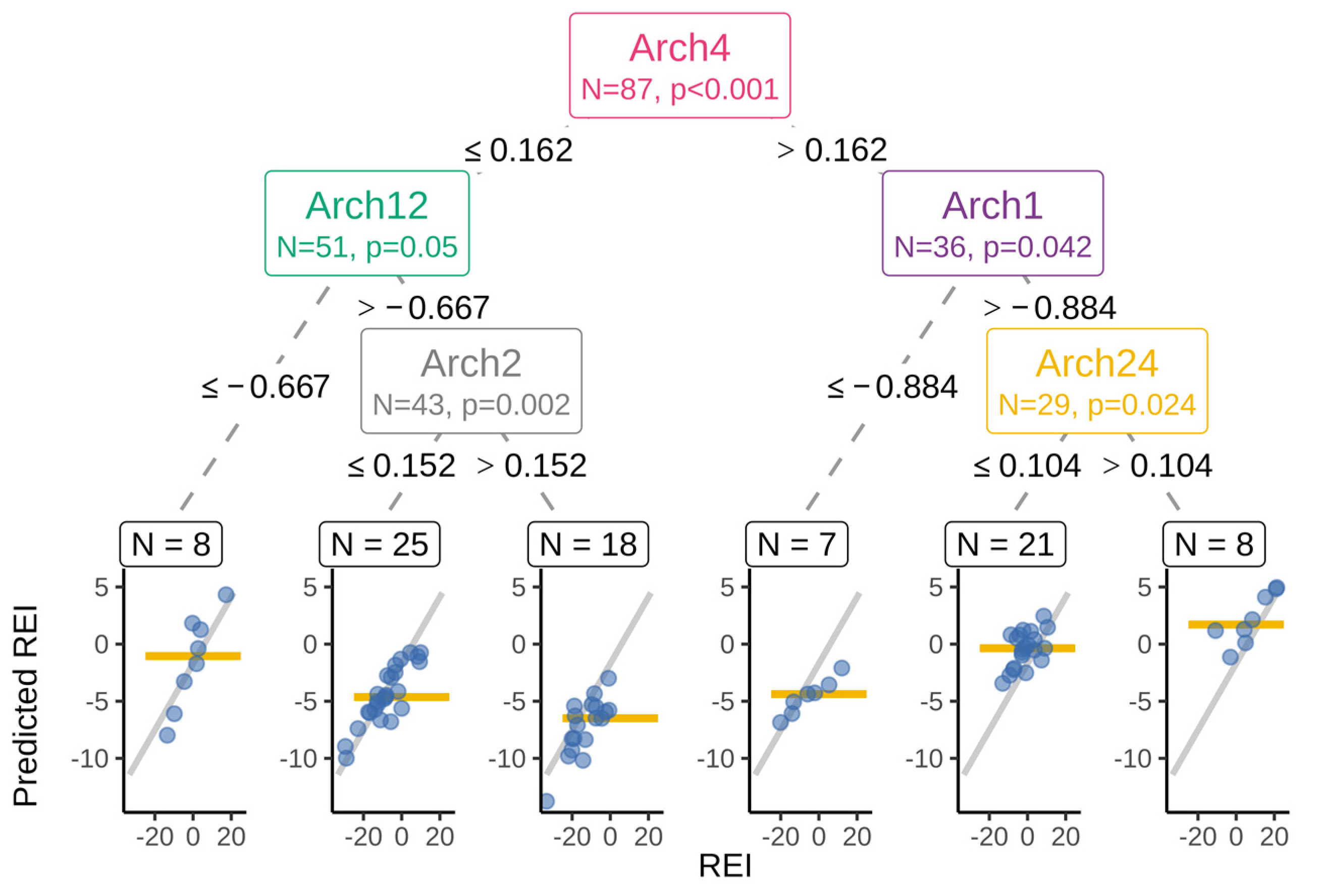
3.2. Predicting REI from Rumen Microbiota
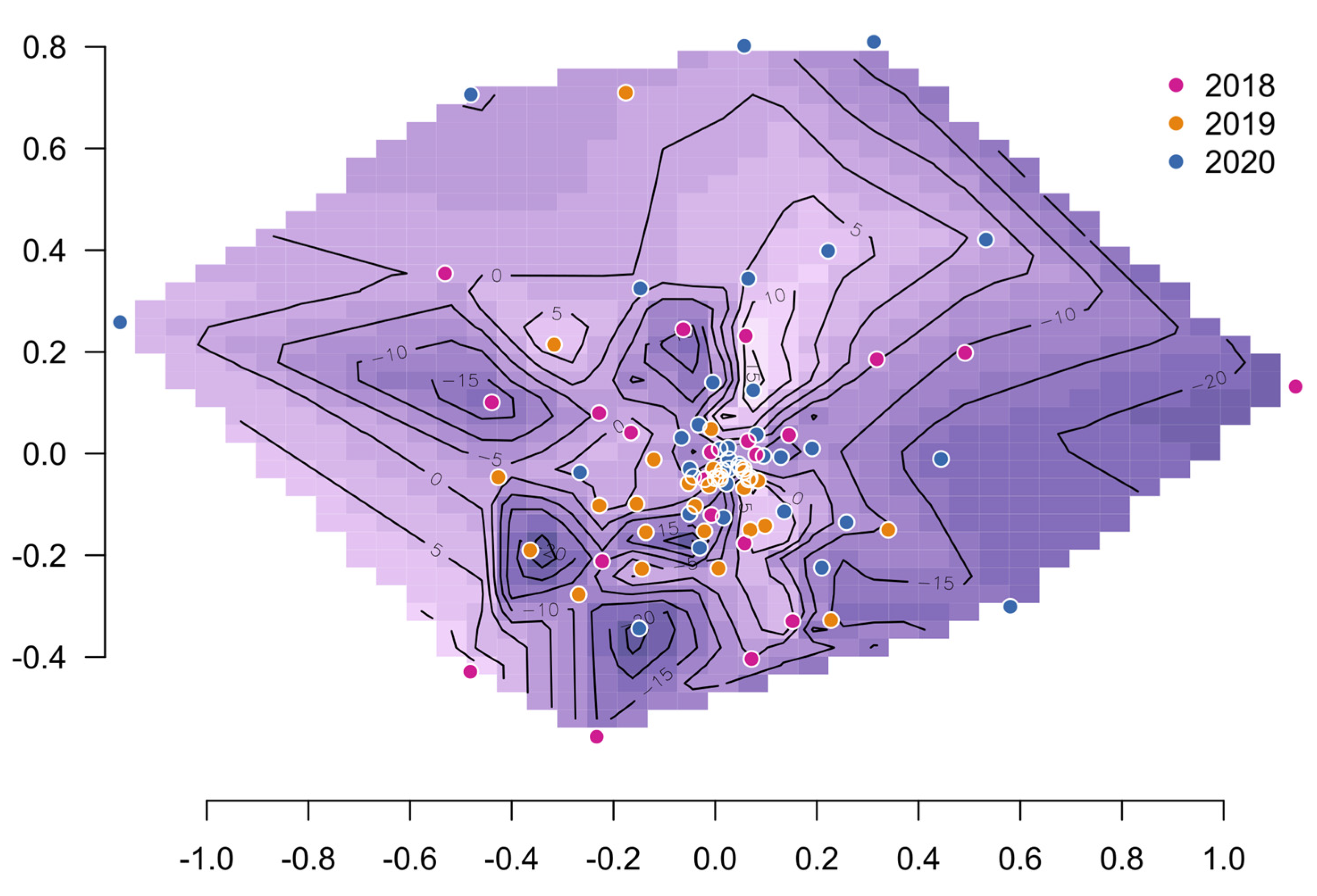
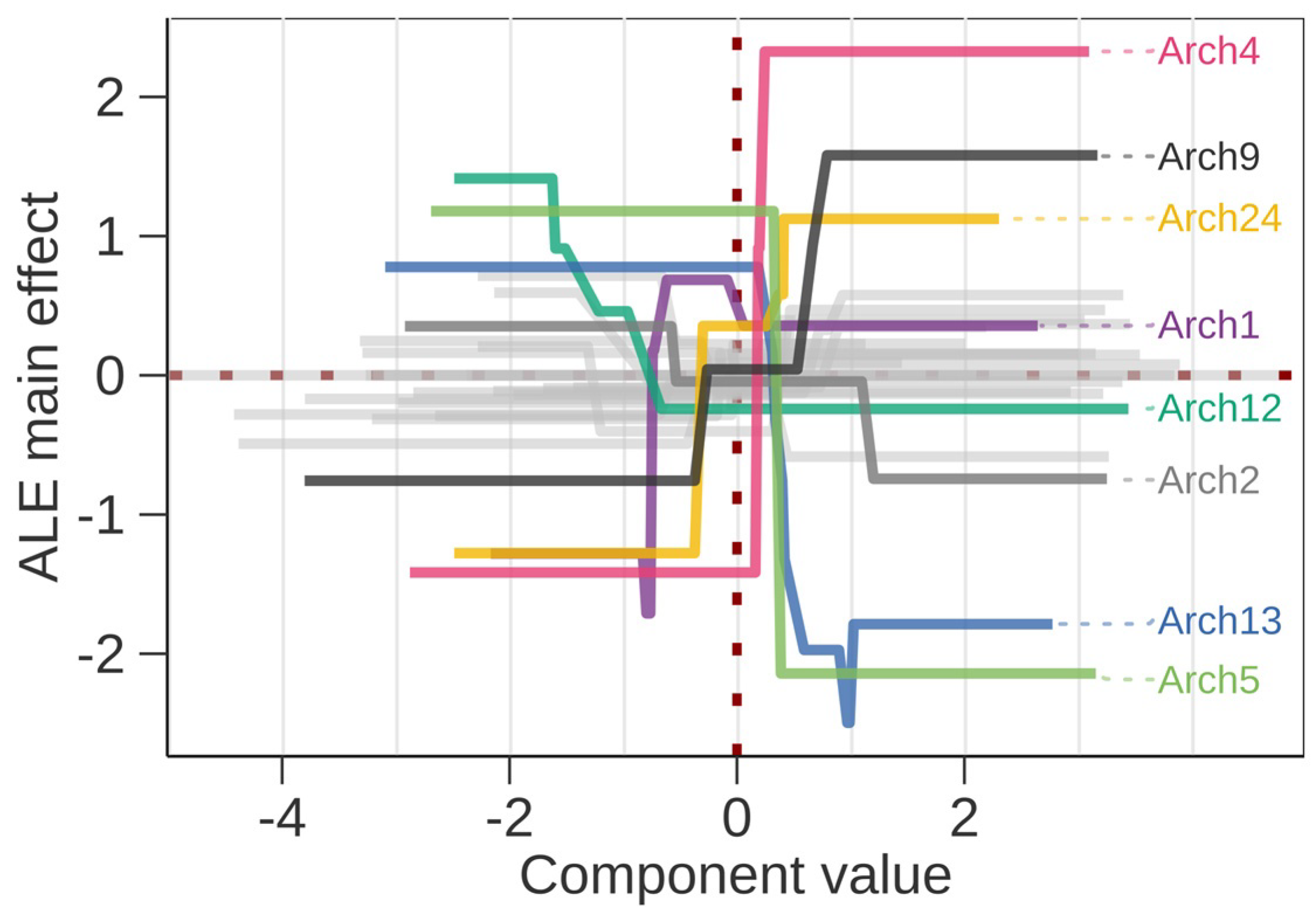
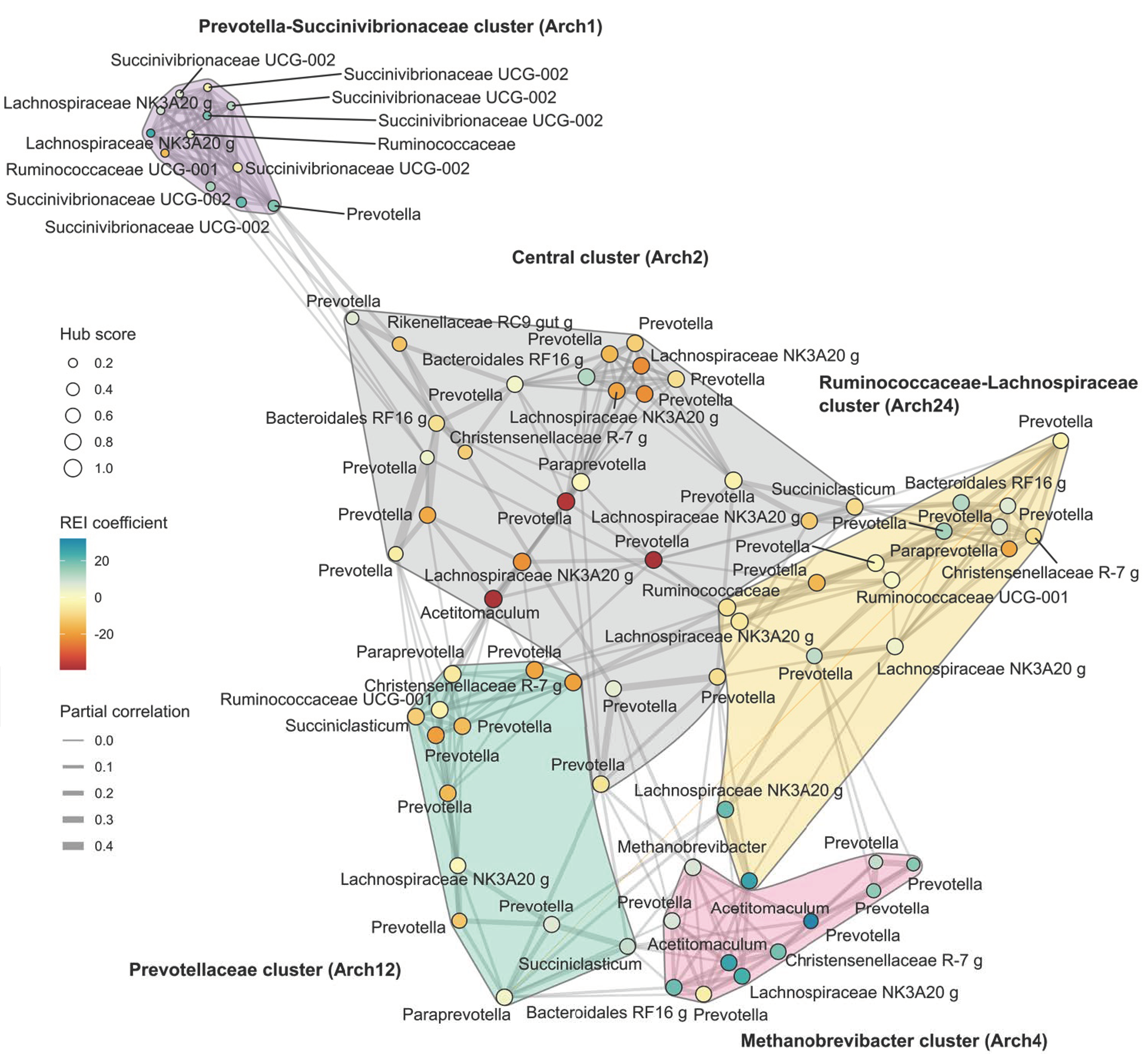
3.3. Interpretation of the Association Results
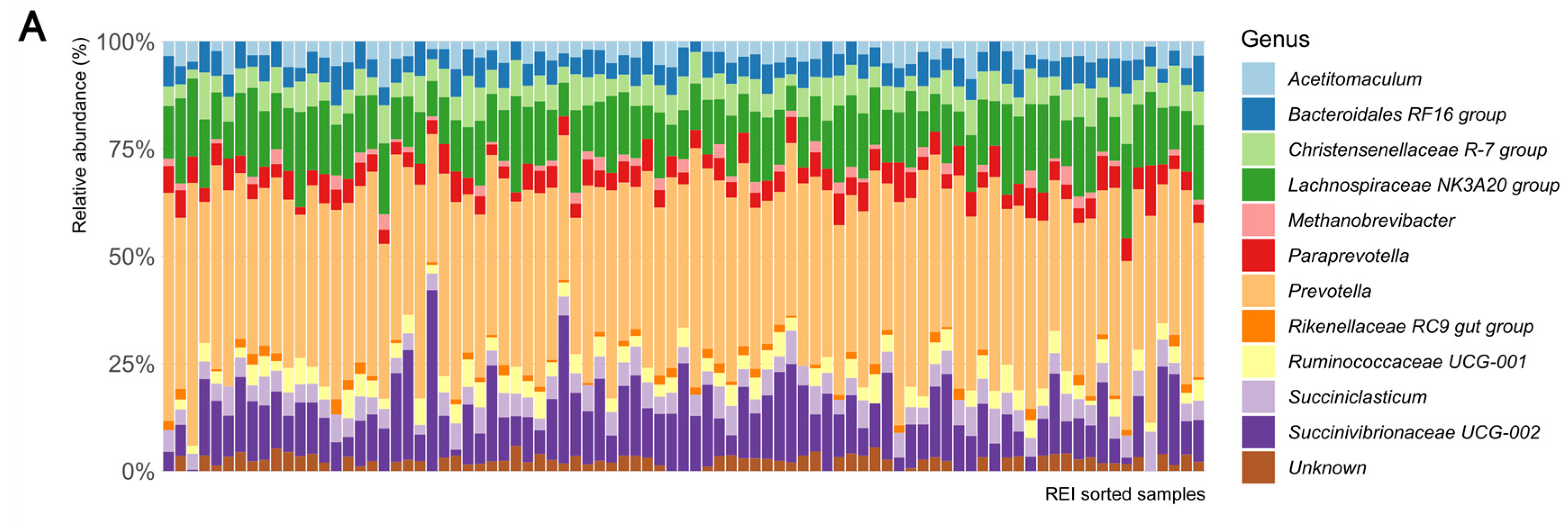
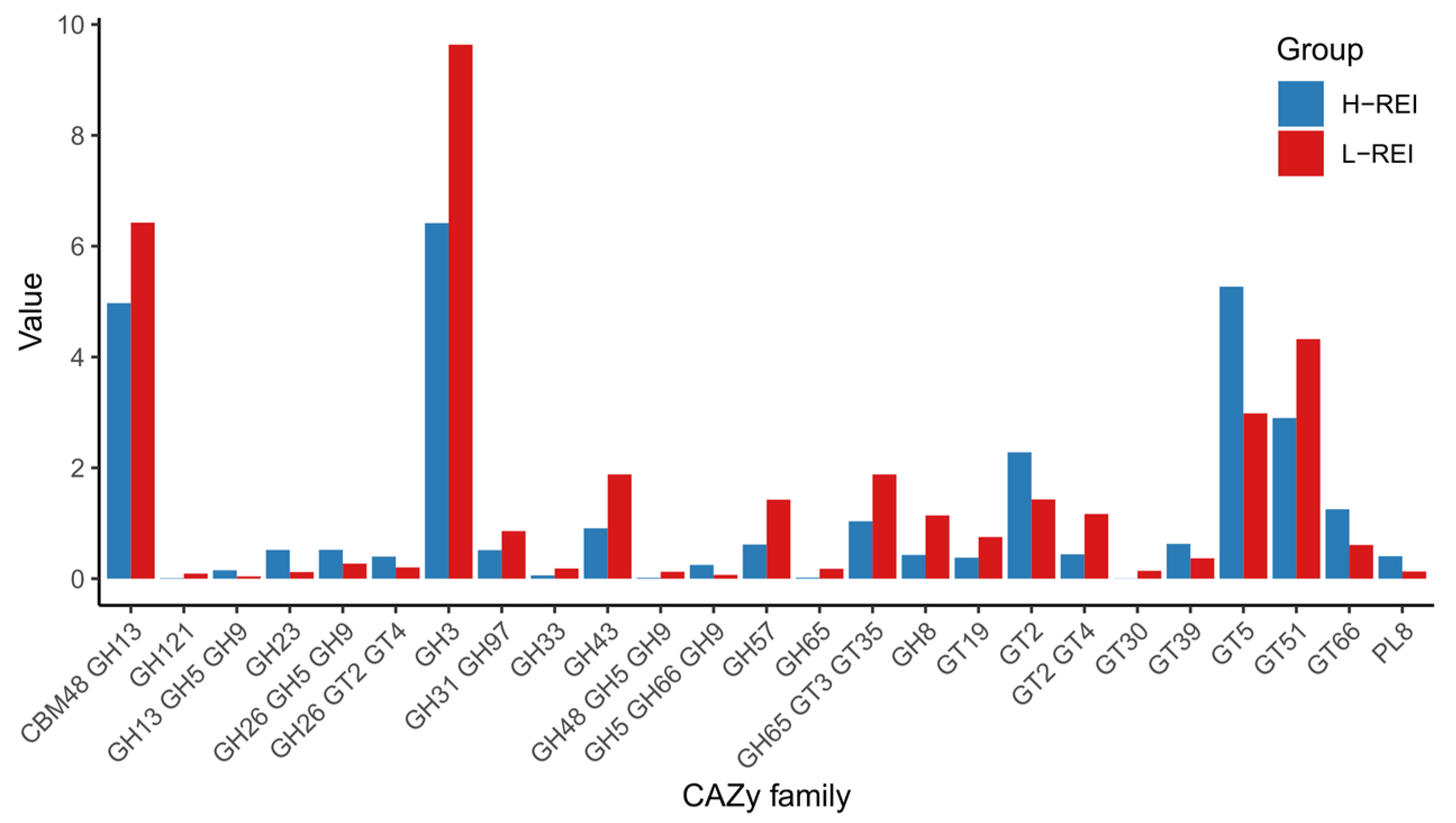
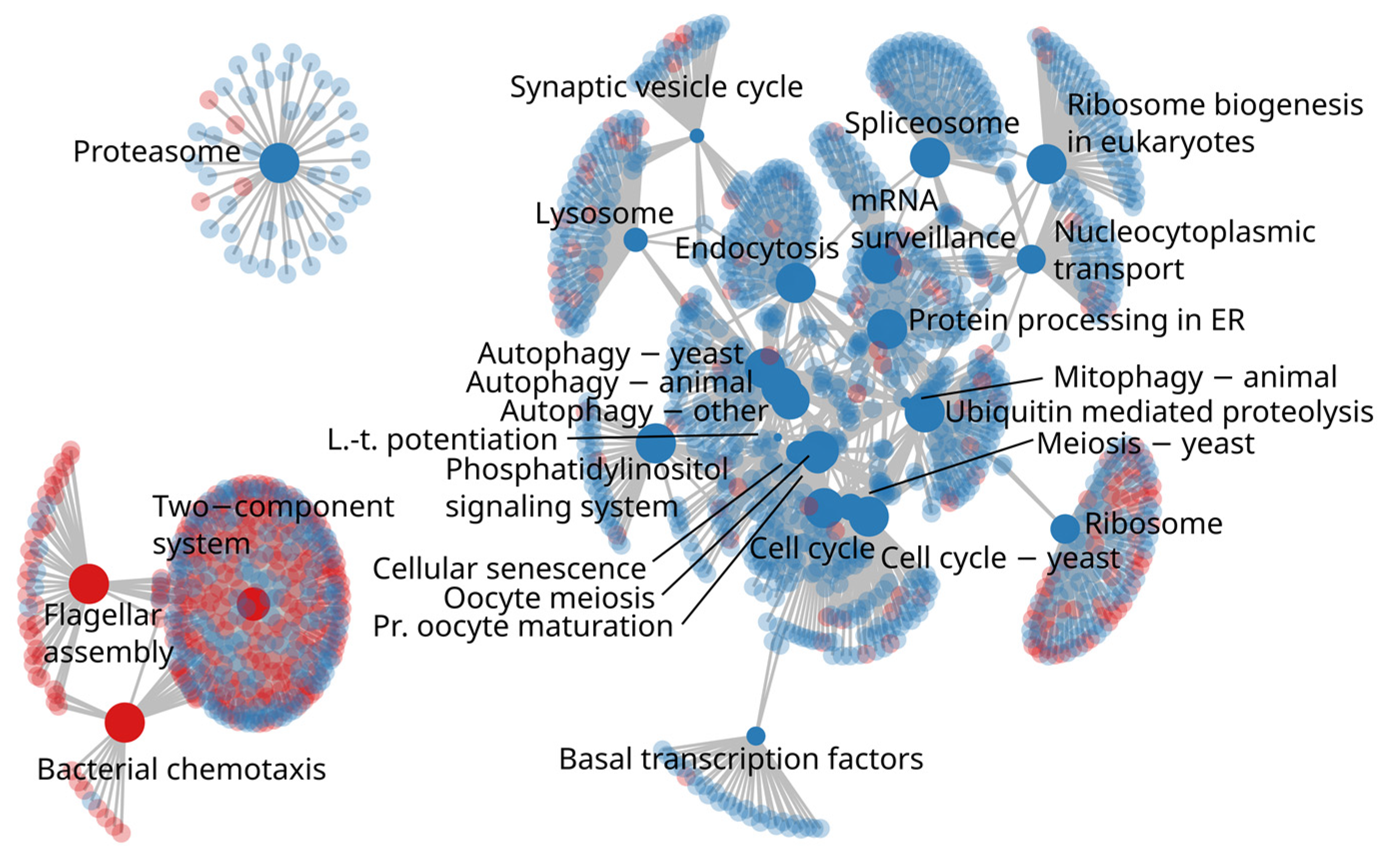
3.4. Differences in Microbial Functional Capacities between H-REI and L-REI Groups
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Capper, J.L.; Cady, R.A.; Bauman, D.E. The Environmental Impact of Dairy Production: 1944 Compared with 20071. J. Anim. Sci. 2009, 87, 2160–2167. [Google Scholar] [CrossRef] [PubMed]
- Knapp, J.R.; Laur, G.L.; Vadas, P.A.; Weiss, W.P.; Tricarico, J.M. Invited Review: Enteric Methane in Dairy Cattle Production: Quantifying the Opportunities and Impact of Reducing Emissions. J. Dairy Sci. 2014, 97, 3231–3261. [Google Scholar] [CrossRef] [PubMed]
- Connor, E.E. Invited Review: Improving Feed Efficiency in Dairy Production: Challenges and Possibilities. Animal 2015, 9, 395–408. [Google Scholar] [CrossRef]
- Liinamo, A.-E.; Mäntysaari, P.; Lidauer, M.H.; Mäntysaari, E.A. Genetic Parameters for Residual Energy Intake and Energy Conversion Efficiency in Nordic Red Dairy Cattle. Acta Agric. Scand. Sect.—Anim. Sci. 2015, 65, 63–72. [Google Scholar] [CrossRef]
- Hardie, L.C.; VandeHaar, M.J.; Tempelman, R.J.; Weigel, K.A.; Armentano, L.E.; Wiggans, G.R.; Veerkamp, R.F.; De Haas, Y.; Coffey, M.P.; Connor, E.E.; et al. The Genetic and Biological Basis of Feed Efficiency in Mid-Lactation Holstein Dairy Cows. J. Dairy Sci. 2017, 100, 9061–9075. [Google Scholar] [CrossRef]
- Mehtiö, T.; Negussie, E.; Mäntysaari, P.; Mäntysaari, E.A.; Lidauer, M.H. Genetic Background in Partitioning of Metabolizable Energy Efficiency in Dairy Cows. J. Dairy Sci. 2018, 101, 4268–4278. [Google Scholar] [CrossRef] [PubMed]
- Løvendahl, P.; Difford, G.F.; Li, B.; Chagunda, M.G.G.; Huhtanen, P.; Lidauer, M.H.; Lassen, J.; Lund, P. Review: Selecting for Improved Feed Efficiency and Reduced Methane Emissions in Dairy Cattle. Animal 2018, 12, s336–s349. [Google Scholar] [CrossRef] [PubMed]
- Brito, L.F.; Oliveira, H.R.; Houlahan, K.; Fonseca, P.A.S.; Lam, S.; Butty, A.M.; Seymour, D.J.; Vargas, G.; Chud, T.C.S.; Silva, F.F.; et al. Genetic Mechanisms Underlying Feed Utilization and Implementation of Genomic Selection for Improved Feed Efficiency in Dairy Cattle. Can. J. Anim. Sci. 2020, 100, 587–604. [Google Scholar] [CrossRef]
- Mäntysaari, P.; Liinamo, A.-E.; Mäntysaari, E.A. Energy Efficiency and Its Relationship with Milk, Body, and Intake Traits and Energy Status among Primiparous Nordic Red Dairy Cattle. J. Dairy Sci. 2012, 95, 3200–3211. [Google Scholar] [CrossRef]
- Henderson, G.; Cox, F.; Ganesh, S.; Jonker, A.; Young, W.; Janssen, P.H. Rumen Microbial Community Composition Varies with Diet and Host, but a Core Microbiome Is Found across a Wide Geographical Range. Sci. Rep. 2015, 5, 14567. [Google Scholar] [CrossRef]
- Tapio, I.; Fischer, D.; Blasco, L.; Tapio, M.; Wallace, R.J.; Bayat, A.R.; Ventto, L.; Kahala, M.; Negussie, E.; Shingfield, K.J.; et al. Taxon Abundance, Diversity, Co-Occurrence and Network Analysis of the Ruminal Microbiota in Response to Dietary Changes in Dairy Cows. PLoS ONE 2017, 12, e0180260. [Google Scholar] [CrossRef]
- Wallace, R.J.; Sasson, G.; Garnsworthy, P.C.; Tapio, I.; Gregson, E.; Bani, P.; Huhtanen, P.; Bayat, A.R.; Strozzi, F.; Biscarini, F.; et al. A Heritable Subset of the Core Rumen Microbiome Dictates Dairy Cow Productivity and Emissions. Sci. Adv. 2019, 5, eaav8391. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Hitch, T.C.A.; Chen, Y.; Creevey, C.J.; Guan, L.L. Comparative Metagenomic and Metatranscriptomic Analyses Reveal the Breed Effect on the Rumen Microbiome and Its Associations with Feed Efficiency in Beef Cattle. Microbiome 2019, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- Daghio, M.; Ciucci, F.; Buccioni, A.; Cappucci, A.; Casarosa, L.; Serra, A.; Conte, G.; Viti, C.; McAmmond, B.M.; Van Hamme, J.D.; et al. Correlation of Breed, Growth Performance, and Rumen Microbiota in Two Rustic Cattle Breeds Reared under Different Conditions. Front. Microbiol. 2021, 12, 652031. [Google Scholar] [CrossRef]
- Roehe, R.; Dewhurst, R.J.; Duthie, C.-A.; Rooke, J.A.; McKain, N.; Ross, D.W.; Hyslop, J.J.; Waterhouse, A.; Freeman, T.C.; Watson, M.; et al. Bovine Host Genetic Variation Influences Rumen Microbial Methane Production with Best Selection Criterion for Low Methane Emitting and Efficiently Feed Converting Hosts Based on Metagenomic Gene Abundance. PLoS Genet. 2016, 12, e1005846. [Google Scholar] [CrossRef]
- Bayat, A.R.; Tapio, I.; Vilkki, J.; Shingfield, K.J.; Leskinen, H. Plant Oil Supplements Reduce Methane Emissions and Improve Milk Fatty Acid Composition in Dairy Cows Fed Grass Silage-Based Diets without Affecting Milk Yield. J. Dairy Sci. 2018, 101, 1136–1151. [Google Scholar] [CrossRef]
- Difford, G.F.; Plichta, D.R.; Løvendahl, P.; Lassen, J.; Noel, S.J.; Højberg, O.; Wright, A.-D.G.; Zhu, Z.; Kristensen, L.; Nielsen, H.B.; et al. Host Genetics and the Rumen Microbiome Jointly Associate with Methane Emissions in Dairy Cows. PLoS Genet. 2018, 14, e1007580. [Google Scholar] [CrossRef]
- Jewell, K.A.; McCormick, C.A.; Odt, C.L.; Weimer, P.J.; Suen, G. Ruminal Bacterial Community Composition in Dairy Cows Is Dynamic over the Course of Two Lactations and Correlates with Feed Efficiency. Appl. Environ. Microbiol. 2015, 81, 4697–4710. [Google Scholar] [CrossRef]
- Shabat, S.K.B.; Sasson, G.; Doron-Faigenboim, A.; Durman, T.; Yaacoby, S.; Berg Miller, M.E.; White, B.A.; Shterzer, N.; Mizrahi, I. Specific Microbiome-Dependent Mechanisms Underlie the Energy Harvest Efficiency of Ruminants. ISME J. 2016, 10, 2958–2972. [Google Scholar] [CrossRef]
- McLoughlin, S.; Spillane, C.; Claffey, N.; Smith, P.E.; O’Rourke, T.; Diskin, M.G.; Waters, S.M. Rumen Microbiome Composition Is Altered in Sheep Divergent in Feed Efficiency. Front. Microbiol. 2020, 11, 1981. [Google Scholar] [CrossRef] [PubMed]
- Paz, H.A.; Hales, K.E.; Wells, J.E.; Kuehn, L.A.; Freetly, H.C.; Berry, E.D.; Flythe, M.D.; Spangler, M.L.; Fernando, S.C. Rumen Bacterial Community Structure Impacts Feed Efficiency in Beef Cattle. J. Anim. Sci. 2018, 96, 1045–1058. [Google Scholar] [CrossRef]
- Li, F.; Guan, L.L. Metatranscriptomic Profiling Reveals Linkages between the Active Rumen Microbiome and Feed Efficiency in Beef Cattle. Appl. Environ. Microbiol. 2017, 83, e00061-17. [Google Scholar] [CrossRef]
- Auffret, M.D.; Stewart, R.D.; Dewhurst, R.J.; Duthie, C.-A.; Watson, M.; Roehe, R. Identification of Microbial Genetic Capacities and Potential Mechanisms Within the Rumen Microbiome Explaining Differences in Beef Cattle Feed Efficiency. Front. Microbiol. 2020, 11, 1229. [Google Scholar] [CrossRef] [PubMed]
- Tarrah, A.; Callegaro, S.; Pakroo, S.; Finocchiaro, R.; Giacomini, A.; Corich, V.; Cassandro, M. New Insights into the Raw Milk Microbiota Diversity from Animals with a Different Genetic Predisposition for Feed Efficiency and Resilience to Mastitis. Sci. Rep. 2022, 12, 13498. [Google Scholar] [CrossRef]
- McCormack, U.M.; Curião, T.; Metzler-Zebeli, B.U.; Magowan, E.; Berry, D.P.; Reyer, H.; Prieto, M.L.; Buzoianu, S.G.; Harrison, M.; Rebeiz, N.; et al. Porcine Feed Efficiency-Associated Intestinal Microbiota and Physiological Traits: Finding Consistent Cross-Locational Biomarkers for Residual Feed Intake. mSystems 2019, 4, e00324-18. [Google Scholar] [CrossRef]
- Xue, M.-Y.; Xie, Y.-Y.; Zhong, Y.; Ma, X.-J.; Sun, H.-Z.; Liu, J.-X. Integrated Meta-Omics Reveals New Ruminal Microbial Features Associated with Feed Efficiency in Dairy Cattle. Microbiome 2022, 10, 32. [Google Scholar] [CrossRef] [PubMed]
- Clemmons, B.A.; Martino, C.; Powers, J.B.; Campagna, S.R.; Voy, B.H.; Donohoe, D.R.; Gaffney, J.; Embree, M.M.; Myer, P.R. Rumen Bacteria and Serum Metabolites Predictive of Feed Efficiency Phenotypes in Beef Cattle. Sci. Rep. 2019, 9, 19265. [Google Scholar] [CrossRef]
- Delgado, B.; Bach, A.; Guasch, I.; González, C.; Elcoso, G.; Pryce, J.E.; Gonzalez-Recio, O. Whole Rumen Metagenome Sequencing Allows Classifying and Predicting Feed Efficiency and Intake Levels in Cattle. Sci. Rep. 2019, 9, 11. [Google Scholar] [CrossRef]
- Zhou, Y.-H.; Gallins, P. A Review and Tutorial of Machine Learning Methods for Microbiome Host Trait Prediction. Front. Genet. 2019, 10, 579. [Google Scholar] [CrossRef]
- Song, K.; Wright, F.A.; Zhou, Y.-H. Systematic Comparisons for Composition Profiles, Taxonomic Levels, and Machine Learning Methods for Microbiome-Based Disease Prediction. Front. Mol. Biosci. 2020, 7, 610845. [Google Scholar] [CrossRef] [PubMed]
- Messad, F.; Louveau, I.; Koffi, B.; Gilbert, H.; Gondret, F. Investigation of Muscle Transcriptomes Using Gradient Boosting Machine Learning Identifies Molecular Predictors of Feed Efficiency in Growing Pigs. BMC Genom. 2019, 20, 659. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Alexandre, P.A.; Ribeiro, G.; Fukumasu, H.; Sun, W.; Reverter, A.; Li, Y. Identification of Predictor Genes for Feed Efficiency in Beef Cattle by Applying Machine Learning Methods to Multi-Tissue Transcriptome Data. Front. Genet. 2021, 12, 619857. [Google Scholar] [CrossRef]
- Mäntysaari, P.; Mäntysaari, E.A.; Kokkonen, T.; Mehtiö, T.; Kajava, S.; Grelet, C.; Lidauer, P.; Lidauer, M.H. Body and Milk Traits as Indicators of Dairy Cow Energy Status in Early Lactation. J. Dairy Sci. 2019, 102, 7904–7916. [Google Scholar] [CrossRef]
- Huhtanen, P.J.; Blauwiekel, R.; Saastamoinen, I. Effects of Intraruminal Infusions of Propionate and Butyrate with Two Different Protein Supplements on Milk Production and Blood Metabolites in Dairy Cows Receiving Grass Silage-Based Diet. J. Sci. Food Agric. 1998, 77, 213–222. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Lozupone, C.A.; Turnbaugh, P.J.; Fierer, N.; Knight, R. Global Patterns of 16S RRNA Diversity at a Depth of Millions of Sequences per Sample. Proc. Natl. Acad. Sci. USA 2011, 108, 4516–4522. [Google Scholar] [CrossRef] [PubMed]
- Huuki, H.; Ahvenjärvi, S.; Lidauer, P.; Popova, M.; Vilkki, J.; Vanhatalo, A.; Tapio, I. Fresh Rumen Liquid Inoculant Enhances the Rumen Microbial Community Establishment in Pre-Weaned Dairy Calves. Front. Microbiol. 2022, 12, 758395. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, Interactive, Scalable and Extensible Microbiome Data Science Using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-Resolution Sample Inference from Illumina Amplicon Data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA Ribosomal RNA Gene Database Project: Improved Data Processing and Web-Based Tools. Nucleic Acids Res. 2012, 41, D590–D596. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An Ultra-Fast All-in-One FASTQ Preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Li, D.; Liu, C.-M.; Luo, R.; Sadakane, K.; Lam, T.-W. MEGAHIT: An Ultra-Fast Single-Node Solution for Large and Complex Metagenomics Assembly via Succinct de Bruijn Graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef] [PubMed]
- Hyatt, D.; Chen, G.-L.; LoCascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic Gene Recognition and Translation Initiation Site Identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef] [PubMed]
- Cantalapiedra, C.P.; Hernández-Plaza, A.; Letunic, I.; Bork, P.; Huerta-Cepas, J. EggNOG-Mapper v2: Functional Annotation, Orthology Assignments, and Domain Prediction at the Metagenomic Scale. Mol. Biol. Evol. 2021, 38, 5825–5829. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast Gapped-Read Alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. FeatureCounts: An Efficient General Purpose Program for Assigning Sequence Reads to Genomic Features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Mölder, F.; Jablonski, K.P.; Letcher, B.; Hall, M.B.; Tomkins-Tinch, C.H.; Sochat, V.; Forster, J.; Lee, S.; Twardziok, S.O.; Kanitz, A.; et al. Sustainable Data Analysis with Snakemake. F1000Research 2021, 10, 33. [Google Scholar] [CrossRef] [PubMed]
- Fischer, D. Fischuu/Snakebite-Metagenomics: Stable Release Version 0.4. 2023.
- Sjaunja, L.O.; Baevre, L.; Junkkarine, L.; Pedersen, J.; Setälä, J. A Nordic Proposal for an Energy Corrected Milk (ECM) Formula. In Proceedings of the 27th Session International Committee for Recording and Productivity of Milk Animals, Paris, France, 2–6 July 1990; pp. 156–157. [Google Scholar]
- Lahti, L.; Shetty, S. Microbiome R Package. 2012. Available online: http://microbiome.github.io (accessed on 19 September 2022).
- McMurdie, P.J.; Holmes, S. Phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef]
- Oksanen, J.; Kindt, R.; Legendre, P.; O’Hara, B.; Simpson, G.L.; Solymos, P.; Stevens, M.H.H.; Wagner, H. Vegan: Community Ecology Package. 2020. Available online: https://cran.microsoft.com/snapshot/2021-08-04/web/packages/vegan/vegan.pdf (accessed on 24 March 2023).
- Jukes, T.H.; Cantor, C.R. Evolution of Protein Molecules. In Mammalian Protein Metabolism; Elsevier: Amsterdam, The Netherlands, 1969; pp. 21–132. ISBN 978-1-4832-3211-9. [Google Scholar]
- Paradis, E.; Schliep, K. Ape 5.0: An Environment for Modern Phylogenetics and Evolutionary Analyses in R. Bioinformatics 2019, 35, 526–528. [Google Scholar] [CrossRef]
- Udell, M.; Horn, C.; Zadeh, R.; Boyd, S. Generalized Low Rank Models. Found. Trends® Mach. Learn. 2016, 9, 1–118. [Google Scholar] [CrossRef]
- LeDell, E.; Gill, N.; Aiello, S.; Fu, A.; Candel, A.; Click, C.; Kraljevic, T.; Nykodym, T.; Aboyoun, P.; Kurka, M.; et al. H2O: R Interface for the “H2O” Scalable Machine Learning Platform. 2023. Available online: https://mran.microsoft.com/snapshot/2021-08-04/web/packages/h2o/index.html (accessed on 24 March 2023).
- Kuhn, M. Caret: Classification and Regression Training. 2022. Available online: https://cran.microsoft.com/snapshot/2022-02-28/web/packages/caret/caret.pdf (accessed on 24 March 2023).
- Chen, T.; Guestrin, C. XGBoost: A Scalable Tree Boosting System. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, San Francisco, CA, USA, 13–17 August 2016; pp. 785–794. [Google Scholar]
- Chen, T.; He, T.; Benesty, M.; Khotilovich, V.; Tang, Y.; Cho, H.; Chen, K.; Mitchell, R.; Cano, I.; Zhou, T.; et al. Xgboost: Extreme Gradient Boosting. 2023. Available online: https://cran.microsoft.com/snapshot/2022-02-07/web/packages/xgboost/xgboost.pdf (accessed on 24 March 2023).
- Liu, Y.; Just, A. SHAPforxgboost: SHAP Plots for “XGBoost”. 2019. Available online: https://cran.r-project.org/web/packages/SHAPforxgboost/SHAPforxgboost.pdf (accessed on 24 March 2023).
- Apley, D. ALEPlot: Accumulated Local Effects (ALE) Plots and Partial Dependence (PD) Plots. 2018. Available online: https://cran.r-project.org/web//packages//ALEPlot/ALEPlot.pdf (accessed on 24 March 2023).
- Molnar, C. Iml: An R Package for Interpretable Machine Learning. J. Open Source Softw. 2018, 3, 786. [Google Scholar] [CrossRef]
- Borkovec, M.; Madin, N. Ggparty: “ggplot” Visualizations for the “Partykit” Package. 2019. Available online: https://cran.r-project.org/web//packages/ggparty/ggparty.pdf (accessed on 24 March 2023).
- Epskamp, S.; Cramer, A.O.J.; Waldorp, L.J.; Schmittmann, V.D.; Borsboom, D. Qgraph: Network Visualizations of Relationships in Psychometric Data. J. Stat. Softw. 2012, 48, 1–18. [Google Scholar] [CrossRef]
- Pedersen, T.L. Ggraph: An Implementation of Grammar of Graphics for Graphs and Networks. 2022. Available online: https://cran.r-project.org/web/packages/ggraph/ggraph.pdf (accessed on 24 March 2023).
- Csardi, G.; Nepusz, T. The Igraph Software Package for Complex Network Research. Int. J. Complex Syst. 2006, 1695, 1–9. [Google Scholar]
- Langfelder, P.; Zhang, B.; Horvath, S. DynamicTreeCut: Methods for Detection of Clusters in Hierarchical Clustering Dendrograms. 2016. Available online: https://cran.r-project.org/web//packages//dynamicTreeCut/dynamicTreeCut.pdf (accessed on 24 March 2023).
- Langfelder, P.; Horvath, S. WGCNA: An R Package for Weighted Correlation Network Analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. Fast R Functions for Robust Correlations and Hierarchical Clustering. J. Stat. Softw. 2012, 46, i11. [Google Scholar] [CrossRef]
- Dowle, M.; Srinivasan, A. Data.Table: Extension of ‘Data.Frame’. 2021. Available online: https://cloud.r-project.org/web/packages/data.table/data.table.pdf (accessed on 24 March 2023).
- Fischer, D.; Oja, H.; Schleutker, J.; Sen, P.K.; Wahlfors, T. Generalized Mann-Whitney Type Tests for Microarray Experiments: Generalized Mann-Whitney Type Tests. Scand. J. Stat. 2014, 41, 672–692. [Google Scholar] [CrossRef]
- Fischer, D.; Oja, H. Mann-Whitney Type Tests for Microarray Experiments: The R Package gMWT. J. Stat. Softw. 2015, 65, 1–19. [Google Scholar] [CrossRef]
- Fischer, D. The R-Package GenomicTools for Multifactor Dimensionality Reduction and the Analysis of (Exploratory) Quantitative Trait Loci. Comput. Methods Programs Biomed. 2017, 151, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Ulgen, E.; Ozisik, O.; Sezerman, O.U. PathfindR: An R Package for Comprehensive Identification of Enriched Pathways in Omics Data through Active Subnetworks. Front. Genet. 2019, 10, 858. [Google Scholar] [CrossRef]
- Muller, E.E.L. Determining Microbial Niche Breadth in the Environment for Better Ecosystem Fate Predictions. mSystems 2019, 4, e00080-19. [Google Scholar] [CrossRef]
- Kenters, N.; Henderson, G.; Jeyanathan, J.; Kittelmann, S.; Janssen, P.H. Isolation of Previously Uncultured Rumen Bacteria by Dilution to Extinction Using a New Liquid Culture Medium. J. Microbiol. Methods 2011, 84, 52–60. [Google Scholar] [CrossRef]
- Kelly, W.J.; Leahy, S.C.; Altermann, E.; Yeoman, C.J.; Dunne, J.C.; Kong, Z.; Pacheco, D.M.; Li, D.; Noel, S.J.; Moon, C.D.; et al. The Glycobiome of the Rumen Bacterium Butyrivibrio Proteoclasticus B316T Highlights Adaptation to a Polysaccharide-Rich Environment. PLoS ONE 2010, 5, e11942. [Google Scholar] [CrossRef]
- Vos, P.; Garrity, G.; Jones, D.; Krieg, N.R.; Ludwig, W.; Rainey, F.A.; Schleifer, K.-H.; Whitman, W.B. (Eds.) Bergey’s Manual of Systematic Bacteriology; Springer: New York, NY, USA, 2019. [Google Scholar]
- Morotomi, M.; Nagai, F.; Watanabe, Y. Description of Christensenella minuta gen. nov., sp. nov., Isolated from Human Faeces, Which Forms a Distinct Branch in the Order Clostridiales, and Proposal of Christensenellaceae fam. Nov. Int. J. Syst. Evol. Microbiol. 2012, 62, 144–149. [Google Scholar] [CrossRef]
- Greening, R.C.; Leedle, J.A.Z. Enrichment and Isolation of Acetitomaculum ruminis, gen. nov., sp. nov.: Acetogenic Bacteria from the Bovine Rumen. Arch. Microbiol. 1989, 151, 399–406. [Google Scholar] [CrossRef]
- Carberry, C.A.; Kenny, D.A.; Han, S.; McCabe, M.S.; Waters, S.M. Effect of Phenotypic Residual Feed Intake and Dietary Forage Content on the Rumen Microbial Community of Beef Cattle. Appl. Environ. Microbiol. 2012, 78, 4949–4958. [Google Scholar] [CrossRef] [PubMed]
- Tapio, I.; Snelling, T.J.; Strozzi, F.; Wallace, R.J. The Ruminal Microbiome Associated with Methane Emissions from Ruminant Livestock. J. Anim. Sci. Biotechnol. 2017, 8, 7. [Google Scholar] [CrossRef]
- Matsui, H.; Ogata, K.; Tajima, K.; Nakamura, M.; Nagamine, T.; Aminov, R.I.; Benno, Y. Phenotypic Characterization of Polysaccharidases Produced by Four Prevotella Type Strains. Curr. Microbiol. 2000, 41, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Morotomi, M.; Nagai, F.; Sakon, H.; Tanaka, R. Paraprevotella clara gen. nov., sp. nov. and Paraprevotella xylaniphila sp. nov., Members of the Family “Prevotellaceae” Isolated from Human Faeces. Int. J. Syst. Evol. Microbiol. 2009, 59, 1895–1900. [Google Scholar] [CrossRef]
- Van Gylswyk, N.O. Succiniclasticum ruminis gen. nov., sp. nov., a Ruminal Bacterium Converting Succinate to Propionate as the Sole Energy-Yielding Mechanism. Int. J. Syst. Bacteriol. 1995, 45, 297–300. [Google Scholar] [CrossRef] [PubMed]
- Myer, P.R.; Smith, T.P.L.; Wells, J.E.; Kuehn, L.A.; Freetly, H.C. Rumen Microbiome from Steers Differing in Feed Efficiency. PLoS ONE 2015, 10, e0129174. [Google Scholar] [CrossRef] [PubMed]
- Clemmons, B.A.; Powers, J.B.; Campagna, S.R.; Seay, T.B.; Embree, M.M.; Myer, P.R. Rumen Fluid Metabolomics of Beef Steers Differing in Feed Efficiency. Metabolomics 2020, 16, 23. [Google Scholar] [CrossRef]
- Olijhoek, D.W.; Løvendahl, P.; Lassen, J.; Hellwing, A.L.F.; Höglund, J.K.; Weisbjerg, M.R.; Noel, S.J.; McLean, F.; Højberg, O.; Lund, P. Methane Production, Rumen Fermentation, and Diet Digestibility of Holstein and Jersey Dairy Cows Being Divergent in Residual Feed Intake and Fed at 2 Forage-to-Concentrate Ratios. J. Dairy Sci. 2018, 101, 9926–9940. [Google Scholar] [CrossRef] [PubMed]
- Sekar, K.; Linker, S.M.; Nguyen, J.; Grünhagen, A.; Stocker, R.; Sauer, U. Bacterial Glycogen Provides Short-Term Benefits in Changing Environments. Appl. Environ. Microbiol. 2020, 86, e00049-20. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wise, M.J. Glycogen with Short Average Chain Length Enhances Bacterial Durability. Naturwissenschaften 2011, 98, 719–729. [Google Scholar] [CrossRef] [PubMed]
- Lima, J.; Auffret, M.D.; Stewart, R.D.; Dewhurst, R.J.; Duthie, C.-A.; Snelling, T.J.; Walker, A.W.; Freeman, T.C.; Watson, M.; Roehe, R. Identification of Rumen Microbial Genes Involved in Pathways Linked to Appetite, Growth, and Feed Conversion Efficiency in Cattle. Front. Genet. 2019, 10, 701. [Google Scholar] [CrossRef]
- Balla, T. Phosphoinositides: Tiny Lipids With Giant Impact on Cell Regulation. Physiol. Rev. 2013, 93, 1019–1137. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Abu-Doleh, A.; Plank, J.; Catalyurek, U.V.; Firkins, J.L.; Yu, Z. The Transcriptome of the Rumen Ciliate Entodinium Caudatum Reveals Some of Its Metabolic Features. BMC Genom. 2019, 20, 1008. [Google Scholar] [CrossRef]
- Karmakar, R. State of the Art of Bacterial Chemotaxis. J. Basic Microbiol. 2021, 61, 366–379. [Google Scholar] [CrossRef]
- Stock, A.M.; Robinson, V.L.; Goudreau, P.N. Two-Component Signal Transduction. Annu. Rev. Biochem. 2000, 69, 183–215. [Google Scholar] [CrossRef]
- Kleerebezem, M.; Quadri, L.E.N.; Kuipers, O.P.; De Vos, W.M. Quorum Sensing by Peptide Pheromones and Two-component Signal-transduction Systems in Gram-positive Bacteria. Mol. Microbiol. 1997, 24, 895–904. [Google Scholar] [CrossRef]
- Cao, Q.; Sun, X.; Rajesh, K.; Chalasani, N.; Gelow, K.; Katz, B.; Shah, V.H.; Sanyal, A.J.; Smirnova, E. Effects of Rare Microbiome Taxa Filtering on Statistical Analysis. Front. Microbiol. 2021, 11, 607325. [Google Scholar] [CrossRef]
- Oudah, M.; Henschel, A. Taxonomy-Aware Feature Engineering for Microbiome Classification. BMC Bioinform. 2018, 19, 227. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| L-REI [−33.4, −9.17] | M-REI (−9.17, −0.9] | H-REI (−0.9, 21.3] | All | |
|---|---|---|---|---|
| n | 29 | 30 | 28 | 87 |
| REI * | −14.3 (7.2) | −4.6 (4.8) | 5.1 (7.1) | −4.6 (14.5) |
| ECM | 30.6 (4.9) | 30.7 (4.1) | 29.5 (2.7) | 30.26 (4.3) |
| BW | 614 (107) | 604 (61) | 589 (80) | 601 (81) |
| DMI | 20.1 (2.8) | 20.6 (1.9) | 21.1 (1.5) | 20.7 (2.0) |
| tVFA (mmol/L) | 109 (16) | 109 (17) | 107 (14) | 108 (16) |
| Acetate (mmol/mol) | 650 (20) B | 655 (24) AB | 659 (27) A | 654 (24) |
| Isovalerate (mmol/mol) | 10.8 (3.7) A | 10.4 (3.3) AB | 9 (4.1) B | 10.3 (3.5) |
| Propionate (mmol/mol) | 190 (19) A | 187 (19) AB | 182 (16) B | 187 (18.5) |
| Gini–Simpson | 0.975 (0.008) | 0.979 (0.008) | 0.977 (0.007) | 0.978 (0.009) |
| Evenness | 0.800 (0.065) | 0.823 (0.057) | 0.817 (0.038) | 0.817 (0.050) |
| Beta | 0.353 (0.126) A | 0.316 (0.103) B | 0.339 (0.136) A | 0.335 (0.133) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tapio, M.; Fischer, D.; Mäntysaari, P.; Tapio, I. Rumen Microbiota Predicts Feed Efficiency of Primiparous Nordic Red Dairy Cows. Microorganisms 2023, 11, 1116. https://doi.org/10.3390/microorganisms11051116
Tapio M, Fischer D, Mäntysaari P, Tapio I. Rumen Microbiota Predicts Feed Efficiency of Primiparous Nordic Red Dairy Cows. Microorganisms. 2023; 11(5):1116. https://doi.org/10.3390/microorganisms11051116
Chicago/Turabian StyleTapio, Miika, Daniel Fischer, Päivi Mäntysaari, and Ilma Tapio. 2023. "Rumen Microbiota Predicts Feed Efficiency of Primiparous Nordic Red Dairy Cows" Microorganisms 11, no. 5: 1116. https://doi.org/10.3390/microorganisms11051116
APA StyleTapio, M., Fischer, D., Mäntysaari, P., & Tapio, I. (2023). Rumen Microbiota Predicts Feed Efficiency of Primiparous Nordic Red Dairy Cows. Microorganisms, 11(5), 1116. https://doi.org/10.3390/microorganisms11051116