

Characterization and Association of Rips Repertoire to Host Range of Novel Ralstonia solanacearum Strains by In Silico Approaches
 ,
,  ,
,  , , , , ,
, , , , ,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Genomes Database
2.2. Quality Control and Assembly
2.3. Genome Annotation and T3Es Recovery
2.4. Prediction of the RSSC Pangenome
2.5. Whole-Genome Methods for Taxonomy Insights
3. Results
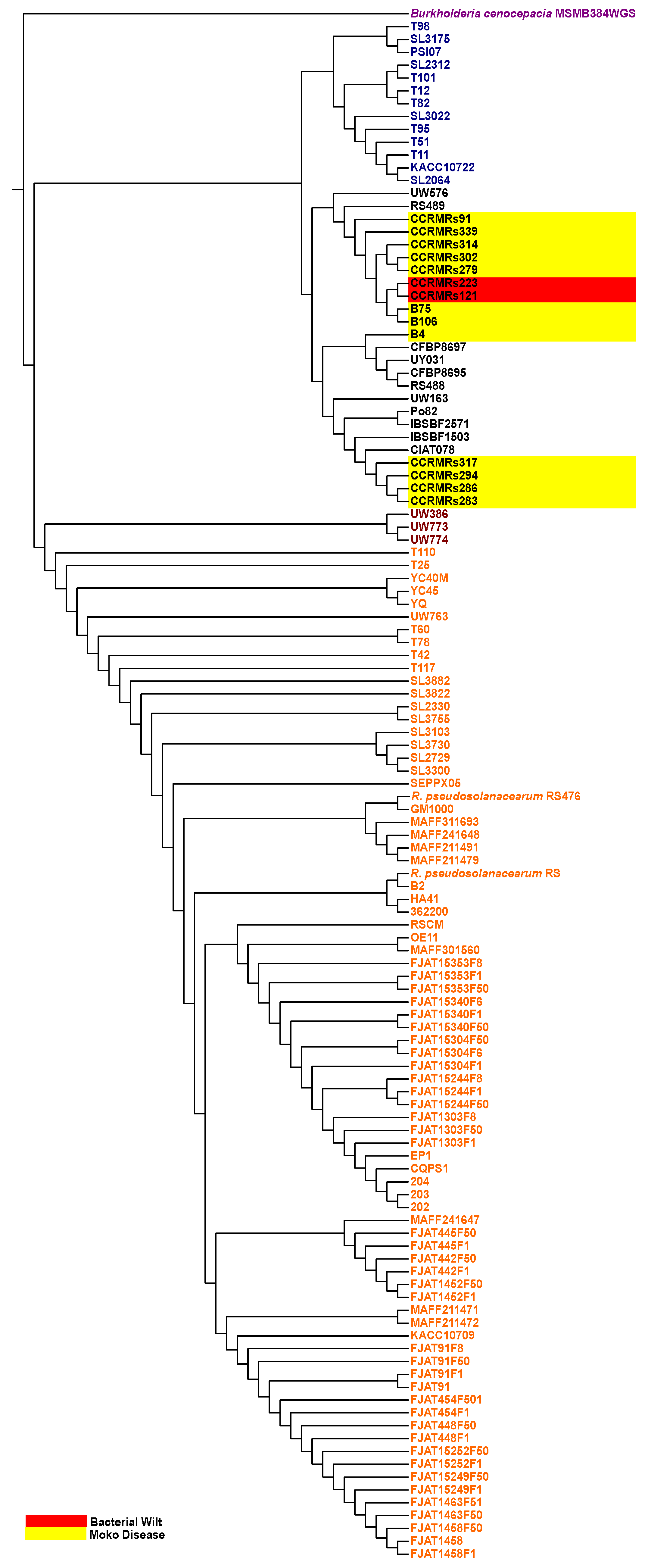
3.1. Genome Sequencing and Characterization of New Brazilian RSSC Isolates
3.2. RSSC Pangenome and Genomic Taxonomy of Newly Sequenced R. solanacearum Isolates
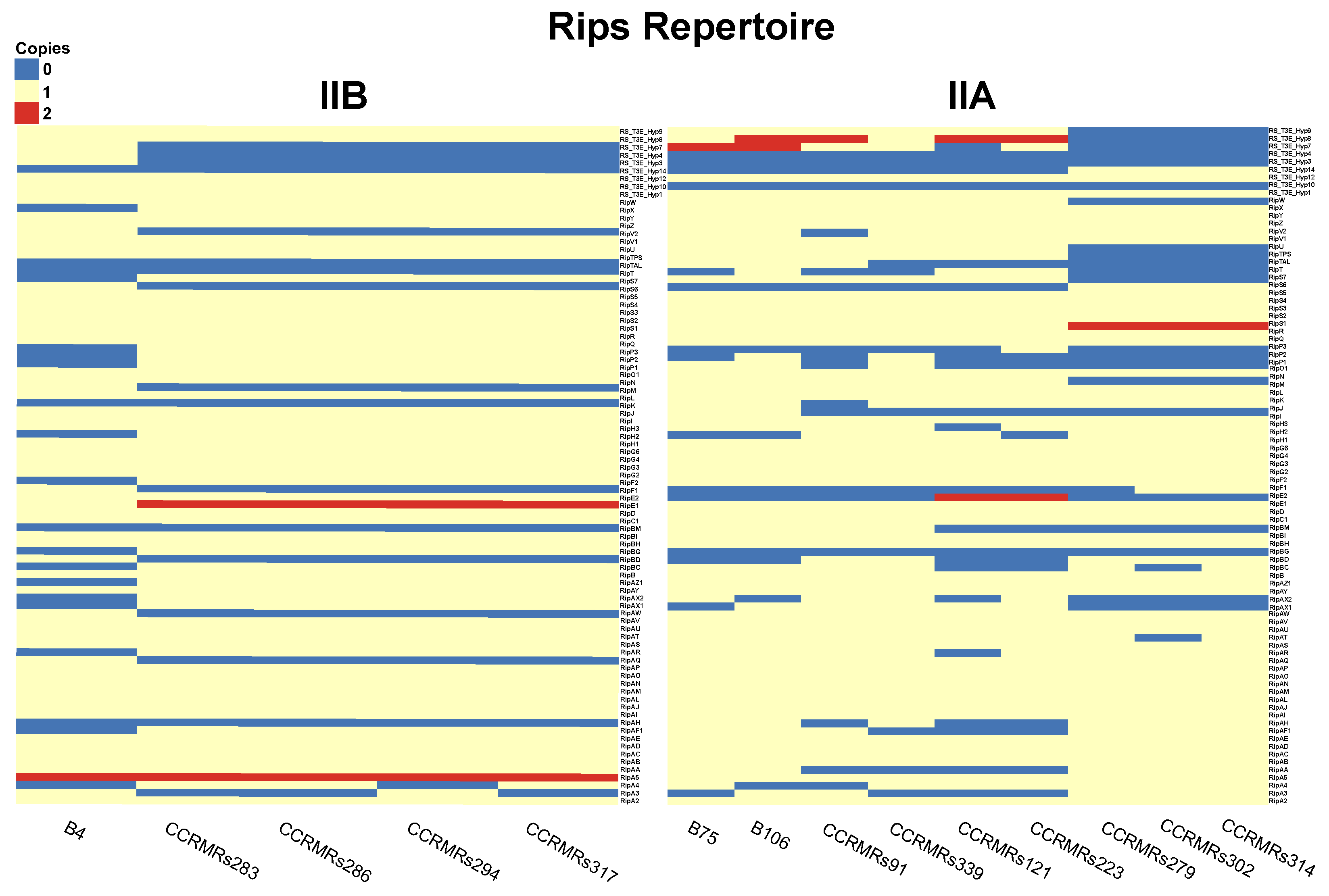
3.3. Prediction of Rips Repertoire of R. solanacearum Strains and Ecotype Correlation
4. Discussion
4.1. Pangenome and Nucleotide Identity Analysis Reveal Global Misclassification of RSSC Isolates in Public Databases and Genetic Diversity of New Brazilian Isolates
4.2. Rips Repertoire of Brazilian Isolates Are More Correlated to Genomic Similarity Rather Than Ecotype
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| BW | Bacterial Wilt |
| Rips | Ralstonia Injected Proteins |
| T3Es | Type III Effectors |
| MDPI | Multidisciplinary Digital Publishing Institute |
| DOAJ | Directory of open access journals |
| TLA | Three letter acronym |
| LD | Linear dichroism |
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strains | GenBank | Bioproject | Origin | Host | Disease |
|---|---|---|---|---|---|
| R. pseudosolanacearum RS | NZ_CP046674 | PRJNA594457 | China (YN) | Tobacco | Bacterial wilt |
| R. pseudosolanacearum RS476 | NZ_CP021762 | PRJNA388859 | Brazil (MA) | Tomato | Bacterial wilt |
| R. solanacearum B106 | JAIVFC000000000 | PRJNA763940 | Benjamin Constant, AM, BR | Banana | Moko disease |
| R. solanacearum B4 | JAIVEX000000000 | PRJNA763940 | Anamã, AM, BR | Banana | Moko disease |
| R. solanacearum B75 | JAIVFE000000000 | PRJNA763940 | Tefé, AM, BR | Banana | Moko disease |
| R. solanacearum CCRMRs121 | JAIVEU000000000 | PRJNA763940 | Belém de São Francisco, PE, BR | Tomato | Bacterial wilt |
| R. solanacearum CCRMRs223 | JAIVEY000000000 | PRJNA763940 | Bezerros, PE, BR | Tomato | Bacterial wilt |
| R. solanacearum CCRMRs279 | JAIVFD000000000 | PRJNA763940 | Manicoré, AM, BR | Banana | Moko disease |
| R. solanacearum CCRMRs283 | JAIVEZ000000000 | PRJNA763940 | Benjamin Constant, AM, BR | Banana | Moko disease |
| R. solanacearum CCRMRs286 | JAIVEV000000000 | PRJNA763940 | Benjamin Constant, AM, BR | Banana | Moko disease |
| R. solanacearum CCRMRs294 | JAIVEW000000000 | PRJNA763940 | Benjamin Constant, AM, BR | Banana | Moko disease |
| R. solanacearum CCRMRs302 | JAIVFA000000000 | PRJNA763940 | Fonte Boa, AM, BR | Banana | Moko disease |
| R. solanacearum CCRMRs314 | JAIVFB000000000 | PRJNA763940 | Tefé, AM, BR | Banana | Moko disease |
| R. solanacearum CCRMRs317 | JAIVFF000000000 | PRJNA763940 | Tefé, AM, BR | Banana | Moko disease |
| R. solanacearum CCRMRs339 | JAIVET000000000 | PRJNA763940 | Coari, AM, BR | Banana | Moko disease |
| R. solanacearum CCRMRs91 | JAIVFG000000000 | PRJNA763940 | Igreja Nova, AL, BR | Banana | Moko disease |
| R. solanacearum 202 | NZ_CP049789 | PRJNA609910 | China (GD) | Tobacco | Bacterial wilt |
| R. solanacearum 203 | NZ_CP049791 | PRJNA609906 | China (GD) | Tobacco | Bacterial wilt |
| R. solanacearum 204 | NZ_CP049793 | PRJNA609905 | China (GD) | Tobacco | Bacterial wilt |
| R. solanacearum 362200 | NZ_CP065531 | PRJNA668065 | China (FJ) | Peanut | Bacterial wilt |
| R. solanacearum B2 | NZ_CP049787 | PRJNA609907 | China (GD) | Tobacco | Bacterial wilt |
| R. solanacearum CFBP8695 | CP047138 | PRJNA596809 | Iran | Potato | Bacterial wilt |
| R. solanacearum CFBP8697 | CP047136 | PRJNA596668 | Iran | Potato | Bacterial wilt |
| R. solanacearum CIAT078 | NZ_CP051295 | PRJNA608676 | Colombia | Plaintain | Moko disease |
| R. solanacearum CQPS1 | NZ_CP016914 | PRJNA331070 | China (SD) | Tobacco | Bacterial wilt |
| R. solanacearum EP1 | NZ_CP015115 | PRJNA288736 | China (GD) | Eggplant | Bacterial wilt |
| R. solanacearum FJAT1303F1 | NZ_CP052128 | PRJNA622642 | China (FJ) | Tomato | Bacterial wilt |
| R. solanacearum FJAT1303F50 | NZ_CP052126 | PRJNA622642 | China (FJ) | Tomato | Bacterial wilt |
| R. solanacearum FJAT1303F8 | NZ_CP052130 | PRJNA622642 | China (FJ) | Tomato | Bacterial wilt |
| R. solanacearum FJAT1452F1 | NZ_CP052124 | PRJNA622642 | China (FJ) | Tomato | Bacterial wilt |
| R. solanacearum FJAT1452F50 | NZ_CP052122 | PRJNA622642 | China (FJ) | Tomato | Bacterial wilt |
| R. solanacearum FJAT1458 | NZ_CP016554 | PRJNA329182 | China (FJ) | Tomato | Bacterial wilt |
| R. solanacearum FJAT1458F1 | NZ_CP052120 | PRJNA622642 | China (FJ) | Tomato | Bacterial wilt |
| R. solanacearum FJAT1458F50 | NZ_CP052118 | PRJNA622642 | China (FJ) | Tomato | Bacterial wilt |
| R. solanacearum FJAT1463F50 | NZ_CP052114 | PRJNA622642 | China (FJ) | Tomato | Bacterial wilt |
| R. solanacearum FJAT1463F1 | NZ_CP052116 | PRJNA622642 | China (FJ) | Tomato | Bacterial wilt |
| R. solanacearum FJAT15244F1 | NZ_CP052112 | PRJNA622642 | China (FJ) | Tomato | Bacterial wilt |
| R. solanacearum FJAT15244F50 | NZ_CP052110 | PRJNA622642 | China (FJ) | Tomato | Bacterial wilt |
| R. solanacearum FJAT15244F8 | NZ_CP059376 | PRJNA647244 | China (FJ) | - | - |
| R. solanacearum FJAT15249F1 | NZ_CP052108 | PRJNA622642 | China (FJ) | Tomato | Bacterial wilt |
| R. solanacearum FJAT15249F50 | NZ_CP052106 | PRJNA622642 | China (FJ) | Tomato | Bacterial wilt |
| R. solanacearum FJAT15252F1 | NZ_CP052104 | PRJNA622642 | China (FJ) | Tomato | Bacterial wilt |
| R. solanacearum FJAT15252F50 | NZ_CP052102 | PRJNA622642 | China (FJ) | Tomato | Bacterial wilt |
| R. solanacearum FJAT15304F1 | NZ_CP052100 | PRJNA622642 | China (FJ) | Tomato | Bacterial wilt |
| R. solanacearum FJAT15304F50 | NZ_CP052098 | PRJNA622642 | China (FJ) | Tomato | Bacterial wilt |
| R. solanacearum FJAT15304F6 | NZ_CP052096 | PRJNA622642 | China (FJ) | Tomato | Bacterial wilt |
| R. solanacearum FJAT15340F1 | NZ_CP052094 | PRJNA622642 | China (FJ) | Tomato | Bacterial wilt |
| R. solanacearum FJAT15340F50 | NZ_CP052092 | PRJNA622642 | China (FJ) | Tomato | Bacterial wilt |
| R. solanacearum FJAT15340F6 | NZ_CP052090 | PRJNA622642 | China (FJ) | Tomato | Bacterial wilt |
| R. solanacearum FJAT15353F1 | NZ_CP052088 | PRJNA622642 | China (FJ) | Tomato | Bacterial wilt |
| R. solanacearum FJAT15353F50 | NZ_CP052086 | PRJNA622642 | China (FJ) | Tomato | Bacterial wilt |
| R. solanacearum FJAT15353F8 | NZ_CP052084 | PRJNA622642 | China (FJ) | Tomato | Bacterial wilt |
| R. solanacearum FJAT442F1 | NZ_CP052082 | PRJNA622642 | China (FJ) | Tomato | Bacterial wilt |
| R. solanacearum FJAT442F50 | NZ_CP052080 | PRJNA622642 | China (FJ) | Tomato | Bacterial wilt |
| R. solanacearum FJAT445F1 | NZ_CP052078 | PRJNA622642 | China (FJ) | Tomato | Bacterial wilt |
| R. solanacearum FJAT445F50 | NZ_CP052076 | PRJNA622642 | China (FJ) | Tomato | Bacterial wilt |
| R. solanacearum FJAT448F1 | NZ_CP052074 | PRJNA622642 | China (FJ) | Tomato | Bacterial wilt |
| R. solanacearum FJAT448F50 | NZ_CP052072 | PRJNA622642 | China (FJ) | Tomato | Bacterial wilt |
| R. solanacearum FJAT454F1 | NZ_CP052070 | PRJNA622642 | China (FJ) | Tomato | Bacterial wilt |
| R. solanacearum FJAT454F501 | NZ_CP060701 | PRJNA622642 | China (FJ) | Tomato | Bacterial wilt |
| R. solanacearum FJAT91 | NZ_CP016612 | PRJNA329188 | China (FJ) | Tomato (healthy) | Bacterial wilt |
| R. solanacearum FJAT91F1 | NZ_CP056083 | PRJNA640736 | China (FJ) | - | - |
| R. solanacearum FJAT91F50 | NZ_CP052068 | PRJNA622642 | China (FJ) | Tomato | Bacterial wilt |
| R. solanacearum FJAT91F8 | NZ_CP056085 | PRJNA622642 | China (FJ) | Tomato | Bacterial wilt |
| R. solanacearum GM1000 | NC_003295 AL646057-AL646075 | PRJNA13 | - | Arabdopsis thaliana | Bacterial wilt |
| R. solanacearum HA41 | NZ_CP022481 | PRJNA392775 | China (HB) | Peanut | Bacterial wilt |
| R. solanacearum IBSBF1503 | NZ_CP012943 | PRJNA297402 | Brazil | Pepino | NPB (non-pathogenic to banana) |
| R. solanacearum IBSBF2571 | NZ_CP026307 | PRJNA431203 | Brazil (SE) | Plaintain | Moko disease |
| R. solanacearum KACC10709 | NZ_CP016904 | PRJNA314721 | South Korea (GY) | Potato | Bacterial wilt |
| R. solanacearum KACC10722 | NZ_CP014702 | PRJNA314571 | South Korea (JE) | Potato | Bacterial wilt |
| R. solanacearum MAFF211471 | NZ_AP024097 | PRJDB10588 | Japan (Kochi) | Ginger | Bacterial wilt |
| R. solanacearum MAFF211472 | NZ_AP024157 | PRJDB9507 | Japan (Kyushu) | Ginger | Bacterial wilt |
| R. solanacearum MAFF211479 | NZ_AP024099 | PRJDB10588 | Japan (Kochi) | Ginger | Bacterial wilt |
| R. solanacearum MAFF211491 | NZ_AP024101 | PRJDB10588 | Japan (Kochi) | Ginger | Bacterial wilt |
| R. solanacearum MAFF241647 | NZ_AP024105 | PRJDB10588 | Japan (Kochi) | Ginger | Bacterial wilt |
| R. solanacearum MAFF241648 | NZ_AP024107 | PRJDB10588 | Japan (Kochi) | Ginger | Bacterial wilt |
| R. solanacearum MAFF301560 | NZ_AP024103 | PRJDB10588 | Japan (Kochi) | Ginger | Bacterial wilt |
| R. solanacearum MAFF311693 | NZ_AP024161 | PRJDB9507 | Japan (Kyushu) | Wild turmeric | Bacterial wilt |
| R. solanacearum OE11 | NZ_CP009763 | PRJDB4012 | Japan (Kochi) | Eggplant | Bacterial wilt |
| R. solanacearum Po82 | NC_017574 | PRJNA66837 | Mexico | Potato | Bacterial wilt/Moko disease |
| R. solanacearum PSI07 | NC_014310 | PRJEA50683 | - | Tomato | Bacterial wilt |
| R. solanacearum RS488 | NZ_CP021652 | PRJNA388430 | Brazil (PR) | Tomato | Bacterial wilt |
| R. solanacearum RS489 | NZ_CP021766 | PRJNA388980 | Brazil (PR) | Tomato | Bacterial wilt |
| R. solanacearum RSCM | NZ_CP025985 | PRJNA422474 | China (GD) | Pumpkin | Bacterial wilt |
| R. solanacearum SEPPX05 | NZ_CP021448 | PRJNA379485 | China (JX) | Sesame | Bacterial wilt |
| R. solanacearum SL2064 | NZ_CP022798 | PRJNA396777 | South Korea (JE) | Potato | Bacterial wilt |
| R. solanacearum SL2312 | NZ_CP022796 | PRJNA396777 | South Korea (JE) | Potato | Bacterial wilt |
| R. solanacearum SL2330 | NZ_CP022794 | PRJNA396777 | South Korea (JE) | Potato | Bacterial wilt |
| R. solanacearum SL2729 | NZ_CP022792 | PRJNA396777 | South Korea (JE) | Potato | Bacterial wilt |
| R. solanacearum SL3022 | CP023016 | PRJNA396777 | South Korea (JE) | Potato | Bacterial wilt |
| R. solanacearum SL3103 | NZ_CP022790 | PRJNA396777 | South Korea (JE) | Potato | Bacterial wilt |
| R. solanacearum SL3175 | NZ_CP022788 | PRJNA396777 | South Korea (JE) | Potato | Bacterial wilt |
| R. solanacearum SL3300 | NZ_CP022786 | PRJNA396777 | South Korea (JE) | Potato | Bacterial wilt |
| R. solanacearum SL3730 | NZ_CP022784 | PRJNA396777 | South Korea (JE) | Potato | Bacterial wilt |
| R. solanacearum SL3755 | NZ_CP022782 | PRJNA396777 | South Korea (JE) | Potato | Bacterial wilt |
| R. solanacearum SL3822 | NZ_CP022780 | PRJNA396777 | South Korea (JE) | Potato | Bacterial wilt |
| R. solanacearum SL3882 | NZ_CP022778 | PRJNA396777 | South Korea (JE) | Potato | Bacterial wilt |
| R. solanacearum T101 | NZ_CP022757 | PRJNA396777 | South Korea (JE) | Potato | Bacterial wilt |
| R. solanacearum T11 | NZ_CP022776 | PRJNA396777 | South Korea (JE) | Potato | Bacterial wilt |
| R. solanacearum T110 | CP023012 | PRJNA396777 | South Korea (JE) | Potato | Bacterial wilt |
| R. solanacearum T117 | NZ_CP022755 | PRJNA396777 | South Korea (JE) | Potato | Bacterial wilt |
| R. solanacearum T12 | NZ_CP022774 | PRJNA396777 | South Korea (JE) | Potato | Bacterial wilt |
| R. solanacearum T25 | CP023014 | PRJNA396777 | South Korea (JE) | Potato | Bacterial wilt |
| R. solanacearum T42 | NZ_CP022772 | PRJNA396777 | South Korea (JE) | Potato | Bacterial wilt |
| R. solanacearum T51 | NZ_CP022770 | PRJNA396777 | South Korea (JE) | Potato | Bacterial wilt |
| R. solanacearum T60 | NZ_CP022768 | PRJNA396777 | South Korea (JE) | Potato | Bacterial wilt |
| R. solanacearum T78 | NZ_CP022765 | PRJNA396777 | South Korea (JE) | Potato | Bacterial wilt |
| R. solanacearum T82 | NZ_CP022763 | PRJNA396777 | South Korea (JE) | Potato | Bacterial wilt |
| R. solanacearum T95 | NZ_CP022761 | PRJNA396777 | South Korea (JE) | Potato | Bacterial wilt |
| R. solanacearum T98 | NZ_CP022759 | PRJNA396777 | South Korea (JE) | Potato | Bacterial wilt |
| R. solanacearum UW163 | NZ_CP012939 | PRJNA297400 | Peru (NA) | Plaintain | Moko disease |
| R. solanacearum UW386 | NZ_CP039339 | PRJNA531204 | Nigeria | Soil | - |
| R. solanacearum UW576 | NZ_CP051175 | PRJNA591018 | Senegal | Tomato | Bacterial wilt |
| R. solanacearum UW763 | NZ_CP051173 | PRJNA591018 | Senegal | Tomato | Bacterial wilt |
| R. solanacearum UW773 | NZ_CP051171 | PRJNA591018 | Senegal | Tomato | Bacterial wilt |
| R. solanacearum UW774 | NZ_CP051169 | PRJNA591018 | Senegal | Tomato | Bacterial wilt |
| R. solanacearum UY031 | NZ_CP012687 | PRJNA278086 | Uruguay | Wild potato | Brown rot |
| R. solanacearum YC40M | NZ_CP015850 | PRJNA314427 | China (GD) | Galangal | Bacterial wilt |
| R. solanacearum YC45 | CP011997 | PRJNA286156 | China (GD) | Ginger | Bacterial wilt |
| R. solanacearum YQ | NZ_CP059489 | PRJNA648113 | China (ZJ) | Casuarina pine | Bacterial wilt |

References
- Manda, R.R.; Addanki, V.A.; Srivastava, S. Bacterial wilt of solanaceous crops. Int. J. Chem. Stud. 2020, 8, 1048–1057. [Google Scholar] [CrossRef]
- Yuliar, N.; Nion, Y.A.; Toyota, K. Recent trends in control methods for bacterial wilt diseases caused by Ralstonia solanacearum. Microbes Environ. 2015, 30, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Guji, M.J.; Yetayew, H.T.; Kidanu, E.D. Yield loss of ginger (Zingiber officinale) due to bacterial wilt (Ralstonia solanacearum in different wilt management systems in Ethiopia. Agric. Food Secur. 2019, 8, 5. [Google Scholar] [CrossRef]
- Lopes, C.A.; Rossato, M. History and Status of Selected Hosts of the Ralstonia solanacearum Species Complex Causing Bacterial Wilt in Brazil. Front. Microbiol. 2018, 9, 1228. [Google Scholar] [CrossRef]
- Genin, S.; Boucher, C. Lessons learned from the genome analysis of ralstonia solanacearum. Annu. Rev. Phytopathol. 2004, 42, 107–134. [Google Scholar] [CrossRef]
- Costa, K.D.D.S.; dos Santos, P.R.; dos Santos, A.M.M.; Silva, A.M.F.; Chagas, J.T.B.; de Carvalho Filho, J.L.S.; Pereira, J.W.d.L.; Silva, M.D.O.; da Silva, J.R.; Menezes, D. Genetic control of tomato resistance to Ralstonia solanacearum. Euphytica 2019, 215, 136. [Google Scholar] [CrossRef]
- Santiago, T.R.; Lopes, C.A.; Caetano-Anollés, G.; Mizubuti, E.S.G. Phylotype and sequevar variability of Ralstonia solanacearum in Brazil, an ancient centre of diversity of the pathogen. Plant Pathol. 2017, 66, 383–392. [Google Scholar] [CrossRef]
- Genin, S.; Denny, T.P. Pathogenomics of the Ralstonia solanacearum Species Complex. Annu. Rev. Phytopathol. 2012, 50, 67–89. [Google Scholar] [CrossRef]
- Safni, I.; Cleenwerck, I.; De Vos, P.; Fegan, M.; Sly, L.; Kappler, U. Polyphasic taxonomic revision of the Ralstonia solanacearum species complex: Proposal to emend the descriptions of Ralstonia solanacearum and Ralstonia syzygii and reclassify current R. syzygii strains as Ralstonia syzygii subsp. syzygii subsp. nov., R. solanacearum phylotype IV strains as Ralstonia syzygii subsp. indonesiensis subsp. nov., banana blood disease bacterium strains as Ralstonia syzygii subsp. celebesensis subsp. nov. and R. solanacearum phylotype I and III strains as Ralstonia pseudosolanacearum sp. nov. Int. J. Syst. Evol. Microbiol. 2014, 64, 3087–3103. [Google Scholar] [CrossRef]
- Zhang, Y.; Qiu, S. Phylogenomic analysis of the genus Ralstonia based on 686 single-copy genes. Antonie Van Leeuwenhoek 2016, 109, 71–82. [Google Scholar] [CrossRef]
- Santiago, T.R.; Lopes, C.A.; Mizubuti, E.S. Diversidade e Variabilidade de Ralstonia spp. In Estado da Arte em Fitobacterioses Tropicais; EDFRPE-Editora Universitária da UFRPE: Recife, Pernambuco, Brasil, 2016; pp. 243–256. [Google Scholar]
- Albuquerque, G.M.R.; Santos, L.A.; Felix, K.C.S.; Rollemberg, C.L.; Silva, A.M.F.; Souza, E.B.; Cellier, G.; Prior, P.; Mariano, R.L.R. Moko Disease-Causing Strains of Ralstonia solanacearum from Brazil Extend Known Diversity in Paraphyletic Phylotype II. Phytopathology® 2014, 104, 1175–1182. [Google Scholar] [CrossRef] [PubMed]
- Sabbagh, C.R.R.; Carrere, S.; Lonjon, F.; Vailleau, F.; Macho, A.P.; Genin, S.; Peeters, N. Pangenomic type III effector database of the plant pathogenic Ralstonia spp. PeerJ 2019, 7, e7346. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Dahal, A.; Zhang, Y.; Rokunuzzaman, M.; Kiba, A.; Hikichi, Y.; Ohnishi, K. Involvement of avirulence genes avrA and popP1 of Japanese Ralstonia solanacearum strains in the pathogenicity to tobacco. Physiol. Mol. Plant Pathol. 2018, 102, 154–162. [Google Scholar] [CrossRef]
- Bocsanczy, A.M.; Bonants, P.; van der Wolf, J.; Bergsma-Vlami, M.; Norman, D.J. Identification of candidate type 3 effectors that determine host specificity associated with emerging Ralstonia pseudosolanacearum strains. Eur. J. Plant Pathol. 2022, 163, 35–50. [Google Scholar] [CrossRef]
- Chen, L.; Lei, N.; Kiba, A.; Hikichi, Y.; Ohnishi, K. Contribution of RipS type III effector family of Ralstonia solanacearum Japanese strain OE1-1 to disease development in eggplant. J. Gen. Plant Pathol. 2021, 87, 77–82. [Google Scholar] [CrossRef]
- Moon, H.; Pandey, A.; Yoon, H.; Choi, S.; Jeon, H.; Prokchorchik, M.; Jung, G.; Witek, K.; Valls, M.; McCann, H.C.; et al. Identification of RipAZ1 as an avirulence determinant of Ralstonia solanacearum in Solanum americanum. Mol. Plant Pathol. 2021, 22, 317–333. [Google Scholar] [CrossRef]
- Landry, D.; González-Fuente, M.; Deslandes, L.; Peeters, N. The large, diverse, and robust arsenal of Ralstonia solanacearum type III effectors and their in planta functions. Mol. Plant Pathol. 2020, 21, 1377–1388. [Google Scholar] [CrossRef]
- Bioinformatics, B. FastQC: A Quality Control Tool for High Throughput Sequence Data; Babraham Institute: Cambridge, UK, 2011. [Google Scholar]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, D.L.; Ariute, J.C.; Rodrigues, F.; Benko-Iseppon, A.; Barh, D.; Azevedo, V.; Aburjaile, F. PanViTa-Pan Virulence and resisTance Analysis. Front. Bioinform. 2023, 3, 1–4. Available online: https://github.com/dlnrodrigues/panvita (accessed on 26 July 2020). [CrossRef] [PubMed]
- Liu, B.; Zheng, D.; Zhou, S.; Chen, L.; Yang, J. VFDB 2022: A general classification scheme for bacterial virulence factors. Nucleic Acids Res. 2022, 50, D912–D917. [Google Scholar] [CrossRef] [PubMed]
- Pal, C.; Bengtsson-Palme, J.; Rensing, C.; Kristiansson, E.; Larsson, D.J. BacMet: Antibacterial biocide and metal resistance genes database. Nucleic Acids Res. 2014, 42, D737–D743. [Google Scholar] [CrossRef]
- Peeters, N.; Carrère, S.; Anisimova, M.; Plener, L.; Cazalé, A.C.; Genin, S. Repertoire, unified nomenclature and evolution of the Type III effector gene set in the Ralstonia solanacearum species complex. BMC Genom. 2013, 14, 859. [Google Scholar] [CrossRef]
- Ailloud, F.; Lowe, T.; Cellier, G.; Roche, D.; Allen, C.; Prior, P. Comparative genomic analysis of Ralstonia solanacearum reveals candidate genes for host specificity. BMC Genom. 2015, 16, 270. [Google Scholar] [CrossRef]
- Page, A.J.; Cummins, C.A.; Hunt, M.; Wong, V.K.; Reuter, S.; Holden, M.T.; Fookes, M.; Falush, D.; Keane, J.A.; Parkhill, J. Roary: Rapid large-scale prokaryote pan genome analysis. Bioinformatics 2015, 31, 3691–3693. [Google Scholar] [CrossRef]
- Hadfield, J.; Croucher, N.J.; Goater, R.J.; Abudahab, K.; Aanensen, D.M.; Harris, S.R. Phandango: An interactive viewer for bacterial population genomics. Bioinformatics 2018, 34, 292–293. [Google Scholar] [CrossRef]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Costa, S.S.; Guimarães, L.C.; Silva, A.; Soares, S.C.; Baraúna, R.A. First Steps in the Analysis of Prokaryotic Pan-Genomes. Bioinform. Biol. Insights 2020, 14, 1177932220938064. [Google Scholar] [CrossRef] [PubMed]
- Tettelin, H.; Riley, D.; Cattuto, C.; Medini, D. Comparative genomics: The bacterial pan-genome. Curr. Opin. Microbiol. 2008, 11, 472–477. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, L.; Glover, R.H.; Humphris, S.; Elphinstone, J.G.; Toth, I.K. Genomics and taxonomy in diagnostics for food security: Soft-rotting enterobacterial plant pathogens. Anal. Methods 2016, 8, 12–24. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Carbasse, J.S.; Peinado-Olarte, R.L.; Göker, M. TYGS and LPSN: A database tandem for fast and reliable genome-based classification and nomenclature of prokaryotes. Nucleic Acids Res. 2022, 50, D801–D807. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Auch, A.F.; Klenk, H.P.; Göker, M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinform. 2013, 14, 60. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Hahnke, R.L.; Petersen, J.; Scheuner, C.; Michael, V.; Fiebig, A.; Rohde, C.; Rohde, M.; Fartmann, B.; Goodwin, L.A.; et al. Complete genome sequence of DSM 30083T, the type strain (U5/41T) of Escherichia coli, and a proposal for delineating subspecies in microbial taxonomy. Stand. Genom. Sci. 2014, 9, 2. [Google Scholar] [CrossRef]
- Cellier, G.; Remenant, B.; Chiroleu, F.; Lefeuvre, P.; Prior, P. Phylogeny and Population Structure of Brown Rot- and Moko Disease-Causing Strains of Ralstonia solanacearum Phylotype II. Appl. Environ. Microbiol. 2012, 78, 2367–2375. [Google Scholar] [CrossRef]
- Paudel, S.; Dobhal, S.; Alvarez, A.M.; Arif, M. Taxonomy and Phylogenetic Research on Ralstonia solanacearum Species Complex: A Complex Pathogen with Extraordinary Economic Consequences. Pathogens 2020, 9, 886. [Google Scholar] [CrossRef]
- Sharma, P.; Johnson, M.A.; Mazloom, R.; Allen, C.; Heath, L.S.; Lowe-Power, T.M.; Vinatzer, B.A. Meta-analysis of the Ralstonia solanacearum species complex (RSSC) based on comparative evolutionary genomics and reverse ecology. Microb. Genom. 2022, 8, 000791. [Google Scholar] [CrossRef]
- Geng, R.; Cheng, L.; Cao, C.; Liu, Z.; Liu, D.; Xiao, Z.; Wu, X.; Huang, Z.; Feng, Q.; Luo, C.; et al. Comprehensive Analysis Reveals the Genetic and Pathogenic Diversity of Ralstonia solanacearum Species Complex and Benefits Its Taxonomic Classification. Front. Microbiol. 2022, 13, 854792. [Google Scholar] [CrossRef]
- Mergeay, M.; Monchy, S.; Vallaeys, T.; Auquier, V.; Benotmane, A.; Bertin, P.; Taghavi, S.; Dunn, J.; van der Lelie, D.; Wattiez, R. Ralstonia metallidurans, a bacterium specifically adapted to toxic metals: Towards a catalogue of metal-responsive genes. FEMS Microbiol. Rev. 2003, 27, 385–410. [Google Scholar] [CrossRef] [PubMed]
- Pugazhendhi, A.; Boovaragamoorthy, G.M.; Ranganathan, K.; Naushad, M.; Kaliannan, T. New insight into effective biosorption of lead from aqueous solution using Ralstonia solanacearum: Characterization and mechanism studies. J. Clean. Prod. 2018, 174, 1234–1239. [Google Scholar] [CrossRef]
- Dang, F.; Lin, J.; Chen, Y.; Li, G.X.; Guan, D.; Zheng, S.J.; He, S. A feedback loop between CaWRKY41 and H2O2 coordinates the response to Ralstonia solanacearum and excess cadmium in pepper. J. Exp. Bot. 2019, 70, 1581–1595. [Google Scholar] [CrossRef] [PubMed]
- Sarowar, S.; Kim, Y.J.; Kim, E.N.; Kim, K.D.; Hwang, B.K.; Islam, R.; Shin, J.S. Overexpression of a pepper basic pathogenesis-related protein 1 gene in tobacco plants enhances resistance to heavy metal and pathogen stresses. Plant Cell Rep. 2005, 24, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Wicker, E.; Lefeuvre, P.; de Cambiaire, J.C.; Lemaire, C.; Poussier, S.; Prior, P. Contrasting recombination patterns and demographic histories of the plant pathogen Ralstonia solanacearum inferred from MLSA. ISME J. 2012, 6, 961–974. [Google Scholar] [CrossRef] [PubMed]
- Santiago, T.R.; Lopes, C.A.; Caetano-Anollés, G.; Mizubuti, E.S.G. Genetic Structure of Ralstonia solanacearum and Ralstonia pseudosolanacearum in Brazil. Plant Dis. 2020, 104, 1019–1025. [Google Scholar] [CrossRef]
- de Pedro-Jové, R.; Puigvert, M.; Sebastià, P.; Macho, A.P.; Monteiro, J.S.; Coll, N.S.; Setúbal, J.C.; Valls, M. Dynamic expression of Ralstonia solanacearum virulence factors and metabolism-controlling genes during plant infection. BMC Genom. 2021, 22, 170. [Google Scholar] [CrossRef]
- Ghosh, S.; O’Connor, T.J. Beyond Paralogs: The Multiple Layers of Redundancy in Bacterial Pathogenesis. Front. Cell. Infect. Microbiol. 2017, 7, 467. [Google Scholar] [CrossRef]
- Nakano, M.; Mukaihara, T. Comprehensive Identification of PTI Suppressors in Type III Effector Repertoire Reveals that Ralstonia solanacearum Activates Jasmonate Signaling at Two Different Steps. Int. J. Mol. Sci. 2019, 20, 5992. [Google Scholar] [CrossRef]
- Solé, M.; Popa, C.; Mith, O.; Sohn, K.H.; Jones, J.D.G.; Deslandes, L.; Valls, M. The awr Gene Family Encodes a Novel Class of Ralstonia solanacearum Type III Effectors Displaying Virulence and Avirulence Activities. Mol.-Plant-Microbe Interact. 2012, 25, 941–953. [Google Scholar] [CrossRef]
- Cho, H.; Song, E.S.; Heu, S.; Baek, J.; Lee, Y.K.; Lee, S.; Lee, S.W.; Park, D.S.; Lee, T.H.; Kim, J.G.; et al. Prediction of Host-Specific Genes by Pan-Genome Analyses of the Korean Ralstonia solanacearum Species Complex. Front. Microbiol. 2019, 10, 506. [Google Scholar] [CrossRef] [PubMed]
- Ailloud, F.; Lowe, T.M.; Robène, I.; Cruveiller, S.; Allen, C.; Prior, P. In planta comparative transcriptomics of host-adapted strains of Ralstonia solanacearum. PeerJ 2016, 4, e1549. [Google Scholar] [CrossRef] [PubMed]
- Cellier, G.; Prior, P. Deciphering phenotypic diversity of Ralstonia solanacearum strains pathogenic to potato. Phytopathology 2010, 100, 1250–1261. [Google Scholar] [CrossRef] [PubMed]
- Ramírez, M.; Moncada, R.N.; Villegas-Escobar, V.; Jackson, R.W.; Ramírez, C.A. Phylogenetic and pathogenic variability of strains of Ralstonia solanacearum causing moko disease in Colombia. Plant Pathol. 2020, 69, 360–369. [Google Scholar] [CrossRef]











| Isolate | Size (Mb) | Contigs | N50 | L50 |
|---|---|---|---|---|
| B106 | 5.50 | 46 | 399,454 | 5 |
| B4 | 5.85 | 50 | 574,994 | 5 |
| B75 | 5.42 | 77 | 333,179 | 6 |
| CCRMRs121 | 5.36 | 35 | 504,573 | 4 |
| CCRMRs223 | 5.57 | 53 | 296,540 | 5 |
| CCRMRs279 | 5.70 | 441 | 46,716 | 34 |
| CCRMRs283 | 5.46 | 81 | 204,913 | 8 |
| CCRMRs286 | 5.46 | 81 | 185,753 | 9 |
| CCRMRs294 | 5.47 | 81 | 204,912 | 8 |
| CCRMRs302 | 5.64 | 380 | 37,222 | 42 |
| CCRMRs314 | 5.69 | 381 | 37,222 | 42 |
| CCRMRs317 | 5.50 | 80 | 205,138 | 7 |
| CCRMRs339 | 5.50 | 249 | 238,614 | 8 |
| CCRMRs91 | 5.46 | 81 | 105,718 | 18 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ariute, J.C.; Felice, A.G.; Soares, S.; da Gama, M.A.S.; de Souza, E.B.; Azevedo, V.; Brenig, B.; Aburjaile, F.; Benko-Iseppon, A.M. Characterization and Association of Rips Repertoire to Host Range of Novel Ralstonia solanacearum Strains by In Silico Approaches. Microorganisms 2023, 11, 954. https://doi.org/10.3390/microorganisms11040954
Ariute JC, Felice AG, Soares S, da Gama MAS, de Souza EB, Azevedo V, Brenig B, Aburjaile F, Benko-Iseppon AM. Characterization and Association of Rips Repertoire to Host Range of Novel Ralstonia solanacearum Strains by In Silico Approaches. Microorganisms. 2023; 11(4):954. https://doi.org/10.3390/microorganisms11040954
Chicago/Turabian StyleAriute, Juan Carlos, Andrei Giachetto Felice, Siomar Soares, Marco Aurélio Siqueira da Gama, Elineide Barbosa de Souza, Vasco Azevedo, Bertram Brenig, Flávia Aburjaile, and Ana Maria Benko-Iseppon. 2023. "Characterization and Association of Rips Repertoire to Host Range of Novel Ralstonia solanacearum Strains by In Silico Approaches" Microorganisms 11, no. 4: 954. https://doi.org/10.3390/microorganisms11040954
APA StyleAriute, J. C., Felice, A. G., Soares, S., da Gama, M. A. S., de Souza, E. B., Azevedo, V., Brenig, B., Aburjaile, F., & Benko-Iseppon, A. M. (2023). Characterization and Association of Rips Repertoire to Host Range of Novel Ralstonia solanacearum Strains by In Silico Approaches. Microorganisms, 11(4), 954. https://doi.org/10.3390/microorganisms11040954